
X-Linked Sideroblastic Anemia: Identification of the Mutation
From www.bloodjournal.org by guest on February 6, 2015. For personal use only. X-Linked Sideroblastic Anemia: Identification of the Mutation in the Erythroid-Specific S-Aminolevulinate Synthase Gene (ALAS2) in the Original Family Described by Cooley By Philip D. Cotter, Donald L. Rucknagel, and David F. Bishop In 1945, Thomas Cooley described the firstcases ofX-linked sideroblastic anemia (XLSA) in two brothers from a large family in which the inheritance of the disease was documented through six generations. Almost 40 years later the enzymatic defect in XLSA was identified as the deficient activity of the erythroid-specific form of S-aminolevulinate synthase (ALAS2). the firstenzyme in the heme biosynthetic pathway. To determine the nature of the mutation in the ALAS2 gene causing XLSA in Cooley's original family, genomic DNAs were isolated from two affected hemizygotes, and each ALAS2exon was PCR amplified and sequenced. A single transversion(A t o C) was identified in exon 5. The mutation predicted the substitution of leucine for phenylalanine at residue 165 (F165L) in the first highlyconserved domain of the ALAS2 catalytic core shared by all species. No other nucleotide changes were found by sequencing each of the x -LINKED sideroblastic anemia (XLSA) is the most common of several inherited forms of sideroblastic anemia. The first report of this metabolic disorder was by Thomas Cooley in 1945.' The same family was further described in 1946 by Rundles and Falls' and is the subject of this report. Since this time, more than 100 cases of XLSA have been de~cribed,~ demonstrating that XLSA is a clinically heterogeneous disorder of erythropoiesis characterized by hypochromic, microcytic erythrocytes; anemia; splenomegaly; elevated tissue and serum iron; and ringed sideroblasts in the marrow. Onset is typically in infancy or early childhood, and X-linked inheritance of the disorder has been established by pedigree analysis. Affected heterozygotes have been identified: as expected for lyonization in X-linked disorders. Carrier detection requires careful determination of erythrocyte size distribution if lyonization is markedly skewed toward activation of the normal allele. Many patients with XLSA respond to oral pyridoxine therapy of 5 to 500 mg/d. Hemoglobin levels increase by varying degrees but rarely return to normal levels. Even with near normal hemoglobin concentrations, microcytic erythrocytes remain. Although transfusions are often given to correct the severe anemia, the lack of effective erythropoiesis typically leads to iron overload, requiring chelation therapy and possibly interfering with the benefits of pyridoxine therapy by disturbing mitochondrial function. Notably, phlebotomy has proven effective in obviating the iron overload without exacerbating the anemia in several case^.^.^ 6-Aminolevulinate synthase (succinyl-coenzyme A [CoA]:glycine C-succinyltransferase [decarboxylating]; EC 2.3.1.37; [ALAS]) catalyzes the first step of heme biosynthesis and requires the B6 vitamer pyridoxal 5"phosphateto accomplish the condensation of succinyl CoA and glycine to form 6-aminolevulinic acid in the mitochondria of animal cells. Two isozymes of ALAS exist in vertebrates: a housekeeping isozyme and an erythroid-specific isozyme.8-" These tissue-specific isozymes are encoded by two separate In humans, the erythroid gene ALAS2 has been Blood, Vol 84, No 11 (December l), 1994: pp 3915-3924 11 exons, including intron/exon boundaries, 1 kb of 5"flanking and 350 nucleotides of 3"flanking sequence. The mutation introduced anMse I site and restrictionanalysis of PCRamplified genomic DNA confirmed the presence of thelesion in the two affected brothers and in three obligate heterozygotes from three generations of this family. Carrier diagnosis in one of additional family members identified the mutation of the proband's sisters. After prokaryotic expression and affinity purification of both mutantnormal and ALAS2 fusion proteins, the specific activity of the F165L mutant enzyme was about 26% of normal. The cofactor, pyridoxal 5"phosphate, activated and/or stabilized the purified mutant recombinant enzyme in vitro, consistent with the pyridoxineresponsive anemia in affected hemizygotes from thisfamily. 0 1994 b y The American Society of Hematology. mapped to the distal subregion of chromosome band Xpl l.21.14 The fact that ALAS2 is on the X chromosome and that its B,-dependent enzymatic activity is often reduced in patients with XLSA15316 pointed to ALAS2 as a candidate gene for this disorder. Recently, we reported the first mutation in ALAS2 in a patient with pyridoxine-responsive XLSA.I7 In this patient, a point mutationin exon 9, which contains the predicted pyridoxal 5"phosphate attachment site, resulted in a greater than 95% reduction of the activity of the recombinant mutant enzyme as compared with the recombinant normal enzyme. Because responsiveness of ALAS2 to pyridoxal 5"phosphate was demonstrated in vitro, the X-linked defect in ALAS2 was consistent, both genotypically and phenotypically, with XLSA in the patient. In this report, we identify and characterize a new ALAS2 mutation in three generations of the original XLSA family described by Cooley. This exon 5 point mutation results in reduced recombinant ALAS2 activity and shows activation by pyridoxal 5"phosphate in vitro. These results confirm the conclusion of Cooley and From the Department of Human Genetics, Mount Sinai School of Medicine, New York, and the Cincinnati Comprehensive Sickle Cell Center, Children ' S Hospital Medical Center, Cincinnati, OH. Submitted February 28, 1994; accepted August 3, 1994. Supported in part by a grant (R01 DK40895) from the National Institutes of Health to D.F.B. and a grant (USPHS 5P60-HL 15996) for the Comprehensive Sickle Cell Center, University of Cincinnati to D.L.R. P.D.C.is the recipient of a March of Dimes Birth Defects Foundation Predoctoral Graduate Research Training Fellowship. Address reprint requests to D.F. Bishop, PhD, Department of Human Genetics, Mount Sinai School of Medicine, New York, NY 10029. The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. section 1734 solely to indicate this fact. 0 1994 by The American Society of Hematology. 