
Proceedings of 6th Argentinian Conference on
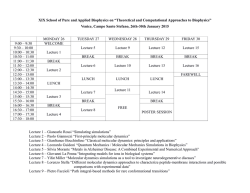
Lorenzetti, Carlos M. Proceedings of the VICAB2 C por A2 B2 C se distribuye bajo una Licencia Creative Commons AtribuciónNoComercial-SinDerivar 4.0 Internacional. Fecha de catalogación: 14/10/2015 Diseño de tapa: Lorenzetti, CM Diagramación: Lorenzetti, CM Sponsors VICAB2 C October 2015 iii 6ta Conferencia Argentina de Bioinformática y Biologı́a Computacional (VICAB2C) 6th Argentinian Conference on Bioinformatics and Computational Biology Program Committee Dr. Dr. Dr. Dr. Dr. Dr. Dr. Dr. Dr. Dr. Dr. Ariel Chernomoretz (Chair) Ana Torresi Dr. Cristina Marino Buslje Cristobal Fresno Dr. Diego Ferreiro Elmer Fernandez Dr. Estefania Mancini Fernan Agüero Dr. Hernán Dopazo Horacio Rotstein Dr. Ignacio Sánchez Leonardo Boechi Dr. Luis Diambra Luis Morelli Dr. Marcelo Marti Marisa Diana Farber Dr. Nuria Campillo Patricio Yankilevich Dr. Paula Fernandez Roxana Elin Teppa Dr. Walter Reartes Steering Committee Dr. Dr. Dr. Dr. Dr. Gustavo Parisi (A2B2C President) Ignacio Ponzoni Cristina Marino Buslje Ariel Chernomoretz Marı́a Victoria Revuelta (RSG Argentina President) Organizing Committee Dr. Ignacio Ponzoni (Chair) Dr. Jessica Carballido Dr. Mónica Dı́az Dr. Rocı́o Cecchini Dr. Cristian Gallo Dr. Carlos Lorenzetti Dr. Ana Maguitman Eng. Fiorella Cravero Eng. Julieta Dussaut Eng. Marı́a Jimena Martı́nez VICAB2 C October 2015 iv A2 B2 C Executive Commission President Vicepresident Secretary Treasure Board memberes Substitute Board memberes Audit VICAB2 C Dr. Dr. Dr. Dr. Dr. Dr. Dr. Dr. Dr. Dr. Gustavo Parisi Fernán Agüero Marı́a Silvina Fornasari Cristina Marino Buslje Elizabeth Tapia Sebastián Fernández Alberti Ariel Chernomoretz Marcel Brun Ajen Ten Have Diego Ferreiro October 2015 v Contents Page Cover Sponsors Program Committee Steering Committe Organizing Committee Executive Commission Program Invited Lectures i iii iv iv iv v viii x Ana Conesa: Unraveling novel transcriptome functional features by NGS technologies. Edgardo Ferrán: Alpha-beta switchability in protein secondary structures. Georgina Stegmayer: Machine learning for *omics data mining. Fernando Carrari: Computational Biology Tools applied to Functional Genomics in a crop model species with Biotechnological purposes. Viviana Echenique: Bioinformatic tools for understanding apomixis in Eragrostis curvula (Schrad) Nees. Silvina Ponce Dawson: Long-range interactions during intracellular Ca2+ signals. Marcelo Costabel: Where computational modelling meets molecular biophysics. Tools and data analysis in structure-function relationship. Ariel Chernomoretz: Network Biology for disease genes in next generation sequencing era. Luis Diambra: Gene networks from expression data. Damiano Piovesan: Protein function prediction in the CAFA era. Giovanni Minervini: The von Hippel-Lindau cancer syndrome: bioinformatics tales from a challenging disease. Hae Kyung Im: Whole genome prediction of gene expression levels and application to disease gene mapping. Marcel Brun: Pattern Recognition Techniques for Genomics Signal Processing. vi 1 2 3 4 5 7 8 9 10 11 12 13 14 Alejandro Giorgetti: Evidence for a Transient Additional Ligand Binding Site in Bitter Taste Receptors. Highlight Papers Oral Sessions 16 21 Sequence Analysis Functional Genomics & Metagenomics Systems Biology & Genome Organization Protein Structure & Function Prediction Poster Session 21 25 28 33 41 Sequence analysis System Biology and Networks Genome Annotation and Organization Evolution, phylogenetics and comparative genomics Genomics, functional genomics and metagenomics Metabolomics and Cheminformatics Proteomics and functional proteomics Protein Structure and Function Prediction Author Index Articles Index VICAB2 C 15 41 49 61 69 77 84 85 88 index-i index-iii October 2015 vii Conference Program Wednesday 14th 08:00 09:15 09:30 10:30 11:00 11:45 12:30 14:00 14:45 15:15 15:45 16:15 17:00 17:30 18:00 19:00 09:30 10:30 11:00 11:45 12:30 14:00 14:45 15:45 16:15 17:00 17:30 18:30 21:00 VICAB2 C Registration Opening Ceremony Opening Lecture: Ana Conesa (Univ. of Florida & CIPF) Coffee Break Invited Lecture: Edgardo Ferrán (IPBS & EBI) Invited Lecture: Georgina Stegmayer (UNL) Lunch Invited Lecture: Fernando Carrari (INTA) Invited Lecture: Viviana Echenique (UNS) Highlights Track: Sanchez-Puerta ”The mitochondrial genome of a hybrid plant” Coffee Break Invited Lecture: Silvina Ponce Dawson (UBA) Invited Lecture: Marcelo Costabel (UNS) Highlights Track: Yimi Amarillo ”Ionic Mechanisms Underlying the Subthreshold Oscillatory Properties of Thalamocortical Neurons” Roundtable Discussions. ”Exploring and redefining the scope of Bioinformatics as discipline” Posters Session & Pizza Party Thursday 15th Papers Session I: Systems Biology & Genome Organization Coffee Break Lecture: Ariel Chernomoretz (F.I.Leloir) Invited Lecture: Luis Diambra (CREG-UNLP) Lunch Invited Lecture: Damiano Piovesan (Univ. Padova) Papers Session II: Prot. Struct. & Function Pred. Coffee Break Invited Lecture: Giovanni Minervini (Univ. Padova) Highlights Track: Javier Iserte ”I-COMS: Interprotein-COrrelated Mutations Server” Papers Session III: Prot. Struct. & Function Pred. Posters Session & Drinks Conference Dinner October 2015 viii 09:30 10:30 11:00 11:45 12:15 14:00 14:45 15:45 16:15 17:15 VICAB2 C Friday 16th Papers Session IV: Sequence Analysis Coffee Break Invited Lecture: Hae Kyung Im (Univ. of Chicago) Highlights Track: Lucı́a Chemes ”Convergent Evolution and Mimicry of Protein Linear Motifs in Host-Pathogen Interactions” Lunch Invited Lecture: Marcel Brun (UNMdP) Papers Session V: Functional Genomics & Metagenomics Coffee Break Closing Lecture: Alejandro Giorgetti (Univ. Verona) Closing Ceremony & Best Posters Awards October 2015 ix Invited Lectures Unraveling novel transcriptome functional features by NGS technologies Ana Conesa Prince Felipe Research Center & Microbiology and Cell Science Department, University of Florida Valencia, Spain Wednesday 14th, 09:30hs, Main Auditorium The last 15 years of genomics research have been much dominated by the development of technologies for transcriptome profiling and for the automated functional annotation of sequence data, that have allowed the extensive study of gene expression in virtually any organism. These technological advances have tremendously expanded out understanding of the complexity of transcriptomes and the dynamics of the genomes. We moved from a rigid conception of the genomes with a large fraction of “junk” DNA and only a small percentage of functional