
2.0 J M2 0.9 - Blood Journal
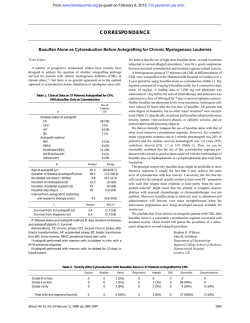
From www.bloodjournal.org by guest on February 6, 2015. For personal use only. 1351 CORRESPONDENCE Factor Vlll Gene Rearrangementin Hemophilia A Carrier Detection: A Word of Caution To the Editor: Carrier detection and prenatal diagnosis is now facilitated by the discovery that a unique inversion of the tip of chromosome X is responsible for 50% of cases of severe hemophilia A.'.*We found a similar frequency (27 of 54) of factor VI11 gene inversions in severe unrelated hemophilia A patients in Israel. The mechanism proposed for the inversion involves disruption of the factor VI11 gene and displacement of exons 1-22 to a site about 1 Mb from the 5' end of the normal gene.',' It is the purpose of this letter to show that, in carriers of the inversion, crossing over between the normal factor VI11 gene and the inverted gene may complicate diagnosis. Coagulation studies performed in 1979 in a family with a sporadic case of severe hemophilia A suggested that the sister and the mother of the patient had significant posterior probabilities of being carriers, ie, 0.7 and 0.2. respectively4 (Fig 1). Reevaluation of this family by DNA analysis of the factor VI11 gene showed that the sister of the patient inherited from her mother the non-hemophilic allele (6.2 kb) at the Xba I polymorphic site of intron 22. This finding strongly suggested that she is not a carrier. In addition, crossing over was detected between the factor VI11 gene and the extragenic markers in the intron 22 homologous regions, at DXS52 and at DXS 15 (Fig 1). More recently, Southern blot analysis for the newly discovered factor VI11 rearrangement showed the inverted factor VI11 gene pattern in the patient, his sister, his mother, and grandmother (Fig 1). However, the latter findings do not define the sister as a carrier in viewofthe crossing over found earlier. Depending on the exact location of the crossover, the sister may be a carrier if the break occurred inside the normal factor VI11 gene, for which the chances are very smatl yet not entirely negligible, particularly in view of her suggestive coagulation tests. If a recombination occurred between the normal and the inverted factor VI11 gene, the sister's maternal X chromosome would bear both the normal and the inverted factor VI11 genes and, consequently, she maynotbe a carrier. Such a recombination should occur in about 2% of such cases because this is the frequency of recombinations between markers in the factor VI11 gene and markers in intron 22 homologous regions that we found in our material (1 of 57 informative meioses) and as reported by others.' Practically, in future pregnancies, fetal blood sampling will be necessary in the sister of the patient for establishing a final diagnosis in male fetuses who will bear the inversion. Thus, the possibility of crossing over between the normal and inverted FVIII gene has to be taken into account when counselling hemophilia A families in which the severe hemophilia A is related to factor VI11 gene inversions. Hava Peretz Sali Usher Uri Martinovitz Uri Seligsohn Institute of Thrombosis and Haemostasis National Haemophilia Center Tel-Hashomer Chemical Pathology Laboratory 1 I 'P1 M1 I En I H 2.0 4.5 5.8 UI J M2 4.8 0.9 2.0 4.5 5.8 M2 4.8 0.9 2.0 4.5 5.8 1.6 5.3 5.8 M2 P2 PIIW 4.8 C.O. 0.9 2.0 4.5 5.8 Fig 1. A pedigreeof a family with a sporadic case of hemophilia A showing the results of DNA analyses. ).1 The hemophilic patient;(01 unaffected males;(0)female carriers;(01suspected carrier. The bars represent parts of the X chromosome containing the normal I O ) I factor Vlll genes. The numbers adjacent to the bars representthe lengths (in kilobases) ofthe polymotphicfragments at the following loci: 0.9/1.2-&/ I site in intron 18; 4.8/6.2-Xbs 1 site in intron 22; 5.4/-extragenic Xb8 I in intron 22 homologous region;1.6/2.O-Msp I and4.5/5.3-Taq I sites at DXS52;and 2.8/5.8--8gl site at DXS15. The family was not informative for the (CAIn polymorphism in intron 13. Two alleles of the (CAln polymorphism in intron22 were in complete