0006-4971/94/8411-0038$3.00/0 3915 From www.bloodjournal.org by guest on February 6, 2015. For personal use only. 3916 COTTER,RUCKNAGEL, AND BISHOP VI1 6 7 8 0 10 l 118 12 1713 19 14 M I Fig 1. Pedigree of the family originally reported by Cooley in 1945.' Those family members marked by an asterisk have been scrwned by PCR and Mse I restriction for the F165L mutation. The number at the upper left of some symbols indicates the age at deeth (inf, died in infancy). his associates that the defect was a congenital inability to synthesize hemoglobin in the face of ample iron stores. CASE REPORTS An extensive kindred with XLSA, now documented through eight generations (Fig l), was the first family with XLSA to be reported, initially by Cooley in 1945' and subsequently as family I1 of Rundles and Falls in 1946.' Affected males of this kindred exhibited the classic features of anemia, peripheral erythrocyte anisocytosis and poikilocytosis, splenomegaly, and a markedly elevated serum iron level.' The first female in this large pedigree was from Holland, whereas most of the American branch of the family now resides in Michigan. Before 1945, affected males in this family usually died in their first year of life, but not later than 17 years of age, due to a severe microcytic, hypochromic anemia. Transfusion therapy was initiated in patients VI-l6 and VI-l9 (Fig l), which resulted in clinical improvement.' At 28 years of age, one of these transfusiondependent males (VI-16) was studied in another laboratory'8 and was found to have hemosiderosis of the marrow with a hemoglobin level of 6.5 g/dL. Although he had been transfusion dependent for the previous 15 years, he was subsequently able to maintain a hemoglobin level of 7.8 to 9.6 g/dL on oral pyridoxine therapy (100 mg/ d)." Patient VII-21. The proband for this report was patient VII-21, a first cousin once removed of the brothers reported by Cooley' (VI16 and VI-19, Fig 1). He wasborn on March 21, 1961, at the University of Michigan Medical Center and received subsequent medical care there. At birth, he weighed 2.18 kg and hada hemoglobin concentration of 7 g/dL. Peripheral blood films showed the classic microcytic, hypochromic erythrocytes with abundant anisocytes and poikilocytes, including ovalocytes and lacrimatocytes (teardrop shaped cells) (Fig 2B). His response to vitamin B, administration and transfusion is summarized in Fig 3. Over the course of his first 21 years, the proband demonstrated a consistent but modest increase in hemoglobin concentration with vitamin B, therapy. Hemoglobin levels decreased to values of 6.5 to 7.0 g/dL when B, was withdrawn and increased to 7.5 to 9.0 g/& when B, therapy was reinstated. The mean corpuscular volume (MCV) deviated little from a median value of 56 fL/cell from 14 years of age to hismost recent evaluation at 32 years of age. His reticulocyte level ranged from 0.5% to 2.2% with no obvious relationship to therapy. At 2 years of age, results of a bone marrow examination were normal. At 19 years of age, the ferritin concentration was 450 pg/L and the serum iron 263 pg/dL, with 100% transfemn saturation. The bone marrow contained large numbers of proerythroblasts, and the bone marrow stain for iron showed abundant classic ringed sideroblasts (Fig 2C). A chromium51 red cell survival time was 21 days (normal 26 to 30 days). Slight splenomegaly wasnoted for the first time at 19 years of age by scanning, giving a sp1een:liverratio of 2.2: 1; compatible with hypersplenism. Fetal hemoglobin concentration was 1.8%, and hemoglobin A2 was 1.8% (normal 2.3% to 3.3%). At 21.5 years of age the liver edge was palpable 6 cm below the costal margin and the span was 10 cm. The spleen descended 6 cm below the left costal margin. Because of exertional dyspnea, a transfusion program was begun to maintain the hemoglobin concentration between 9 and 12 g/dL. At 25 years of age, with a ferritin concentration of 3,700 p g L , desfemoxamine chelation was begun. Three years later, with the development of retinopathy and type I diabetes, the transfusion and chelation therapy were discontinued. At 32 years of age, increasing the pyridoxine to 500 mg/d produced no hematologic improvement. Patient VII-22. Patient VII-22, born 15 months after his brother, VU-21, weighed 2.32 kg and had a hemoglobin concentration of 8 g/dL. He was allegedly placed on pyridoxine intradermally at birth, but as with his brother, by 1 month of age the hemoglobin concentration had decreased to 5.2 g/dL (hematocrit 21.5%, reticulocyte count 1.9%, MCV 56.5 %/cell). He was transfused once and continued to From www.bloodjournal.org by guest on February 6, 2015. For personal use only. INHERITANCE OF ANALAS2 A MUTATION IN XLSA L I - B C 3917 receive intradermal pyridoxine hydrochloride (25 mg/d) until 9 years of age. He did not showas clear a responseto B6 therapyas did his brother.Uptotheageof3years, his hemoglobinconcentration / & with B6 therapy, and from age 3 to 9 years the averaged 9.3 g average hemoglobin was 9.0g/&. In the subsequent8 years without Bstherapy, the average hemoglobin concentration was 8.5 g/&. The reticulocyte level varied between 0.6% and2.4%, with no apparent explanation for the fluctuation. The spleen became palpable at 3.5 years of age. At 4 years of age his height was at the tenth percentile, his weight at the third percentile. At 5 years of age, when an interatrial septal defect was repaired, the spleen was palpablecm4 below the costal margin. At 7 years of age, when an orchiopexy was performed, his height and weight were at the 25th percentile. At age 17, bone marrow examination showed a picture similar to his brother’s witherythroid hyperplasia and large numbers of proerythroblasts and ringed sideroblasts.His serum iron level was 275p&&, saturation 100%. His ferritin level was 