genes, to a much more dynamic view where long range interactions and pervasive expression are part of the active genome biology. The number of RNA biotypes has expanded and the last human genome annotation indicates that the large majority of our genes are non-coding. In the last couple of years, the possibility to measure gene expression at the single-cell level, and the development of long read technologies for RNA-seq have opened new perspectives in transcriptome complexity and diversity in cellular populations, which poses new questions about the relationship between transcriptional regulation and cellular functionality. In this lecture I will cover the journey that transcriptome characterization has undergone during this period and will discuss new functional insights of gene expression made possible by the latest technological developments. Biography Currently Dr. Conesa leads the Genomics of Gene Expression Lab at the Computational Genomics Program of the Centro de Investigaciones Prı́ncipe Felipe, Valencia (Spain) and is Professor of Bioinformatics at the Microbiology and Cell Science Department at the University of Florida in Gainesville. She has recognized expertise in bioinformatics, gene expression data analysis and functional annotation. Her group has developed statistical methods and software tools that analyze the dynamics aspects transcriptomes, integrate these with other types of molecular data and annotate them functionally, with a special focus on Next Generation Sequencing (NGS) data. She is developer of software tools such as the highly-cited, field-standard Blast2GO, a suite for functional annotation of novel sequence data used by thousands scientists worldwide, Paintomics (visualization of integrated pathway data) and Qualimap (quality control of mapped NGS data). I have published statistical tools for gene expression analysis such as maSigPro (time series analysis), minAS (multivariate feature selection), ASCA-genes (multivariate multifactorial gene expression analysis), SEA (user-friendly software and functional analysis of time course gene expression data) and NOIseq (RNA-seq differential expression analysis). These bioinformatics projects have also led to an extensive set of scientific collaborations with researchers in Europe, North America, South America and South Africa to investigate gene expression in both model and non-model organisms. Leading principal investigator of several research projects that use high-throughput sequencing for functional genomic and coordinator of STATegra, a 3-year, 6 million euro (∼10 million US dollar) European FP7 funded project with eleven european and american partners, that is developing statistical tools for the integration of diverse NGS, proteomics and metabolomics data. The DEANN project is a Marie Curie Action involving 12 European and Latin American partners to create a scientific network to the study of genome variation and gene expression in endemic human, plant and animal populations using sequencing data. Other current projects deal with the development of computational approaches for the functional characterization of long noncoding RNAs and the differential functional annotation of transcript isoforms. In addition to this research Dr. Conesa has an extensive record of bioinformatics education and training. During the last five years, she has organized and taught over 20 bioinformatics short courses on five continents and collaborated in several national Master programs. She is also the founder and scientific advisor of BioBam, a bioinformatics software spin-off company that offers professional implementations of some of the tools developed at her lab. VICAB2 C October 2015 1 Alpha-beta switchability in protein secondary structures Edgardo Ferrán IPBS & EBI Wednesday 14th, 11:00hs, Main Auditorium Intriguingly, in some cases the secondary structure of some protein segments may vary between alpha-helix and beta-strand. We developed an algorithm to predict these switchable segments, based solely on the protein sequence and used the extracellular parts of the four human FGF receptors to validate the prediction. For instance, for FGFR2 we predicted that beta4 and beta5 strands of the third Ig-like domain were highly switchable. We found PDB structures confirming at least the switchability prediction for FGFR2 beta5. Interestingly, predicted switchable strands of the FGF receptors have typically a high number of somatic mutations associated with cancer. We then evaluated if chemical compounds blocking the FGFR2 receptor in one of the switchable conformations of beta5 could modulate FGFR2 signaling. To this end we selected 32 compounds from a virtual screening of a library containing 1.4 million of chemical compounds and tested them experimentally. Proliferation assays with FGF7-stimulated SNU-16 cells and a FGFR2-dependent Erk1/2 phosphorylation assay with FGFR2-transfected L6 cells, revealed that many of the selected compounds were in fact novel activators and inhibitors of FGFR2. I will finally shortly comment other possible studies based on alpha-beta switchability. References 1. Diaz C., Leplatois P., Angelloz-Nicoud P., Lecomte M., Josse A., Delpech M., Pecceu F., Loison G., Shire D., Pascal M., Ferrara P., & Ferrán E. Differential Virtual Screening (DVS) with Active and Inactive Molecular Models for Finding and Profiling GPCR Modulators: Case of the CCK1 Receptor. Mol. Inf., 30, 345-358 (2011). 2. Raby A.-C., Holst B., Le Bouder E., Diaz C., Ferrán E., Conraux L., Guillemot J-C., Coles B., Kift-Morgan A., Colmont C.S., Szakmany T., Ferrara P., Hall J.E., Topley N. & Labéta M.O. Targeting the TLR co-receptor CD14 with TLR2-derived peptides modulates immune responses to pathogens. Science Translation Medicine, Vol 5, Issue 185, 185ra64, https://doi.org/10.1126/scitranslmed.3005544 (cover article) (2013). 