linkage dimquilibtiumwith the intron 22 Xba I polymorphism. P1, P2 and M1, M2 denote the paternal and meternal haplotypes, respectively. The arrow points to the npparent region on the X chromosomes wherathe crouing over (CO) could have occurred to produce the P1/M2 haplotype. From www.bloodjournal.org by guest on February 6, 2015. For personal use only. 1352 CORRESPONDENCE False-Positive Residual Disease Assessment After Bone Marrow Transplant in Acute Lymphoblastic Leukemia To the Editor: Polymerase chain reaction(PCR) of gene rearrangements provides a sensitive widely applicable technique for the detection of minimal residual disease (MRD) in childhood acute lymphoblastic leukemia (ALL).' Generally, remission bone marrow (BM) DNA is amplified. dot blotted. and hybridized with tumor-specific probes. The signal obtained is compared with that generated by an equivalent amount of normal BM mononuclear cell DNA. Two recent reports in Blood describe the use of such a method.',' In this report. we highlight a potential pitfall of this approach that may occur at times of rebound polyclonallymphocytosissuchas that seenafterBMtransplant (BMT)." A retrospective analysis of MRD after BMT was performed in a 2-year-old boy with common ALL. He presented with a white cell @ P A treated accordingtothe count of 125 X lO"/L andwasinitially Medical Research Council UKALL x D protocol. Marrowremission was documented at day 28. A fully matched unmanipulated sibling therallograft was performed4 months after diagnosis. Conditioning apy was cyclophosphamide at 120 m g k p and single fraction total body irradiation. Cyclosporin alone was administered as graft-versus-hostdisease(GVHD)prophylaxis.Thetransplantcoursewas uneventfulandhedid not haveanymajorepisodes of sepsis or require treatment for GVHD. He remains well and in remission 3 years after BMT. 28 postinduction.at BM DNAobtainedatpresentation.atday BM harvest. and at 3.6. 12, and 18 months post-BMT was analyzed for MRD by Ig heavy chain (IgH) PCR as previously described.' Five microliters of each product was dot blotted and probed with a D28 BMH 3 6 9 12 18-2 -3 -4 -5 N N C 100bp> 82bp > P B D28 BMH 3 6 9 12 18 -2 -3 -4 -S N N C C P D28 BMH 3 6 918 12 -3 -4 -5 N N c Fig 1. PAGE analysis and hybridization. (A) An 846 nondenaturing PAGE gel showing amplified BM DNA samples collected at presentation, at day 28, at BM harvest (BMHI and at 3, 6, 12, and 18 months post-BMT. Logarithmic dilutions of tumor DNA in normal BM DNA (-2 t o -51. normal BM DNA (NI, and a no template DNA control (C) are included. (B) Dot blot analvsis of duplicate products hybridized with a '*P-labeled 20-base oligonucleotide DNJ regionprobe. (C) Electroblot analysis. Products were resolved by 8% nondenaturing PAGE and electrophoretically transferred t o nylon membranes (Hybond N+; Amersham International, Amenham, UK) with a Millipore "MilliBlot" apparatus at a currentof 4 mAlcm' for 30 minutes. Membranes were fixed in 0.4 N NaOH, neutralized in 2 x SSC, and air-dried before probe hybridization. The second band observed in the dilution lanes is artefactual and does not compromise residual disease assessment. From www.bloodjournal.org by guest on February 6, 2015. For personal use only. 1994 84: 1351-1352 Factor VIII gene rearrangement in hemophilia A carrier detection: a word of caution [letter] H Peretz, S Usher, U Martinovitz and U Seligsohn Updated information and services can be found at: http://www.bloodjournal.org/content/84/4/1351.citation.full.html Articles on similar topics can be found in the following Blood collections Information about reproducing this article in parts or in its entirety may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requests Information about ordering reprints may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#reprints Information about subscriptions and ASH membership may be found online at: http://www.bloodjournal.org/site/subscriptions/index.xhtml Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036. Copyright 2011 by The American Society of Hematology; all rights reserved.











© Copyright 2026