240 p@. At 18 years of age, regular transfusions were begun because of exercise intolerance. Two years later, liver and spleen were palpable 10 cm below the costal margins. Because of dramatically increased transfusion requirements, splenectomy was performed at 20.5 years of age. Subsequently his hemoglobin decreased from 8.5 to6.7 g/&. He then experienced repeated episodesof deep venous thrombosis, although his platelet count was only 575,000cells/&. He transferred his medical care elsewhere from the Universityof Michigan and continued to have venous thrombosis and pulmonary emboli, some of which were life threatening. A bone marrow transplant was performed at another institution, and hedied from graft-versus-host disease at 23 years of age. Other relatives. During the past 20 years the blood of over 25 members of this family were studied by one of us (D.L.R.). Eight obligate heterozygotes had normal hemoglobin concentrations, although their erythrocytes contained a microcytic, hypochromic subpopulation.Forexample,theproband‘smother(VI-5)showeda dimorphic film with only a few pale, small cells and an occasional g / & and an MCV poikilocyte, with a hemoglobin level around 12.3 of101 %/cell (Fig 2A). By automated red cell size analyses, the microcytes comprised approximately 10% of the red cell mass with an MCV of 50 &/cell. MATERIALS AND METHODS Fig 2. Morphology of peripheral erythrocytes and marrow erythroblasts in XLSA. (A) Peripheral blood film from VI-5, the mother of the proband, showing a dimorphic erythrocyte population. The majority of erythrocytes are normal with two microcytic, hypochromic cells visible (original magnification x 313). (B) Peripheral blood film from the probandWI-21. The erythrocytes are characteristically hypochromic and microcytic.Note the presence of ovalocytes, lacrimatocytes, and anisocytes (original magnification x 313). (C) Bone marrow film from the proband, Vll-21, stained with Ped‘s. Shown are characteristic ringed sideroblasts. These are erythroblastswith red-stained nudei andPrussianblue-stainedirongranulescorresponding to iron-ladenmitochondriain a perinuclear distribution (originalmagnification x313, panel C isprinted with 1 . 4 ~more magnification than panelsA and B). Histochemistry. PeripheralfilmswerepreparedwithWright’s stain, and the bone marrow film was analyzed for iron by Perl’s stain. In the latter, potassium ferrocyanide reacts withfemc iron to form Prussian blue, and neutral red dye counterstains the nuclei red. The cytoplasm and hemoglobin are not stained. Photography was on Ektachrome or Fuji IS0 64tungsten film using a Nikon Labophot2 microscope with a Fluor 100/1.30 oil immersion objective, a2.5X projection lens and 1.2% tube magnification for 3X magnification 13 before photographic enlargement. Molecular analysisof the ALAS2 gene. Genomic DNA from the proband (W-21) and otherfamilymemberswasextractedfrom peripheral blood, cultured lymphoblasts, and other tissues according to standard techniques.lg DNA from the proband’s deceased brother (VU-22)wasextractedfromhisspleenwhichhadbeenkeptat -20°Cby one ofus(D.L.R.) since his splenectomy.Subcloning and sequencing of the polymerase chain reaction (PCR)-amplified as preALAS2geneexonsandflankingregionswasperformed viously described.” Each 100-pL PCR reaction mixture contained 1 pg of genomic DNA, 100 pm01 each of the sense and antisense oligonucleotides containingEcoRI sites, 50 pmol/L each dNTP, 50 mmom KCI, 10 mmol/L Tris-HC1 (pH 9.0), 0.1% Triton X-100, 1.5 mmol/L MgCl,, and 2.5 U Taq polymerase (Promega, Madison, From www.bloodjournal.org by guest on February 6, 2015. For personal use only. 3918 COTTER, RUCKNAGEL, AND BISHOP 20 1 3 25 4 .. 24 5 23 6 22 19 21 20 Age (YO Fig 3. Hematologic profile of the proband, Vll-21, from 0 to 6 and l 9 to 25 years of age. For vitamin Bs administration the solid bars are 25 mgld intradermal or oral PLP and the grey bars are 25 mgld intradermal pyridoxine. Transfusion of 2 U every 2 to 4 weeks are indicated by dark grey bars. WI). The reaction mixture was incubated at 94°C for 4 minutes, then 30 cycles of amplification were performed in a PTC-100 Programmable Thermal Cycler (MJ Research, Watertown, MA) with denaturation at 94°Cfor 30 seconds, primer annealing at the melting temperature (t,) of the oligonucleotide pairs for 30 seconds, and extension at 72°C for I minute. All PCR products were restricted and subcloned into pGEM-4Z (Promega). ALAS2 exon 5 was amplified (annealing temperature 62°C) with the following primers: sense 5' AGACTAGCCAGGGAGAGACT 3' and antisense 5' GCCGCCGAATTCTTTCCATGTGTGGTT"C 3'. Sequences were confirmed in multiple subclones if differences were found from our published sequence." The numbering of the complementary DNA (cDNA) previously usedt7has been corrected by adding five nucleotides (nt) to include an upstream in-frame methionine identified in the genomic sequence (Bishop, unprlhlished observations) and not present in the reported cDNA clone." Primer extension studies (Bishop, unpublished observations) identified this as the first methionine after the CAP site, confirming the result of Cox et al.'3 Products from PCR amplification of ALAS2 exon 5 were restricted with 4 U Mse I (New England Biolabs, Beverly, MA). All restriction digests were electrophoresed in 2% agarose (Ultra Pure grade, GIBCO BRL, Gaithersburg, MD) gels containing 0.1 pg/ml ethidium bromide and photographed on a Fotodyne UV Transilluminator with Polaroid Type 57 film. Protein homology comparisons and secondary structure predictions were performed withthe PILEUP program of the Genetics Computer Group (GCG) Sequence Analysis Software Package"'and with the Predictprotein program,*' respectively. Expression ofthe nomulandF165LAUS2 enzymes. Recombinantnormal and mutant ALAS2 were generated using the pMAL fusion protein expression system (New England Biolabs). resulting in maltose binding protein (MBP) attached tothe N-terminus of ALAS2 with a Factor Xa-cleavable linkage. The EcoRl fragment from pGEM4Z-AE2" was initially subcloned into EcoRI-restricted