3. Diaz C., Herbert C., Vermat T., Alcouffe C., Bozec T., Sibrac D., Herbert JM., Ferrara P., Bono F., & Ferrán E. Virtual Screening on an Alpha-Helix to Beta-Strand Switchable Region of the FGFR2 Extracellular Domain Revealed Positive and Negative Modulators. Proteins, 82:2982–2997 (2014). Biography Dr. Ferrán is a key opinion leader and a recognized expert on computational biology, bioinformatics & systems biology. He worked for 24 years at Sanofi, one of the world’s top-5 pharmaceutical companies, after working 14 years in academia in Argentina. He is now visiting scientist at the Institute of Pharmacology and Structural Biology (IPBS, Toulouse, France), the Centre for Genomic Regulation (CRG, Barcelona Spain) and the European Bioinformatics Institute (EBI, Hinxton, UK). VICAB2 C October 2015 2 Machine learning for *omics data mining Georgina Stegmayer Research Institute for Signals, Systems and Computational Intelligence, sinc(i) FICH-UNL, CONICET Wednesday 14th, 11:45hs, Main Auditorium Biology has experienced an important data explosion. The technical advances achieved by the genomics, metabolomics, transcriptomics and proteomics technologies in recent years have significantly increased the amount of data available to study different aspects of an organism. The need to “mine” information from such data has once again become a challenge. This requires novel computational techniques and models to automatically perform data mining tasks such as integration, clustering, classification, knowledge inference and discovery, among others. In this talk, I will present the machine learning models and tools that we have developed in our research group for several *omics data mining problems: multiple sources data fusion with clustering ensembles, gene function inference with GO labels and clustering, miRNAs prediction with deep self-organizing maps, de-novo metabolic pathways synthesis with KEGG and evolutionary algorithms, gene regulatory network discovery with artificial neural networks, among others. Biography Georgina Stegmayer received the Engineering degree in Information Systems from Universidad Tecnologica Nacional Facultad Regional Santa Fe, Argentina, in 2000, and the Ph.D. degree in Electronic Devices from Politecnico di Torino, Italy, in 2006. Since 2007 she is Professor at the Department of Informatics in UTN-FRSF and UNL. She is currently Adjunct Researcher at the National Scientific and Technical Research Council (CONICET) Argentina. She is author and co-author of numerous papers on journals, book chapters and conference proceedings in artificial neural networks. Her current research interests are in the fields of data mining and pattern recognition with application to bioinformatics. VICAB2 C October 2015 3 Computational Biology Tools applied to Functional Genomics in a crop model species with Biotechnological purposes Fernando Carrari CICVyA-INTA CONICET, Universidad de Buenos Aires Wednesday 14th, 14:00hs, Main Auditorium A suite of bioinformatics tools have proved valuable to understand important traits such as crop yield and fruit development and ripening in tomato. Since the completion of the tomato genome, this species became a model for functional genomics studies. By using available computational suites we have developed a set of resources (i.e iRNA transgenic plants, VIGS protocols, miRNA databases, metabolite profile repositories) which are now available for breeding programs aiming to improve crop yield and nutritional quality of the fruits. A review of the achievements and products obtained to date will be presented in this talk. Biography Prof. Fernando Carrari is adjunct professor at the Plant Genetics and Breeding Dept of University of Buenos Aires, independent researcher of the National Council of S&T and PI at the Biotech Institute of the National Institute of Agricultural Technology (INTA). He has developed his scientific career in Buenos Aires (1997-2000 and 2005-to date) and as a visiting scientist at the Max-Planck-Institute in Germany (2000-2004). Over the past 15 years he has developed an extensive experience in the plant molecular physiology field. He recently has contributed to the sequencing of the tomato and the wild species S. pennellii genomes. VICAB2 C October 2015 4 Bioinformatic tools for understanding apomixis in Eragrostis curvula (Schrad) Nees Viviana Echenique National Scientific and Technical Research Council CERZOS - CCT Bahia Blanca Wednesday 14th, 14:45hs, Main Auditorium Apomixis, clonal reproduction by seeds, is a puzzling process focused by the scientific community for many years that has interesting evolutionary implications as well as great economical and agricultural potentials, that could significantly change agricultural practices. It probably arose from a few alterations in genes controlling plant sexuality. The switch from sex to apomixis is hypothesized to result from deregulation of developmental pathways leading to sexual seed development, and the trigger for deregulation involves the global genomic effects of hybridization and polyploidy. Our research group has been studying the molecular mechanisms involved in the reproductive behavior of Eragrostis curvula, an apomictic perennial grass widely naturalized in semiarid regions or Argentina. Among the strategies that have been chosen to dissect apomixis regulation are transcriptome sequencing and the use bioinformatic tools. Thus, initial studies were conducted on 12,295 expressed sequence tags (ESTs) generated from near-isogenic lines with different ploidy levels and reproductive modes, leading to genome coverage of 22%. ESTs were clustered and assembled in 8,864 unigenes, 79% of which were functionally categorized by BLASTX analysis against public databases, but only 38% could be classified according to Gene Ontology. A comparative expression analysis between libraries revealed that 112 unigenes were modulated by the reproductive mode or by ploidy. These genes were in silico mapped onto maize genome to identify candidates mapping to the region syntenic to the diplospory locus. As next generation sequencing (NGS) technologies became available, a reference transcriptome for this grass was obtained through a deeper sequencing by 454 GS FLX+ Roche (INDEAR, Rosario, Argentina), leading to 2,617,197 reads (952 Mbp) with an average length of 364 bp. Due to a lack of E. curvula whole-genome sequence, a de novo assembly of the high quality clean reads was conducted. Using the Newbler assembler software (v 2.6; Roche, IN, USA), 63,763 contiguous sequences were obtained and further assembled into 49,568 isotigs, the bioinformatic equivalent to unique RNA transcripts. Annotation was done by the software BLAST2GO. An in silico differential expression analysis revealed that ∼10% of the transcripts were expressed by the sexual genotype and ∼20% by the apomictic one, constituting a starting point to the in vitro validation of genes involved in apomixis regulation. The use of the software MEGAN allowed us to detect ∼5000 genes that are not represented in other plant species and that are excellent candidates to analyze. The E. curvula reference transcriptome has been also useful to develop probes to microarray design (Agilent) to identify genes expressed