
Tetrahedron Letters No. 36, pp 3315
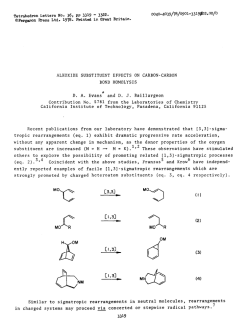
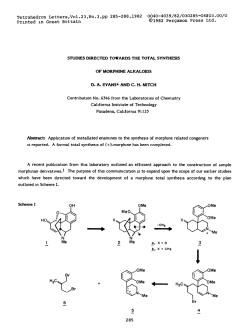
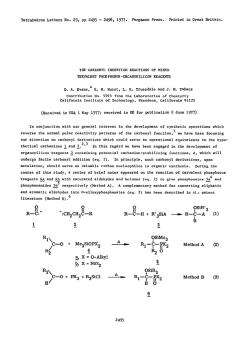
TetrahedronLettersNo. 36, pp 3315 - 3318. OPergamon Ress Ltd. 1978. Printedin Great Britain. INTRINSIC FRAGMENTATION MODES OF PRIMARY ALKOXIDES D. A. Evans* and D. J. Baillargeon Contribution No. 5808 from the Laboratories of Chemistry California Institute of Technology, Pasadena, California 91125 In the previous communication we have provided thermochemical estimates on (eq. 1). 1 The purpose of this communication is to compare the energetics of gas phase homolysis the perturbation of negatively charged oxygen on bond homolysis (eq. 1) with that of heterolysis (eq. Z), and to correlate these results with analogous processes in solution where metal counterions are known to play an important role in the mode of alkoxide fragmentation. DW -OCH2-R - -OCH2-R - -O-t&* + lR (1) + -R (2) AH; O=CH, Alkoxide fragmentation processes, both in solution 2-7 and in the gas phase, 3 have been reported by several investigators. From these studies the fragmentation mode (homolysis vs. heterolysis) appears to be defined not only by substrate alkoxide but also by counterion and solvent. In a classic study Cram demonstrated the importance of alkali metal counterions in dictating the course of alkoxide fragmentation via heterolytic or homolytic modes. (M = K, Na, Li), predominant heterolysis In the case of substrate 1 (eq. 2) was observed for M = K while homolysis (eq. 1) was preferred for M = Li.' Ph CH3 MO-k-f! Ph bh a,,2 I 3315 tvl khet ’ khom K 2.2 Na 1.6 Li 0.05 No. 36 331.6 In conjunction ments, we of metal of reaction have counterion Calculation limited for the of these highly bond dissociation (eq. 2) can employing of in effects modes, can be the primary data generalized Analogous the enthalpy for the thermochemical (EA) to the cycle and bond and the include ionic (AH;). due present for In estimating process in strengths. the fragmentation fragmentation illustrTt;d to time. the procedure absence enthalpies alkoxides to the the phase at ionic rearrange- in gas (DH”) for thermochemical affinities alkoxides radical possible alkoxides. from molecular of by estimating only approach electron Table is relevant energies, known summarized reactivity fragmentation this derived on alkoxide-promoted intrinsic energies substituted be studies the two however, more our and solvent availability principle, of with determined Scheme 1 by The results ’ are I. I Scheme AH; -0-CH,-R 0=CH2 - + +e- -R AH; / I t AH; *O-CH,-R = EA(.0CH2--R) AH; In the solvent, absence by 34, heterolysis in dependent cleavage the free homolytic Due to conclusions. of metal for At metal the of oxygen in the DH” and should are of of radical increase, curtailed coupling process alkoxides lend is not study, alkoxides. to 5 to homolysis the above on the However, as than Our therm0 out. support increasingly bond concluded rather available increments, pairing reported solution they and AH; (MOCH2-R) . by differing ion by Cram’s and magnesium data counterion in ruled bond have heterolytic be over for on the products, via and favored and Mclver’ couldn’t primary but is observation Arnett thermochemical by counterion The preference proceeding latter metal tri-tert-butylcarbinol M+, on DH” (MOCH2-R) As suggested many lithium salt AH: for time both EA1.R) 3-butenoxide with Cram’s 5 Recently, was probably present cations, counterions. preferred absence of and .R - respectively. agrees potassium although DH” and AH: values properties the ethoxide solution. decomposition estimates effect 1 in the influence 28 kcal/mol alkoxides of cleavage, chemical moderating + DH(*OCH2-RI + methoxide, and of DMSO). alkoxide the of 17, fragmentation (25”C, that of heterolysis homolysis O=CH2 - the both donor electronegative may well be 3317 No. 36 Table Calculated I. Fragmentation Primary Alkoxides, R 42 CH3 68 51 CH,CH=CH, 58 30 values 2 (Scheme 2 have been In the radical Wittig in II). to the bond rearrangement pair Scheme the homolysis covalent is such Wittig as well phase, 298OK. those discussed this carbon subject to 12*13 Thus, metal rearrangement issues. in charged alkyls as as heterolytic mechanistic intermediates fragmentation. while well-known examined rearrangement alkoxide b Gas kcal/mol. fragmentations Homolytic l2 intensively carbon-oxygen found alkoxide resemblance ethers in reported I. Mechanistically, for AH; DH”(&H2-R? 76 ‘Ref. evidence for H ‘All a striking Energies a,b -OCH2-R donor effects dissociated which similar metal Finally, have process. substituent counterion modes studies sigmatropic the II M+ C-O-R, II e- - / R2 L RI \ / R2 C=O + transfer M-R3 RI ==s provided 12 that undergo radical I MO-C-R, R2 of facilitates to alkyls + ‘R3 RI\..- bear metallated dissociation Recent [1,2] do not. of above and/or NO. 36 3318 charged intermediates accessible from metallated ethers or alkoxides are simply related by an electron transfer process. This corresponds to the single electron transfer (SET) mechanism proposed by Ashby for the addition of organometallics to 14 carbonyl compounds. Accordingly, those factors identified by Ashby which favor the SET mechanism in organometallic-carbonyl addition should be relevant to defining the course of alkoxide fragmentation. Acknowledgement. _1_-.,___5____q-.* Support from the National Science Foundation is gratefully acknowledged. REFERENCES 1. D. A. Evans and D. J. Baillargeon, Tetrahedron Lett., 0000 (1978) and references cited therein. 2. H. D. Zook, J. March, and D. F. Smith, J. Am. Chem. Sot., 53, 1617 (1959). 3. E. M. Arnett, L. E. Small, R. T. McIver, Jr., and J. S. Miller, J. Org. Chem., 43, 815 (1978) and references cited therein. 4. R: A. Benkeser M. P. Siklosi, and E. C. Mozdzen, J. Am. Chem. Sot., __100, 2134 (1978) ani references cited therein. 5. D. J. Cram, A. Langemann, W. Lwowski, and K. R. Kopecky, ibid., __, 81 5760 (1959). N. Hirota and S. I. Weissman, ibid., SC, 2538 (1964). 6. 7. G. 0. Schenck, G. Matthias, M. Pape, M. Cziesla, and G. von Bt'nau, Liebigs Ann. Chem., __I 719, 80 (1968). 8. Pertinent EA values in kcal/mol are as follow: EA(.H) = 18.9 (ref. 9); = 12.7 (ref. 11). EA(.CH3) = 1.8 (ref. 10); EA(.CH2CH=CH2) 9. D. R. Stull and H. Prophet, Ed., Natl. Stand. Ref. Data Ser., Natl. Bur. Stand ., _3_7(1971). G. B. Ellison, P. C. Engelking, and W. C. Lineberger, J. Am. Chem. Sot., 100, 2556 (1978). 10. 11. 12. 13. 14. A. ---H. Zimmerman and J. I. Brauman, ibid., 9,9,, 3565 (1977). For recent discussions of the different possible mechanisms see: u. Schollkopf, Angew. Chem., Int. Ed. Engl., 2, 763 (1970); J. F. Garst and C. D. Smith, J. Am. Chem. Sot., __ 98, 1526 (1976). H. F. Ebel, V. Db'rr, and B. 0. Wagner, Angew. Chem., Int. Ed. Engl., 2, 163 (1970). I. G. Lopp, J. D. Buhler, and E. C. Ashby, J. Am. Chem. SOC., zz, 4966 (1975) and references cited therein. (Receivedin USA 15 June 1978;received inUK for publication4 July 1978)








© Copyright 2026