pMAL-c2 (New England Biolabs) to yield the clone pMALc2-AEI. The 5' endof the ALAS2 portion of the fusion protein was engi- neered by PCR to start at Asp 79 as determined by homology to the predicted mouse erythroid ALAS N-terminus.",'3 PCR amplification was accomplished as previously described, using I O ng of template DNA (pGEM4Z-AE2) and oligonucleotides: sense 5' GATGGGAAGAGCAAGATTGTG 3' and antisense 5' GCCGCCAAGCTTCTCGAGGGGACAGATGGC 3' (annealing temperature 56°C). PCR products were blunt-ended using T4 DNA polymerase by incubating for 15 minutes at15°Cin 50 mmol/L NaCI, 10 mmol/L Tris-HCI, 10 mmol/L MgClz, 1 mmol/L DTT, 125 pmol/L of each dNTP and 3 U T4 DNA polymerase (New England Biolabs). The enzyme was inactivated by incubating at 100°C for 5 minutes. The blunt-ended PCR products were restricted with Xho 1, gel-purified, and cloned into Xmn IIXho I-restricted andgel-purified pMALc2-AEI. The F165L mutation was initially introduced into the pET5a-AE2I7clone by site-directed mutagenesis using the PCR ovcrlap mcthodZ4to yield the clone pET5a-AE7. The Xtnn YXho 1 fragment from pET5aAE7was cloned into Xmn IIXho I-restricted and gel-purified pMALc2-AE2. The resulting clones, pMALc2-AE2 and pMALc2AE7, were sequence confirmed. The pMALc2-AE2 and pMALc2-AE7 expression constructs were transfected into Escherichiu coli BL2 I , pLysS (Novagen, Madison, WI) and overnight cultures grown in LB (GIBCO BRL) media with 50 pglmL ampicillin (Sigma, St Louis, MO) and 34 pglml chloramphenicol (Sigma). The next day, 100-mL cultures in LBlampicillin media were initiated with a 1:40 dilution of the overnight cultures and grown to 0.4 A, units. Inductionwith 0.4 mmol/L isopropyl 0-D-thiogalactopyranosidewas performed in LBIampicillinmedia for 4 hours at 37°C. Cells were pelleted at 4 , 0 0 0 ~for 15 minutes, resuspended in 5 mL of SO mmol/L HEPESlpH7.5, 1 mmol/L EDTA, 5 mmol/L DTT,and frozen at -7OT for at least 1 hour. MgCIZ,RNase A (Sigma), andDNase I (Sigma) were added to the thawed lysate to final concentrations of 1 mmol/L, 200 pgImL, and 20 pglmL, respectively. The lysate was incubated for 20 minutesat room temperature, then centrifuged at 4°C for 30 minutes at 10,000~. The supernatant was retained, filtered through a 0.45-pm filter (Gelman, Ann Arbor, MI) toyield the crude E coli extract. This was From www.bloodjournal.org by guest on February 6, 2015. For personal use only. INHERITANCE OF AN ALAS2 MUTATION IN XLSA 3919 then diluted with 3 v01 of 50 mmol/L HEPES/pH 7.5, 1 m m o l n EDTA, and 5 mmol/L DTT and applied to an amylose resin affinity column (New England Biolabs). MRP-ALAS2 fusion proteins were eluted from the column with 10 m m o l n maltose, SO mmol/L HEPESlpH 7.5, 1 mmol/L EDTA, and S mmol/L DTT and stored at -20°C. ALAS activity was assayed as previously described" with detection by Ehrlich's reagent after reaction with ethyl acetoacetate." One unit of activity is that amount of enzyme required to catalyze the production of I nmol of 6-aminolevulinate per hour under the conditions of the assay. Protein concentration was determined by a modification of the Ruorescamine method." Both the normal and F16SLfusion proteins were assayed at various concentrations of added pyridoxal S'-phosphate (PLP). RESULTS Characterization the of ALAS2 mutation. Genomic DNA from peripheral blood was isolated from patient VII2 1. Each exon of the ALAS2 gene, including 50 to 150 nt of flanking intron sequence, 1 kb of 5', and 350 nt of 3' flanking sequence, was PCR-amplifiedand sequenced. A single base change in exon 5 was the only difference from the normal sequence. This C-to-A transversion at nt 547 (Fig 4) predicted the substitution of leucine for phenylalanine at residue 165 (F165L) and introduced an Mse I site. PCRamplified exon 5 from 50 unrelated white females was restricted with Mse I and analyzed by agarose gel electrophoresis for detection of the predicted normal 325-bp or mutant 208-bp and 117-bp fragments. The absence of restriction in 1 0 0 alleles demonstrated that the mutation was not a common polymorphism (data not shown). Restriction analysis of exon 5 amplified genomic DNA from family members demonstrated 100% concordance between the presence of the Mse I site and a microcytic, hypochromic red blood cell population in affected hemizygotes and obligate heterozygotes. X-linked inheritance of the F165L mutation was demonstrated in three generations. The proband, his grandmother, mother, aunt, sister, and brother all had beenpreviously diagnosed as hemizygotes or carriers, and all had the mutation as evidenced by 208 bp and 1 17 bp Mse I restriction fragments (Fig 5). Two phenotypically normal sisters and an unaffected nephew all lacked the mutation as demonstrated by uncut 325-bp fragments (Fig 5 ) . Expression and characterization of recombinant normal and mutant fusion proteins. Recombinant normal and F165L ALAS2 proteins were expressed in E coli as fusion proteins with the maltose-binding protein (MBP) using the pMAL expression system. Previous expression of recombinant ALAS2 in the pET5a system" resulted in only a small fraction (less than 0.1%) of soluble recombinant ALAS2,the remainder being localized in inclusion bodies. Expression of recombinant ALAS2 as a fusion with the 43-kD MBP resulted in greater than 10% of the predicted 90-kD ALAS2 fusion protein remaining soluble in the supernatant. The MBP moiety also allowed partial purification by affinity chromatography using an amylose resin (Fig 6). Crude extract and affinity-purified normal and mutant ALAS2 fusion proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis in 10% gels with a 3% stacking gel according to the method of Laemmli and Favre." Approximately 1 0 0 pg of crude extract and 4 pg of affinitypurified fusion protein were applied to each well. The 14to 94-kD protein molecular weight standards (Pharmacia, Piscataway, NJ) were in the firstandlast wells (Fig 6). Although similar amounts of both recombinant normal and mutant 90-kD fusion proteins were visible in the gel for the crude extracts, the amount of F165L mutant fusion protein was reduced relative to normal for the affinity-purified material. Extracts from cells lacking the pMAL fusion construct lacked the 90-kD band (data not shown). The specific activity of the purified mutant ALAS2 was about 26% of the purified normal enzyme. Because the 90-kD mutant band was also reduced in intensity to about one third of the normal band in the affinity-purified lanes (Fig 6), it is possible that the mutation affects stability. The presence of the fusion protein on the aminoterminus of the ALAS2 protein apparently does not inhibit activity because the specific activity was unchanged after complete cleavage by factor Xa (data not shown). 