according reproductive mode though hybridization with mRNA from ovules. Complementary to such strategies, four small RNAs libraries were constructed though NGS from apomictic and sexual genotypes, to elucidate their relevance in apomixis expression. A total of 163.728.389 reads were obtained with a size average of 23 nt, being overrepresented as expected the sRNAs of 21 and 24nt. The software CLC Genomics Workbench (6.0.3) set to a minimum sampling count of 3, lead to 1,869,631 and 2,596,410 different tags for the sexual and apomictic libraries, respectively. The BLAST analysis of such sequences against miRNA databases revealed that 0.4% of the tags correspond to miRNAs being identified matches with only the 10% of the sequences deposited in such database. Bioinformatic tools are very useful tools for our research and will help to understand the nature of apomixis in plants. Biography Viviana Echenique obtained her degree and PhD in Biology at the Universidad Nacional del Sur, Bahia Blanca, Argentina and received postdoctoral training in different foreign Institutions like the Swiss Federal Institute of Technology (ETH) in Zürich, Switzerland, the Plant Biotechnology Centre, Australia and UCDavis, California, US. Currently is Director and Principal Investigator of CERZOS (Centro de Recursos Naturales Renovables de la Zona Semiárida CCT – CONICET Bahı́a Blanca) and Full Professor, Departamento de Agronomı́a, Universidad Nacional del Sur. The group leaded by Prof. Viviana Echenique is focused on the study of the reproductive mode of Eragrostis curvula, a polymorphic grass native to Southern Africa, member of the Poaceae and naturalized in different regions of Argentina. The group adopted several VICAB2 C October 2015 5 strategies aimed at the elucidation of genetic and/or epigenetic mechanisms involved in apomixis pathways. Another research line is aimed at the development of biotechnological tools to assist wheat breeding programs in Argentina. The group is constituted by other four researchers, two technicians and four PhD Students. In the last five years Echenique’s lab has published 20 articles in peer reviewed journals, 3 books and 8 book chapters, three patents and received funds from National (CONICET, ANPCyT, UNS) and International Institutions (DAAD, ECOS, EU). Recently the group had gained some expertise in bioinformatics by different collaborations, especially as a member of the Wheat International Sequencing Consortium. VICAB2 C October 2015 6 Long-range interactions during intracellular Ca2+ signals Silvina Ponce Dawson Departamento de Fisica FCEN-UBA e IFIBA, CONICET-UBA Wednesday 14th, 16:15hs, Main Auditorium Intracellular Ca2+ signals are used by almost all cell types for a large variety of purposes. It is believed that the information they convey is stored in the space and time distribution of the cytosolic Ca2+ concentration. In many occasions extracellular stimuli give rise to sequences of Ca2+ spikes that are usually the consequence of the (coupled) Ca2+ release from the endoplasmic reticulum through channels which open probability is an increasing function of cytosolic Ca2+. Thus, the cytosol behaves as an excitable system that is driven by the stimulus. It is then believed that the intensity of the stimulus is somehow encoded in the frequency of the spikes. The sequences of spikes, however, are not completely regular. How can the information be stored reliably in such a case? It has recently been proposed that in this type of signals the decoding of the information involves some type of fold-change detection and that there is a global feedback mechanism that reduces noise. In this talk I will focus on the latter. I will discuss to what extent cytosolic or luminal Ca2+ can provide some long range interaction that could underlie this global feedback. I will show the results of numerical simulations of a simple model and experimental results on the dynamics of Ca2+ inside the endoplasmic reticulum that indicate that there is a rapid exchange of ions in the lumen that wipes out spatial heterogeneities relatively fast during Ca2+ global release events. Biography Silvina Ponce Dawson obtained her Licenciatura in Physics and her PhD at the University of Buenos Aires working on astrophysical plasmas and turbulence. She was then post-doc at the University of Maryland and at Los Alamos National Laboratory where she worked on nonlinear dynamics and chaos and on pattern formation. She came back to Argentina where she became Professor at the Physics Department of the School of Exact and Natural Sciences, University of Buenos Aires, and researcher of CONICET. She started to do research in biological physics over fifteen years ago, from the point of view of mathematical modeling at first and more recently, doing optical experiments. Her current interests include cell signaling and information transmission in cells. Silvina has been deeply involved in the issue of women in physics. In particular, she has been part of the Working Group on Women in Physics of the International Union of Pure and Applied Physics first as the representative of Latin America and later on as chair. VICAB2 C October 2015 7 Where computational modelling meets molecular biophysics. Tools and data analysis in structure-function relationship Marcelo Costabel Universidad Nacional del Sur Departamento de Fisica Wednesday 14th, 17:00hs, Main Auditorium How do biological macromolecules perform their function? Which are the structural components responsible for this? How can we simulate these highly complex and irregular mechanisms efficiently? Progress in the aknowledgement of protein structures and advances in the understanding of the physics of macromolecules enables, nowadays, more realistic simulation techniques, which allow the study of a great number of biological mechanisms in detail. In short, there is a close interplay between theory, algorithmic progress and application. In this talk, basic principles of some aspects of computational molecular modelling are exposed, and interlinked with results obtained in our laboratory. Biography Dr. Marcelo Costabel received his Physics degree from the Universidad Nacional del Sur, Bahia Blanca and obtained a PhD at the Universidad Nacional de La Plata. After that, he did a postdoctoral stay working in protein crystallography at the Institut Pasteur, Paris, France. Back to Argentine he had a position as professor at the Physics Department of the UNS. From 2007 is Group Director of the Biophysics Group at DF-UNS/IFISUR, focusing his research in structurefunction relationships of biological macromolecules using computational tools. In particular he is interested in analysis of electrostatic interactions and Molecular Dynamics simulations, working on different collaboration projects with national colleagues and scientists abroad. VICAB2 C October 2015 8 Network Biology for disease genes in next generation sequencing era Ariel Chernomoretz University of Buenos Aires Department of Physics (FCEN) Thursday 15th, 11:00hs, Main Auditorium Complex networks are useful tools to characterize biologically sensible molecular interaction patterns and to unravel molecular mechanisms for a variety of human diseases. In this talk I will discuss some results in connection with the tolerance to deleterious germline variants and network multi-scale signatures displayed by different groups of disease genes in the context of protein interaction networks. Biography Dr. Chernomoretz obtained his PhD in Physics at the University of Buenos Aires. He spent 3 years as a Genome-Quebec postdoctoral fellow at the Bioinformatic platform of the Molecular Endocrinology and Cancer Research Center of the Laval University Medical Center (Quebec Canada). Back in Buenos Aires in 2006 he pursued his career as a full time CONICET researcher at the School of Science’s Physics Department of the University of Buenos Aires, where he currently holds a Computational Physics’s professor position. Since 2011 he also leads the Integrative Systems Biology Unit at Leloir Institute Foundation. His main interests are high-throughput transcriptomics, functional genomics, network biology and drug repurposing. VICAB2 C October 2015 9 Gene networks from expression data Luis Diambra Lab. de Biologia de Sistemas Centro Regional de Estudios Genomicos–UNLP Thursday 15th, 11:45hs, Main Auditorium One of the key issue in modern biology is the elucidation of the structure and function of gene regulatory circuits. To address this challenge many efforts have been devoted to develop computational methods able to infer gene regulatory networks. The structure of a network is defined as a graph whose nodes are associated with genes, and whose edges represent the interactions between the nodes. The task of uncovering the GRN architecture from gene-expression profiles, associated with cell states, represents a very complex inverse problem that has become central in functional genomics. In this talk I will review some emerging tools to embed steady states, and phenotypic transitions between these steady states in response to environmental cues, into the dynamics of a gene network model. Furthermore, applications for modeling parasite’s life cycle, and cell differentiation will be also discussed. Biography Luis Diambra is currently a researcher of the National Council for Scientific and Technical Research (CONICET). He graduated in 1993 at the Universidad Nacional de La Plata (UNLP) in Physics. By 1996, he obtained his PhD in Neural Networks also at the UNLP. From 2007 is Group Director of the Systems Biology Lab at CREG-UNLP. His research area is the development and application of computer simulation tools oriented to System Biology. In particular, he is interested in modeling at different scales, gene expression regulation, biological rhythms, and pattern formation. VICAB2 C October 2015 10 Protein function prediction in the CAFA era Damiano Piovesan University of Padova Department of Biomedical Sciences - DSB Thursday 15th, 14:00hs, Main Auditorium Identifying protein functions can be useful for numerous applications in biology. The prediction of Gene Ontology (GO) functional terms from sequence remains however a challenging task, as shown by the recent CAFA (Critical Assessment of protein Function Annotation, http://biofunctionprediction.org) experiments. The best methods to predict protein function were shown to rely on sequence similarity searches for conserved regions or homologous proteins. INGA (Interaction Network GO Annotator, http://protein.bio.unipd.it/inga) is one of the top ten predictors according to the CAFA-2 assessment. INGA is able to achieve a better accuracy by extending homology search with methods that use different sources of information like proteinprotein interactions or domain architecture knowledge. However, even if molecular function can be accurately predicted, the biological role of a protein remains very problematic for all automatic methods. In the future, when entire organism interactomes will become available, good quality data will result in a better capacity to generate new hypotheses. Biography After his Ph.D. research under Rita Casadio at University of Bologna, Damiano came to the Biocomputing UP group in 2013 under Silvio Tosatto at University of Padua. From 2014 he became project leader for computational methods development in the same lab. His research is focused on structural biology and protein function prediction. His work is mainly based on network analysis including proteinprotein interaction to predict protein function, residue-interaction to reveal key residues in protein structures and large scale sequence similarity graphs to infer protein relationships. VICAB2 C October 2015 11 The von Hippel-Lindau cancer syndrome: bioinformatics tales from a challenging disease Giovanni Minervini University of Padova Department of Biomedical Sciences - DSB Thursday 15th, 16:15hs, Main Auditorium Von Hippel-Lindau (VHL) syndrome is a hereditary condition predisposing to the development of different cancer forms, related to germline inactivation of the homonymous tumor suppressor pVHL. The best characterized function of pVHL is as the ubiquitin E3 ligase promoting degradation of Hypoxia Inducible Factor (HIF) via the proteasome. It acts as molecular hub, in several radically different cellular pathways, with more than 200 different interactions. While a pVHL crystal structure is available for several years, molecular details of its function remain largely unknown. Here, we present how bioinformatics can be used to shed light on the main pVHL functions. Systems biology model was used to describe the pVHL biochemical environment, while the combination of conservation analysis, molecular dynamics and phatogenicity predictions were used to characterize the molecular details of several pVHL interactors. We found that the variability of different VHL manifestations is largely correlated to the concomitant inactivation of different metabolic pathways, as well as with defects in pVHL regulator proteins. This study shows how bioinformatics can be effectively