3' 3' A T C i4rA G Fig4.Sequenceanalysis of ALAS2 exon 5of the proband (Vll-21) demonstrating the C-toA transversion atnt 547,predicting the substitution of leucine forphenylalanineat residue 165 (F165L). G 5' /g /% A G $f C G A T C Normal '\% C 5' From www.bloodjournal.org by guest on February 6, 2015. For personal use only. 3920 COTER, RUCKNAGEL, AND BISHOP v Q “I b8 VI11 U Std. bp 1 1 A 2 3 4 5 6 7 8 Effect of PLP on normal and mutantALAS2 activity. The affinity-purified recombinant normal and F165L fusion proteins were assayed with 0 or 5 pmol/L PLP after two and three freeze-thaw cycles (Table 1). The residual specific activity of the F165L enzyme at 0 pmol/L added PLP was 13% that of the normal enzyme and at 5 pmoVL PLP averaged 26%. The specific activity of the normal MBP-ALAS2 protein increased about 9% from 0 to 5 pmoVL added PLP. In contrast, the specific activity of the F165L MBP-ALAS2 fusion protein increased over twofold in the presence of 5 pmol/L PLP. The addition of protease inhibitors in the assay had no effect, suggesting that PLP activated or stabilized the enzymes rather than protected them against proteolysis. Homology comparison and secondary structure prediction. Homology comparisons of human ALAS2 exon 5 with 14 other ALAS sequences, using the PILEUP program of the GCG package,*’ demonstrated that the mutation occurred in a region that was well conserved (Fig 7) and that the mutated phenylalanine residue was invariant in all but the chick erythroid sequence. The beginning of exon 5 corresponds to the aminoterminus of prokaryotic ALAS sequences, suggesting that exons 5 through 1 1 constitute the essential catalytic region of the enzyme. The F165L mutation occurs within 22 residues of the beginning of exon 5 and is the most aminoterminal mutation found to date in patients with XLSA. Secondary structure predictions for the region of exon 5 surrounding the mutation were obtained with version 11.93 of the PHD program of Rost and Sander.” The expected accuracy was about 64% because the program was run without comparison to ALAS homologues. Although the normal sequence predicted a transition from &sheet to a-helix structure in the region of F165 (data not shown), the F165L mutation resulted in a prediction of all a-helix structure in this region. Although this predicted structural change was consistent with the altered phenotype, accurate correlation awaits tertiary structure determination. 9 10 Fig 5. Mse I digestionof PCR products from exon 5 of the ALAS2 gene from the proband (Vll-21) and members of his family. After Mse I digestion of PCR products from normal individuals a 325-bp band is visible. The presence ofthe F165L mutation resulted in two fragments of 208 and 117 bp. Lane 1 is the size standard (Hae 111 digestion of /X174). Lanes 2 to 4 and 7 are carrier females. Lanes 5 and 6 are the hemizygous males (Vll-22 and Vll-21). Lanes 8 to 10 are normal individuals.Pedigreenumberscorresponding to Fig 1 are above each symbol. DISCUSSION In 1945, Cooley described “a severe type of hereditary anemia with elliptocytosis.”’ This first reported case of what is now known as XLSA has been shown here to be caused by a point mutation in the ALAS2 gene. An F165L mutation was identified in the proband (VII-21) and his affected brother (VII-22). descendants of the family members originally studied by Cooley.’ In this family, the Mse I site created by the mutation provided definitive heterozygote diagnosis. All tested female family members with dimorphic erythrocyte populations were confirmed heterozygous for the F165L mutation, andall individuals tested in this pedigree with normal red cell populations lacked the F165L mutation. The X-linked inheritance of this specific ALAS2 mutation was established through three generations of this large kindred. Normaland mutant ALAS2 were expressed from the pMAL-c2 vector in E coli as maltose binding protein (MBP)ALAS2 fusion proteins and affinity purified for enzymatic characterization. Perhaps due to the fact that a substantial part of the aminoterminal region may be unnecessary for catalytic activity (absent in prokaryotes), the presence of the maltose binding protein did not significantly reduce ALAS2 catalysis: it could be cleaved with Factor Xa with no change in ALAS2 activity (data not shown). The fusion protein helped maintain the enzyme in a soluble form and facilitated affinity purification with an amylose resin column. The final specific activity of the normal enzyme, which was estimated to be about 70% pure (Fig 6), was about 20,000 U h g . This was over 100-fold more active than the homogeneous murine erythroid ALAS22’ and may reflect loss of murine ALAS2 activity during purification involving denaturation and renaturation from inclusion bodies. The effect of PLP on ALAS2 enzymatic activity wascharacterized for the normal and mutant recombinant enzymes. Assayed inthe presence of 5 p m o l n PLP, the F165L enzyme had about 26% residual specific activity relative to the normal enzyme after affinity purification.The presence of muta- From www.bloodjournal.org by guest on February 6, 2015. For personal use only. 3921 INHERITANCE OF AN ALAS2 MUTATION IN XLSA Crude Extract Affinity Purified kD 94 - 67 43 Fig 6. SDS-PAGE gel of recombinant normal and F165LMBP-ALAS2 fusion proteins. Lanes 1 and 6 are molecular weight standards. Lanes 2 and 3 are total crude lysate supernatants after induction of E coli transformed with the normal and F165L expression constructs, respectively. Lanes 4 and 5 are the purified normal and Fl65L fusion proteins after amylose affinity chromatography. ! 