used to study a complex multi-factorial disease. Biography Giovanni Minervini is currently a postdoctoral researcher at the University of Padova. After his Ph.D. research under Prof. Fabio Polticelli at University of Roma Tre, he came to the Biocomputing UP group in 2012 under Silvio Tosatto at University of Padua. His current research projects focus on interdisciplinary applications of bioinformatics to the broad areas of cancer research. Dr. Minervini areas of expertise include molecular dynamics simulation and analysis, ab initio protein structure prediction, protein function characterization and analysis. Dr. Minervini has coauthored numerous journal publications, conference articles and book chapter in the aforementioned topics, and has received awards and grants from funding agencies including the AIRC (Italian Association for Cancer Research). VICAB2 C October 2015 12 Whole genome prediction of gene expression levels and application to disease gene mapping Hae Kyung Im University of Chicago Section of Genetic Medicine Friday 16th, 11:00hs, Main Auditorium Genome wide association studies have been very successful in identifying tens of thousands of genetic variants robustly associated with complex diseases and other traits. Most of these variants lie in non coding regions. However the biological mechanism underlying the majority of these mechanisms are not well understood. One possible mechanism by which genotype can affect phenotype is through the regulation of gene expression levels. Recent reporting that a substantial portion of the phenotypic variability is explained by variants that determine chromatin accessibility gives strong support to this idea. Motivated by these facts, we propose a method called PrediXcan that takes advantage of this mechanism to discover genes implicated in the etiology of the trait. More specifically, PrediXcan uses genetically predicted levels of gene expression and correlates them with the phenotype of interest. The prediction models of the transcriptome are trained using reference datasets where both gene expression and genotype data are available. Application of this method to the WTCCC found recapitulated many known genes but also identified novel ones. References 1. Gamazon, Eric R., Heather E. Wheeler, Kaanan P. Shah, Sahar V. Mozaffari, Keston Aquino-Michaels, Robert J. Carroll, Anne E. Eyler et al. “A gene-based association method for mapping traits using reference transcriptome data.” Nature genetics 47, no. 9 (2015): 1091-1098. Biography Hae Kyung Im is a researcher (Assistant Professor rank) at the University of Chicago developing models to sift through vast amounts of genomic data and generate knowledge that can be ultimately translated to improve the health of people. She received her doctoral degree in Statistics from the Department of Statistics at the University of Chicago focusing on spatial and environmental statistics. She also holds MS degrees in Physics (Instituto Balseiro) and Financial Mathematics (University of Chicago). In the spatial statistics area, she developed methods such as a semi-parametrically estimation method of spectral densities from irregularly spaced environmental datasets, a novel approach to efficiently approximate complex large-scale air quality model outputs, and a spatio-temporal approach for interpolating temperatures in the metropolitan Chicago area. Since 2009 she has worked on biomedical research enabling discoveries through a distinct statistical modeling perspective. Her current research focuses on prediction and dissection of complex traits integrating large-scale data from multiple sources. She collaborates actively with large national and international consortia such as Genotype Tissue Expression (GTEx) and T2D-GENES to identify scientific questions that are most pressing. VICAB2 C October 2015 13 Pattern Recognition Techniques for Genomics Signal Processing Marcel Brun Universidad Nacional de Mar del Plata Departamento de Matemática (F. de Ingenierı́a) Friday 16th, 14hs, Main Auditorium A key aspect of GSP (Genomics Signal Processing) is the correct modeling of the biological problem, for the application of pattern recognition techniques suitable to the problem to be solved. Pattern Recognition is the discipline that deals with the generation of machines that solve perceptual tasks, for which human perception is efficient, but that contain such formal complexity that require the use of models that are both descriptive and predictive. One of the most important sub-disciplines in Pattern Recognition is the classification of objects into categories, which is broadly used in GSP, for example to discriminate tissues, based on genotypic information, for diagnosis and prognosis of diseases, and as part of many data processing algorithms. Three aspects of classifier design are significant: a) the design of the decision rule, b) the selection of the features to be used, and c) the estimation of the classifier error. The scientific validity of a classifier depends on the correct design of these three stages, highly related between them. Additionally, non supervised classification techniques, or Clustering, are extensively used in the analysis of genomics data, so its correct usage and validation are of importance for a correct result. In this talk we will give an introduction of the essential concepts of Pattern Recognition, Classification, Feature Selection, Error Estimation, and its application to problems in Genomics Signal Processing. Biography Marcel Brun received his Ph.D. in computer science from Universidad de San Pablo, Brazil, in 2002. Currently, he is Professor of the Department of Mathematics in the Engineering School at UNMDP, Argentina. He does research at the Digital Image Processing Lab, of the School of Engineering, UNMdP. His areas of interest include pattern recognition applied to genomic signal processing and biomedical image processing. His past research experience include postdoctoral stages at Texas A&M University (College Station, Texas), University of Louisville (Louisville, Kentucky) and the Translational Genomics Research Institute, TGen (Phoenix, Arizona). He is the author and coauthor of more than 30 journal articles, directing PhD students in Image Processing and Bioinformatics. VICAB2 C October 2015 14 Evidence for a Transient Additional Ligand Binding Site in Bitter Taste Receptors Alejandro Giorgetti University of Verona Department of Biotechnology Friday 16th, 16:15hs, Main Auditorium Most human G protein coupled receptors (GPCRs) are activated by small molecules binding to their 7transmembrane (7-TM) helix bundle. They belong to basally diverging branches: the 25 bitter taste 2 receptors and most members of the very large rhodopsin-like/class A GPCRs subfamily. Some members of the class A branch have been suggested to feature not only an orthosteric agonist-binding site but also a more extracellular or “vestibular” site, involved in the binding process. Here we use dedicated bioinformatics tools in combination with a hybrid molecular mechanics/coarse-grained (MM/CG) molecular dynamics approach on bitter taste receptors. Three ∼ 1µs molecular simulation trajectories find two sites hosting the agonist, which together elucidate experimental data measured previously and in this work. This mechanism shares similarities with the one suggested for the evolutionarily distant class A GPCRs. It might be instrumental for the remarkably broad but specific spectrum of agonists of these chemosensory receptors. Biography Dr. Alejandro Giorgetti obtained his degree in Physics at the ’Universidad Nacional del Sur’, Bahia Blanca, Argentina. He obtained a Master in Medical Physics at the University of Buenos Aires, Argentina and a Ph.D. in Physical and Statistical Biology at the International School for Advanced Studies (SISSA), Trieste, Italy. After that, he spent three years in the Biocomputing lab. at the Universidad di Roma ’La Sapienza’ before accepting his current academic position. Alejandro Giorgetti has been appointed as Assistant Professor (tenured) of Biochemistry at the University of Verona, Italy since 2007. Since his appointment, he is the Principal Investigator of the Applied Bioinformatics Group (http: //molsim.sci.univr.it/bioinfo). He has authored/co-authored 35 peer-reviewed articles in medium/high impact factor journals. He has also been involved as co-author in the writing of five book chapters. He has also served as a reviewer for several journals. His research interests regard the protein structural bioinformatics area, in which he applies the latter computational techniques to biochemical/biological systems of utmost importance. The group is actively involved in different collaboration projects both within the national territory and with scientists abroad. The last years, he has been also appointed as Adjunct Visiting scientist (2011-2015) at the German Research School for Simulation Sciences (GRS), Juelich, Germany. VICAB2 C October 2015 15 Author Index Author Index Adrián Turjanski, 61 Agustina Arroyuelo, 94 Agustina Pascual, 25 Alejandro Ariel Icazatti Zuñiga, 40 Alejandro Fendrik, 49 Alejandro Giorgetti, 15 Alejandro Mechaly, 38 Alejandro Nadra, 71 Alejandro Soba, 51 Alexander Monzon, 33, 88 Ana Conesa, 1 Analia Seravalle, 26 Andrea Mangano, 45, 46 Andres Romanowski, 83 Anette Stryhn, 21 Angela Tissone, 17 Ariel Aptekmann, 71 Ariel Chernomoretz, 9, 27 Ariel E. Mechaly, 38 Arjen ten Have, 43, 44, 74, 92 Bianca Brun, 26 Carolina Lia Gandini, 69 Cecilia Suárez, 51 Cristian A. Yones, 23, 42 Cristian Rohr, 33 Cristina Marino-Buslje, 19, 75, 80, 87 Damiano Piovesan, 11 Daniel Koile, 63 Daniela Megrian, 72 Darı́o Fernández Do Porto, 61 David Moi, 72 Diego Anfossi, 75 Diego H. Milone, 23, 42 Diego J. Zea, 19, 75, 80, 87, 88 Diego Manuel Luna, 89 Diego U. Ferreiro, 34 Edgardo Ferrán, 2 Edita Karosiene, 21 Elin Teppa, 19 Elmer Fernandez, 82 Emiel Post Uiterweer, 82 Emilio Fenoy, 36 VICAB2 C Emmanuel Luján, 51 Ernesto Caffarena, 90 Ernesto Rotondo, 49 Esteban Hasson, 78 Esteban Hernando, 83 Estefania Mancini, 27, 83 Eugenia Nazzi, 89 Ezequiel Sosa, 61 Fabián Fay, 26 Facundo Orts, 43, 44 Federico Giri, 73 Fernán Agüero, 85 Fernando Carrari, 4 Fiorella Cravero, 84 Flávio Gomes Araújo, 28 Francisco Iaconis, 57 Franco Simonetti, 19 Georgina Stegmayer, 3, 23, 42 Germán Burguener, 61 German Mato, 17 Giovanni Minervini, 12 Guadalupe Méjico, 26 Guilherme Oliveira, 28 Guillermo Benı́tez, 76 Guillermo Marshall, 51 Guillermo Ortı́, 38 Guillermo Pratta, 30 Gustavo Gasaneo, 32, 57 Gustavo M. Somoza, 38 Gustavo Parisi, 33, 35, 48, 76, 88, 93 Gustavo Vazquez, 84 Hae Kyung Im, 13 Ignacio E. Sánchez, 20, 71 Ignacio Eguinoa, 22 Ignacio Enrique Sánchez, 22 Ignacio Ponzoni, 84 Inti A. Pagnuco, 43, 44, 54 Izinara Rosse, 28 Jan van Kan, 74 Javier Iserte, 19 Jeffrey Palmer, 16 Jordi Viñas, 38 Jorge A. Vila, 40, 94 October 2015 index-i Author Index Juan Félix Orlowski, 52 Juan Hurtado, 78 Juan Ignacio Specht, 57 Juan J. Diaz-Montana, 59 Juan Manuel Cabrera, 73 Julia Marchetti, 76 Julian Mensch, 78 Juliana Assis, 28 Julieta Bonacina, 65 Karen Gabriela Scheps, 48 Kirk Conrad, 82 Laura Kamenetzky, 28, 42, 47 Laura Lazzati, 85 Leonardo Dimieri, 32 Leonardo Lucianna, 61 Lilia Romanelli, 49 Liliana Guerra, 51 Lucas Daurelio, 67 Lucas Luciano Maldonado, 28 Lucas Maldonado, 47 Lucia Beatriz Chemes, 20 Luciana Escobar, 77 Lucila Saavedra, 65 Luis Diambra, 10, 77 Luisa Sen, 45, 46 M. Virginia Sanchez-Puerta, 16, 69 Mónica Dı́az, 84 Mónica Fabbro, 26 Marı́a Florencia Fernandez, 45 Marı́a Jimena Martı́nez, 84 Marı́a Luján Freije, 32 Marı́a Mercedes Palomino, 61 Mara Rosenzvit, 28, 47 Marcel Brun, 14, 43, 44, 54 Marcela Cucher, 28, 47 Marcela Nadal, 17 Marcelo Costabel, 8 Marcelo Golemba, 45 Marcelo Martı́, 61 Marcelo Soria, 52 Marcelo Yanovsky, 27, 83 Marcia Anahı́ Hasenahuer, 35, 48 Maria Belén Carbonetto, 26 Maria Florencia Fernandez, 46 Maria Rabaglino, 82 Maria Silvina Fornasari, 33, 48, 76, 88 Maria Victoria Revuelta, 43, 44, 74, 92 Mariana Allievi, 61 Mariano Torres Manno, 67 Mariano Vera, 92 VICAB2 C Mariela Sciara, 26 Martı́n Espariz, 67 Martı́n Váquez, 26 Marta Quaglino, 30 Massimo Andreatta, 21 Maximiliano de Sousa Serro, 63 Maximiliano Distefano, 45, 46 Michael Rasmussen, 21 Miguel A. Ré, 41 Mikhajlo Zubko, 16 Morten Nielsen, 21, 36, 63, 75 Nadia Suarez, 65 Natalia Macchiaroli, 28, 47 Nicolás Moreyra, 78 Nicolás Visacovsky, 51 Nicolas Palopoli, 58 Nicolas Stocchi, 92 Norberto Diaz-Diaz, 59 Osvaldo Martin, 40, 94 Oswaldo Tovar, 38 Pablo Aguilar, 72 Patricio German Barletta, 35 Patricio Yankilevich, 63 Paula Aulicino, 45, 46 Paula Macat, 30 Priscila Ailin, 92 R. Gonzalo Parra, 34, 89 Raúl Grigera, 90 Ricardo Espinosa Silva, 90 Richard Edwards, 58 Rocio Espada, 34 Ruben Gustavo Schlaen, 27, 83 Søren Buus, 21 Sabrina Sanchez, 83 Sandra Ruzal, 61 Santiago Carmona, 85 Santiago Revale, 26 Sebastián Fernández Alberti, 35, 93 Silvina Ponce Dawson, 7 Silvio C. E. Tosatto, 88, 93 Soledad Ochoa, 80 Soledad Romero, 26 Tadeo Enrique Saldaño, 93 Virginia Ballarin, 54 Viviana Echenique, 5 Viviana Varela, 48 Yimy Amarillo, 17 October 2015 index-ii Articles