30 tion-specific lower molecular weight species in this preparation suggested that the mutant enzyme may be more susceptible to degradation during purification. It also may be more susceptible to inactivation by loss of the PLP moiety because addition of PLP resulted in over 110% activation as opposed to a 9% activation for the normal enzyme. Our working hypothesis is that the mutation alters the local secondary structure and possibly perturbs the overall conformation decreasing stability and/or reducing the affinity for PLP. The activating and/or stabilizing effect of PLP may be the explanation for the modest increase in hemoglobin seen in hemizygotes after pyridoxine supplementation. Additional PLP binding studies and stability experiments with homogeneous normal and mutant ALAS2 will be needed to test this hypothesis. Comparison of all reported ALAS sequences demonstrated that the phenylalanine at residue 165 was highly conserved. When the 15 published amino acid sequences for the ALAS2 and ALAS 1 isozymes were aligned in the region of the F165L mutation, phenylalanine 165 was invariant in all sequences except that for the chicken erythroid isozyme (Fig 7). The immediate context was also highly conserved as compared with flanking sequences, suggesting that this domain is important for enzyme structure and/or catalytic activity. Although there are 139 and 193 amino acids in human erythroid and hepatic ALAS I , respectively, before the beginning of the residues encoded by exon 5 , the beginning of all six of the bacterial peptides corresponds to the fourth residue of this exon (Fig 7). Thus, it is expected that phenylalanine 143 defines the minimal aminoterminal boundary of the region of the ALAS2 peptide required for catalytic activity. The F165L mutation is the most aminoterminal ALAS2 mutation identified to date and occurs in the first highly conserved domain of this catalytically competent core. The secondary structure prediction program of Rost and Sander?' predicted that the F165L mutation would introduce a local change from &sheet to a-helix in the region of the mutation. This could provide a mechanism for the observed reduced stability of the F165L recombinant ALAS2 fusion protein. Our case studies confirm and extend the original report From www.bloodjournal.org by guest on February 6, 2015. For personal use only. COTTER, RUCKNAGEL, AND BISHOP 3922 Table 1. Effect of PLP on Normal and F165L Mutant ALAS2 Enzyme Activity ALAS2 Specific Activity (U/ mg)t Experiment* 0 pmollL PLP 5.0 prnol/L PLP Increase (%l* 1 Normal ALAS2 F165L ALAS2 F165L (% of normal) 17,300 2,170 (13) 19,100 4,620 (24) 10 113 Normal ALAS2 F165L ALAS2 F165L (% of normal) 13,700 1,810 (13) 14,600 4,120 (28) 8 128 2 Experiment 1 was conducted after the enzyme samples had been frozen and thawed twice and experiment 2 after a third freeze thaw cycle. t Normal and F165L recombinant ALAS2 fusion proteins were affinity purified. Percentage increase of activity at 5 p n o l / L PLP over 0 pmol/L PLP. * by Cooley'anddemonstrate thatthisfamily is aclassic example of XLSA. Hemizygotes presented atbirth with predominantly microcytic, hypochromic erythrocytes of variable shapes and sizes, and their anemia showed a moderate response to pyridoxine therapy, as was previously demonstrated in this family." Iron overloadbecame significant with time as evidenced by abundant ringedsideroblastsin the marrow, elevated plasma ferritin, and saturated plasma transferrin. The obligate heterozygotes were uniformly dimorphic in peripheral blood cell size, although this was often a small population. This result could be due to skewed lyonization, Mutation F165L impaired hematopoiesis, or diminished survival of the erythrocytes derived from the mutant allele. Diagnosis of XLSA was aided by careful determination of MCV and/or red cell distribution width in mothers and femalerelatives of affected individuals. The moderate in vitro response of the recombinant F16SL enzyme to PLP was mirrored in the partial clinical response of affected males in this family to pyridoxine supplementation and appeared to be compromised by iron overload. For example, patient VII-21 showed repeated response to PLP or pyridoxine therapy and wasable tomaintain a hemoglobin of 8 to 9 g/dL for 16 years on 25 to 50 mg pyridoxineld and even in the absence of pyridoxine for a decade, until he was placed on a transfusion program due to exercise intolerance (Fig 3 ) . The patient's 16-year period with tolerable anemia wassignificant because previouslymales in this family, if untreated by pyridoxine or transfusion, typically died in the first year of life(Fig l). It isnotclearwhy the brothers became so anemic in the first month of life, even while receiving pyridoxine. The fluctuations in hemoglobin concentration,with or withoutpyridoxineadministration, are unexplained, as is the seemingly greater response to pyridoxine of VII-21than his brother. Their splenic involvement also differed, the spleen becoming palpableat least 3.5 years earlier in VI-22and requiringremovalaftertransfusions were initiated. Although transfusions immediately alleviated the anemia, thesubsequent increaseinmitochondrial iron stores may have compromised what limited heme synthetic capacity was available. The cause of death in XLSA patients, other than of severeanemia in infancy, is often dueto progressive iron overload.' Several reports have highlighted the value of phlebotomy to remove excess iron in XLSA patient^.'"^^^ In 7 L 0 . . . 0 0 0 . . 