Index Articles Index 1. Invited Speakers Alpha-beta switchability in protein secondary structures, 2 Bioinformatic tools for understanding apomixis in Eragrostis curvula (Schrad) Nees, 5 Computational Biology Tools applied to Functional Genomics in a crop model species with Biotechnological purposes, 4 Evidence for a Transient Additional Ligand Binding Site in Bitter Taste Receptors, 15 Gene networks from expression data, 10 Long-range interactions during intracellular Ca2+ signals, 7 Machine learning for *omics data mining, 3 Network Biology for disease genes in next generation sequencing era, 9 Pattern Recognition Techniques for Genomics Signal Processing, 14 Protein function prediction in the CAFA era, 11 The von Hippel-Lindau cancer syndrome: bioinformatics tales from a challenging disease, 12 Unraveling novel transcriptome functional features by NGS technologies, 1 Where computational modelling meets molecular biophysics. Tools and data analysis in structure-function relationship, 8 Whole genome prediction of gene expression levels and application to disease gene mapping, 13 2. Highlight Papers Convergent Evolution and Mimicry of Protein Linear Motifs in Host-Pathogen Interactions, 20 I-COMS: Interprotein-COrrelated Mutations Server, 19 Ionic Mechanisms Underlying the Subthreshold Oscillatory Properties of Thalamocortical Neurons, 17 The mitochondrial genome of a hybrid plant, 16 3. Oral Session A step towards a unifying model for eye movements, 32 Active site pocket dynamics in the Epidermal Growth Factor Receptor Kinase, 35 ASpli: an integrative R package for the analysis of alternative splicing using RNA-Seq, 27 Bioinformatic Analysis of the Argentine Human Microbiome - A pilot study, 26 CoDNaS 2.0: a database of conformational diversity of native state in proteins, 33 Comparative and functional genomics of Chagas disease vectors, 25 Development and comparison of stability and affinity based networks, 36 Heterosis for tomato fruit polypeptide profiles assessed by permutational MANOVA, 30 Local Frustration and the Energy Landscapes of Ankyrin Repeat Proteins, 34 miRNAss: a semi-supervised approach for microRNA prediction, 23 New high-quality genome of a platyhelminth parasite: Comparative genomics of three species of the genus Echinococcus, 28 PATENA: an algorithm for the design of protein linker sequences, 22 Phylogenetic and Gene/Protein Structure Analysis of Two Paralogous Kisspeptin Receptors (Kiss2R And Kiss3R) in Pejerrey (Odontesthes bonariensis), 38 Prediction of peptide-MHC class II binding affinity with improved binding core identification; implications for the interpretation of T cell cross-reactivity, 21 Validation and Determination of nucleic acid structures from NMR 13C chemical shifts, 40 4. Poster Session A sequence and structure based model to explain information content relation with growth temperature, 71 An Integral framework for QSAR Modelling using Computational Intelligence and Visual Analytics, 84 VICAB2 C October 2015 index-iii Articles Index Analyses of foreign plastid sequences in plant mitochondria, 69 Analyses of present proteins with reduce number of amino acids support the origin of first proteins from random sequences, 76 Annotation and metabolic network construction of the Probiotic Strain Lactobacillus acidophilus ATCC 4356. Visualization in X-OMEQ platform, 61 BoLA (Bovine Leukocyte Antigens) class 1 typing from next-generation-sequence data, 63 Cluster Specific Dunn index for co-expression detection, 54 Clustering of Bacillus strains with potential plant growth promoting activity by searching specific genetic determinants, 67 Development of a computer application for the construction, visualization and analysis of glycan molecules, 94 Development of an algorithm to predict HIV-1 tropism from V3 loop sequence for BF subtype, 45 Differential expression analysis of cold tolerance adaptation in D. buzzatii by RNA-seq de novo approach, 78 Essential dynamics of cold denaturation proteins: Model frataxin Yfh1, 90 Estimation of independence between discrete and continuous data sets, 41 Evolution and Structure-Function Prediction of Eqolisins in Fungi, 92 Evomorph: Evolutionary morphometrics tool using Aegla singularis data, 73 Exploratory search strategies and Phylogenetic Analysis of Fusogenic Proteins in Eukariota, 72 Gaps matter Could protein multiple sequence alignment gaps predict protein contacts?, 87 Genome-wide analysis of acute effects of ligth on the regulation of alternative splicing in Arabidopsis thaliana, 83 Genome-wide identification of microRNAs and their targets in the zoonotic parasite Echinococcus canadensis, 47 Genomic insights into Enterococcus faecalis strains isolated from meat , 65 GFD-Net: a novel apporach for analyzing the functional dissimilarity of gene networks, 59 Improved pipeline to determine HIV-1 tropism for pyrosequencing reads using geno2pheno, 46 In silico generation of tumor invasion patterns, 51 Large-scale prediction of short linear motifs using structural information from protein-protein interactions, 58 Mathematical model for self renewal of epithelial intestinal tissues, 49 miRNAfe: a tool for feature extraction in pre-miRNA prediction, 42 MIToS.jl: Mutual Information Tools for prOtein Sequence analysis in Julia, 75 Molecular characterization of novel β-globin variants associated to dominant β-thalassemia, 48 Mutational patterns of somatic mutations for a functional classification of human cancers, 80 Positive Selection is shaping the Evolution of two Aspartic Proteinases in phytopathogenic Botrytis species, 74 Protein disorder promotes protein conformational diversity, 88 Relation between dynamically relevant residues and topological networks parameters, 93 Role of endometrium in preeclampia: A molecular signature Supported Vector Machine (SVM) based model for endometrial maturation prediction, 82 Search for new short peptide in “non-coding sequences, 77 Sequence-Structure Analysis of the WD40 Protein Family, 89 Software for creating efficient high-density tiling peptide chip designs and performing analysis of peptide microarray data for the identification of antibody epitopes, 85 The alignment of protein superfamily sequences Part I: Identifying Cluster Specific Subsequences (CSS) in Protein Families, 43 The alignment of protein superfamily sequences Part II: Multiple Alignment Touch-up Engine (MATE), 44 The mechanics of fading in the visual system, 57 Using co-occurrence networks techniques: Meta-analysis of soil metagenomic data, 52 VICAB2 C October 2015 index-iv







© Copyright 2026