0 0 0 H-ry-X56352 Musery-M15268 Opsery-L02632 Humhep-X56351 Rathep-J04044 Chkhep"24366 Aspnid-X64170 Saccer"26329 Chkery-M24367 Brajap-M16751 Agrrad-P26505 Rhosph-LO7489 Rhimel-X02853 Rhocap-X53309 Rhosph-LO7490 Exon W5 Boundary Exon 5/6 Boundary 3 Fig 7. Homology between the human ALASZ'*,35 residues encoded by exon 5 and the corresponding residues in all the published ALAS sequences. From top to bottom are human erythroid mouse ALAS2,= op?anus tau erythroid ALAS"(GenBankAccession No L026321, human housekeeping ALASl'2,'3,36rat ALASl,"" chicken ALAS1,'O Aspergillusnidulans,"Saccharomycesceravisiae,u chicken erythroid ALAS2," Bradyrhizobium japonicum," Agrobscterium radiobacter/" Rhodobacter sphaeroides," Rhizobium meliloti," Rhodobacter capsula?us,"' and a secondALAS isozyme from Rhodobacter sphaeroides." The shaded region indicates identity to thenormal human ALAS2 sequence. The solid circles indicate that the amino acid is invariant and the open circles indicate one or two differences in the 15 sequences. From www.bloodjournal.org by guest on February 6, 2015. For personal use only. INHERITANCE OF AN ALAS2 MUTATION IN XLSA these cases, repeated phlebotomy resulted in reduced iron levels without adversely affecting hemoglobin concentration, indicating that stored iron was capable of being mobilized for bone marrow hemoglobin production and that the bone marrow was capable of responding to blood loss despite the limitation to e r y t h r o p ~ i e s i s . ~Significantly, ”~~~ patients who were previously refractory to pyridoxine therapy became responsive after the iron stores were d e ~ l e t e d . ~ . ~ In this and our previous study” we have demonstrated an in vitro response to PLP of the mutant recombinant enzymes consistent with the clinical response to pyridoxine seen in the patients. In addition, we have reported in abstracts the existence of four additional unrelated XLSA families with unique pyridoxine-responsive ALAS2 mutation^,^^.^' and others have reported in abstracts four mutations in five families, one of which was suggested not to be pyridoxine respon~ i v e . ~ Thus, ‘ . ~ ~ mutations causing this disorder seem to be largely private. If X-linkage is not demonstrated, hereditary sideroblastic anemia refractory to pyridoxine is most likely due to some other cause, and we recently reported exclusion of X-linkage for pyridoxine-refractory hereditary sideroblastic anemia, with evidence for autosomal i n h e r i t a n ~ e . ~ ~ In conclusion, we have reported a newmutationin the erythroid-specific ALAS2 gene in a large kindred with moderately pyridoxine-responsive XLSA and have demonstrated an in vitro activation of the F165L recombinant mutant enzyme by PLP. This was the first XLSA family to be described in the literature,’.’ and their mutation has now been identified and confirms the involvement of the X-chromosomal ALAS2 gene in the etiology of XLSA. ACKNOWLEDGMENT We thank Drs Ron Bishop, Robert Cody, Ruth Heyn, and Mark Roth at the University of Michigan for excellent care of the patients reported herein. The authors appreciate the gift of pyridoxal 5’phosphate from Dr John D. Hines. REFERENCES 1. Cooley TB: A severe type of hereditary anemia with elliptocytosis. Interesting sequence of splenectomy. Am J Med Sci 209561, 1945 2. Rundles RW, Falls H F Hereditary (?sex linked) anemia. Am J Med Sci 211:641, 1946 3. Bottomley SS: Sideroblastic anaemia. Clin Haematol 11:389, 1982 4. Lee GR, MacDiamid WD, Cartwright GE, Wintrobe MM: Hereditary, X-linked, sideroachrestic anemia. The isolation of two erythrocyte populations differing in Xg” blood type and porphyrin content. Blood 3259, 1968 5. Vogler WR, Mingioli ES: Porphyrin synthesis and heme synthetase activity in pyridoxine-responsive anemia. Blood 32:979, 1968 6. Verloop MC, Bierenga M, Diezeraad-Njoo A: Primary or essential sideroachrestic anaemias. Pathogenesis and therapy. Acta Haematol 27: 129, 1962 7. Weintraub LR, Conrad ME, Crosby WH: Iron-loading anemia. Treatment with repeated phlebotomies and pyridoxine. N Engl J Med 275:169, 1966 8. Bishop DF, Kitchen H, Wood WA: Evidence for erythroid and 3923 nonerythroid forms of 6-aminolevulinate synthetase. Arch Biochem Biophys 206:380, 1981 9. Watanabe N. Hayashi N, Kikuchi G: 6-Aminolevulinate synthase isozymes in the liver and erythroid cells of chicken. Biochem Biophys Res Commun 113:377, 1983 10. Yamamoto M, Fujita H, Watanabe N, Hayashi N, Kikuchi G: An immunochemical study of 6-aminolevulinate synthase and 6aminolevulinate dehydratase in liver and erythroid cells of rat. Arch Biochem Biophys 245:76, 1986 11. Fernindez J, Gonzilez 0, Martin M, Ruiz Ami1 M: Erythroid 5-aminolevulinate synthetase from trout (Salmo gairdneri R.). Comp Biochem Physiol [B] 85675, 1986 12. Bishop D F Two different genes encode 6-aminolevulinate synthase in humans: Nucleotide sequences of cDNAs for the housekeeping and erythroid genes. Nucleic Acids Res 18:7187, 1990 13. Cox TC, Bawden MJ, Martin A, May B K Human erythroid 5-aminolevulinate synthase: Promoter analysis and identification of an iron-responsive element in the mRNA. EMBO J 1 0 1891, 1991 14. Cotter PD, Willard HF, Gorski JL, Bishop DF: Assignment of human erythroid 6-aminolevulinate synthase (ALAS2) to a distal subregion of band Xp11.21 by PCR analysis of somatic cell hybrids containing X; autosome translocations. Genomics 13:211, 1992 15. Aoki Y, Urata G , Takaku F: 6-Aminolevulinic acid synthtase activity in erythroblasts of patients with primary sideroblastic anemia. Acta Haematol Jpn 36:74, 1973 16. Aoki Y, Urata G , Wada 0,Takaku F: Measurement of 6aminolevulinic acid synthetase activity inhuman erythroblasts. J Clin Invest 53:1326, 1974 17. Cotter PD, Baumann M, Bishop DF: Enzymatic defect in “Xlinked” sideroblastic anemia: Molecular evidence for erythroid 6aminolevulinate synthase deficiency. Proc Natl Acad Sci USA 89:4028, 1992 18. Bishop RC, Bethell W:Hereditary hypochromic anemia with transfusion hemosiderosis treated with pyridoxine. Report of a case. N Engl J Med 261:486, 1959 19. Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. 2nd ed, v01 1-3. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, 1989 20. Devereux J, Haeberli P, Smithies 0: A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res 12:387, 1984 21. Rost B, Sander C: A prediction of protein secondary structure at better than 70% accuracy. J Mol Biol 232584, 1993 22. Dierks P Molecular biology of eukaryotic 5-aminolevulinate synthase, in Dailey HA (ed): Biosynthesis of Heme and Chlorophylls. New York, McGraw-Hill, 1990, p 201 23. Ferreira GC, Dailey HA: Expression of mammalian 5-aminolevulinate synthase in Escherichia coli. Overproduction, purification, and characterization. J Bioi Chem 268:584, 1993 24. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR: Sitedirected mutagenesis by overlap extension using the polymerase chain reaction. Gene 7751, 1989 25. Bishop DF, Wood WA: An assay for 6-aminolevulinic acid synthase based on a specific, semiautomatic determination of picomole quantities of 6-[’4]aminolevulinate. Anal Biochem 80:466, 1977 26. Mauzerall D, Granick S: The occurrence and determination of 6-aminolevulinic acid and porphobilinogen in urine. J Bioi Chem 219:435, 1956 27. Bishop DF, Wampler DE, Sgouris JT, Bonefeld RI, Anderson DK, Hawley MC, Sweeley CC: Pilot scale purification of u-galactosidase A from Cohn fraction IV-l of human plasma. Biochim Biophys Acta 524:109, 1978 From www.bloodjournal.org by guest on February 6, 2015. For personal use only. 3924 28. Laemmli UK, Favre M: Maturation of the head of bacteriophage T4 I. DNA packing events. J Mol Biol 80:575, 1973 29.Bottomley SS, Healy HM, Brandenburg MA, May BK: 5Aminolevulinate synthase in sideroblastic anemias: mRNA and enzyme activity levels in bonemarrow cells. Am J Hematol 41:76, I992 30. Cotter PD, Baumann M, Rucknagel DL, Fitzsimons EJ, May A, Bishop DF: Heterogeneity in X-linked sideroblastic anemia due to unique mutations in the erythroid 6-aminolevulinic acid synthase gene. Am J Hum Genet 51:A45, 1992 3 1. Cotter PD, Bishop DF: Congenital sideroblastic anemia: Correlation of the microcytic, pyridoxine-responsive phenotype with coding region mutations in the erythroid 6-aminolevulinate synthase gene. Am J Hum Genet 53:145A, l993 32. Cox TC, Bottomley SS, Wiley JS, MayBK: Erythroid 5aminolevulinate synthase deficiency due to a point mutation in a kindred with X-linked sideroblastic anemia. Blood 80:341A, 1992 33. Bottomley SS, Wise PD, Whetsell LH, Schaefer FV: Heterogeneity of molecular defects of erythroid 5-aminolevulinate synthase in X-linked sideroblastic anemia. Blood 82:433A, 1993 34. Jardine PE, Cotter PD, Johnson SA, Fitzsimons EJ, Tyfield L, Lunt PW, Bishop D F Pyridoxine refractory congenital sideroblastic anemia with evidence for autosomal inheritance: Exclusion of linkage to ALAS2 at Xp11.21 by polymorphism analysis. J Med Genet 31:213, 1994 35. May BK, Bhasker CR, Bawden MJ, Cox TC: Molecular regulation of 5-aminolevulinate synthase. Diseases related to heme biosynthesis. Mol Biol Med 7:405, 1990 36. Schoenhaut DS, Curtis PJ: Nucleotide sequence of mouse 5aminolevulinic acid synthase cDNA and expression of its gene in hepatic and erythroid tissues. Gene 48:55, 1986 37. Bruning G, Ferkowicz M, Cornell N: Heme biosynthesis in fish and land vertebrates: Enzyme and cDNA comparisons. Biol Bull 185:327, 1993 38. Yamamoto M, Kure S, Engel JD, Hiraga K: Structure, turnover, and heme-mediated suppression of the level of mRNA encod- COTTER, RUCKNAGEL, AND BISHOP ing rat liver 6-aminolevulinate synthase. J Biol Chem 263: 15973, 1988 39. Srivastava G, Borthwick IA, Maguire DJ, Elferink CJ, Bawden MJ, Mercer JF, May BK: Regulation of 5-aminolevulinate synthase mRNA in different rat tissues. J Biol Chem 263:5202, 1988 40. Borthwick IA, Srivastava G, Day AR, Pirola BA, Snoswell MA, May BK, Elliott WH: Complete nucleotide sequence of hepatic 5-aminolaevulinate synthase precursor. Eur J Biochem 150:48 I , 1985 41. Bradshaw RE, Dixon SWC, RaittDC, Pillar TM: Isolation and nucleotide sequence of the 5-aminolevulinate synthase gene from Aspergillus nidulans. Curr Genet 23:501, 1993 42. Urban-Grimal D, Volland C, Gamier T, DehouxP, LabbeBois R: The nucleotide sequence of the HEM1 gene and evidence for a precursor form of the mitochondrial 5-aminolevulinate synthase in Succharomyces cerevisiae. Eur J Biochem 156:511. 1986 43. Riddle RD, Yamamoto M, Engel JD: Expression of b-aminolevulinate synthase in avian cells: Separate genes encode erythroidspecific and nonspecific isozymes. Proc Natl Acad Sci USA 86:792, 1989 44. McClung CR, Somerville JE, Guerinot ML, Chelm BK: Structure of the Bradyrhizobium japonicum gene hemA encoding 5aminolevulinic acid synthase. Gene 54: 133, 1987 45. Drolet M, Sasarman A: Cloning and nucleotide sequence of MolGen Genet the hernA gene of Agrobucteriumradiobucter. 226:250, 1991 46. Neidle EL, Kaplan S: Expression of the Rhodobacter sphaeroides hemA and hemT genes, encoding two 5-aminolevulinate acid synthase isozymes. J Bacteriol 175:2292, 1993 47. Leong SA, Williams PH, Ditta GS: Analysis of the 5’ regulatory region of the gene for 6-aminolevulinic acid synthetase of Rhizobium melilofi. Nucleic Acids Res 13:5965, 1985 48. Hornberger U, Liebetanz R, Tichy HV, Drews G: Cloning and sequencing of the hemA gene of Rhodobucter cupsulatus and isolation of a 6-aminolevulinic acid-dependent mutant strain. Mol Gen Genet 221:371, 1990 From www.bloodjournal.org by guest on February 6, 2015. For personal use only. 1994 84: 3915-3924 X-linked sideroblastic anemia: identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley PD Cotter, DL Rucknagel and DF Bishop Updated information and services can be found at: http://www.bloodjournal.org/content/84/11/3915.full.html Articles on similar topics can be found in the following Blood collections Information about reproducing this article in parts or in its entirety may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requests Information about ordering reprints may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#reprints Information about subscriptions and ASH membership may be found online at: http://www.bloodjournal.org/site/subscriptions/index.xhtml Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036. Copyright 2011 by The American Society of Hematology; all rights reserved.


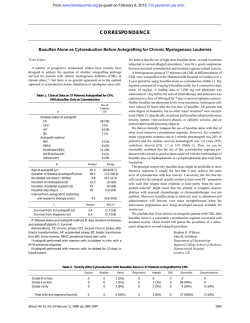









© Copyright 2026