
Revista Farmaindustrial-63 - Dirección de Innovación y Calidad
FI VALIDACIONES Y CERTIFICACIONES CLIMATIZACIÓN FILTRACIÓN ESTERILIZACIÓN LIMPIEZA DE EQUIPOS TRATAMIENTO DE AGUAS lifescienceslab BIOINFORMÁTICA AÑO VIII NOVIEMBRE/DICIEMBRE 2014 Nº 63 Revista Profesional para Proveedores de la Industria Farmacéutica y Tecnología de Laboratorio FI Revista Profesional para Proveedores de la Industria Farmacéutica y Tecnología de Laboratorio Estimado lector: Encaramos ya la recta final de este 2014. Pocos días quedan para alcanzar el año próximo y, para estas fechas tan especiales, traemos una edición preparada con mimo, como no podía ser de otra forma. En lo que a la revista se refiere, el equipo de Farmespaña Industrial, basándose en el feedback de los profesionales del sector, ya ha definido el programa editorial para 2015, un programa más pulido y ajustado a las demandas de los lectores, en el que hemos tratado, esperamos con éxito, de cubrir todos los temas de interés. Es muy previsible que en los próximos años asistamos a numerosos cambios (esperemos que vengan de la mano de la tan ansiada recuperación económica). Vivimos tiempos convulsos, pero estos períodos suelen ser también ricos en oportunidades; saber aprovecharlas es uno de los motores del desarrollo de cualquier sector y el farmacéutico no es una excepción. Además de las secciones especiales que dedicaremos en cada uno de los números regulares a los principales eventos del sector, hemos querido añadir una edición especial, que se publicará en noviembre y que estará centrada en logística farmacéutica, un tema de relevancia en el que incluiremos contenidos sobre envase y embalaje, etiquetado y codificación, logística y distribución… El próximo año nos traerá novedades, como la segunda edición de FarmaForum, en la que Farmespaña Industrial está implicada de forma importante y que ya ha ultimado un programa ambicioso donde se ha potenciado al máximo la participación de las principales asociaciones y empresas del sector. Desde el equipo de Farmespaña Industrial, deseamos que pasen unas felices fiestas y que entren con buen pie en el nuevo año que, seguro, nos traerá buenas noticias. Les esperamos en la próxima edición. FI TCI AÑO VIII NOVIEMBRE/DICIEMBRE 2014 Nº 63 Revista Profesional para Proveedores de la Industria Farmacéutica y Tecnología de Laboratorio VALIDACIONES Y CERTIFICACIONES CLIMATIZACIÓN FILTRACIÓN ESTERILIZACIÓN C/ Berlín, nº45. Polígono Ind. Ciudad de Parla 28983 – Parla (Madrid) Tlf.: 91 664 46 37 | Fax: 91 605 11 07 [email protected] | www.tci-ingenieria.es F FI LIMPIEZA DE EQUIPOS TRATAMIENTO DE AGUAS lifescienceslab BIOINFORMÁTICA Bienvenidos a Farmespaña Industrial AGENDA EMPRESAS, EQUIPAMIENTOS Y SERVICIOS AEPIMIFA ISPE CLIMATIZACIÓN TRATAMIENTO DE AGUAS FILTRACIÓN ESTERILIZACIÓN PUBLIRREPORTAJE CASO PRÁCTICO LIMPIEZA DE EQUIPOS VALIDACIONES Y CERTIFICACIONES NORMATIVA lifescienceslab SECTOR BIOINFORMÁTICA GUÍA DE SERVICIOS ÍNDICE DE ANUNCIANTES Revista Profesional para Proveedores de la Industria Farmacéutica y Tecnología de Laboratorio FARMESPAÑA INDUSTRIAL no se hace responsable de las opiniones emitidas por los autores, colaboradores y anunciantes, cuyos trabajos publicamos, sin que esto implique necesariamente compartir sus opiniones. Queda prohibida la reproducción parcial o total de los originales publicados sin autorización expresa por escrito. ISSN-1699-4205 DL: M-15766-2005 Editor: Eugenio Pérez de Lema Consejo Asesor: Coordinación: Gisela Bühl D. Sergi Roura, Director Gral. Grifols. Director Financiero: Carlos Fernández D. Antonio de la Sotilla, Director Gral. Vestilab S.A. Redacción: Dr. Alejandro Gesteira Tel. + 34 986 19 04 88 [email protected] D. Rogelio Pardo, Director Gerente Madrid NETWORK BIOCLUSTER. Director Comercial: Marcos Muiños Tel. + 34 986 19 12 93 [email protected] D. Eduardo Gaspar, Director ADF Tecnogen. y Circagen y miembro de BioMadrid. D. Eduardo Sanz, Senior Advisor. D. Aser Alonso Responsable de Ingeniería de Proceso de Alcon España. Publicidad: Fernanda Darriba Tel: + 34 91 630 85 91 [email protected] Mª Teresa Martín Tel: + 34 91 302 53 00. [email protected] Diseño y maquetación: Daniel C. Bernardo Edita: OMNIMEDIA S.L. · C/ Rosa de Lima 1 bis. Edificio Alba, oficina 104 28290 Las Matas (Madrid) · Tel: 902 36 46 99 · Fax: 91 630 85 95 [email protected] · www.grupo-omnimedia.com Farmespaña Industrial es miembro de la Asociación Española de Editoriales de Publicaciones Periódicas, que a su vez es miembro de FIPP. 4 6 18 22 24 28 38 42 46 48 52 56 62 67 68 72 74 82 AGENDA PAPERLESS LAB ACADEMY 2015 FARMAFORUM 2015 EXPOSÓLIDOS DESCRIPCIÓN: Salón Internacional de la tecnología y el procesamiento de sólidos ORGANIZADORES: Profei s.l LUGAR DE CELEBRACIÓN: La Farga de L’Hospitalet, Barcelona FECHA DE CELEBRACIÓN: 17 al 19 de febrero de 2015 MÁS INFORMACIÓN: www.exposolidos.com DESCRIPCIÓN: Foro de la industria farmacéutica, biofarmacéutica, cosmética y tecnología de laboratorios ORGANIZADORES: FarmaForum LUGAR DE CELEBRACIÓN: Madrid, Centro de Congresos Príncipe Felipe FECHA: 4 y 5 marzo de 2015 MÁS INFORMACIÓN: www.farmaforum.es, [email protected] DESCRIPCIÓN: Congreso enfocado en la gestión de los procesos de cambio en laboratorio ORGANIZADORES: Industrial Lab Automation y NL42 Management Consulting LUGAR DE CELEBRACIÓN: Barcelona, Gran Conference Hotel Rey Don Jaime FECHA DE CELEBRACIÓN: 14 y 15 de abril de 2015 MÁS INFORMACIÓN: www. industriallabautomation.com ACHEMA HISPACK DESCRIPCIÓN: Salón dedicado al envase, embalaje y la PLV ORGANIZADORES: Fira de Barcelona LUGAR DE CELEBRACIÓN: Recinto de Gran Vía, Barcelona FECHA DE CELEBRACIÓN: 21 al 14 de abril de 2015 MÁS INFORMACIÓN: www.hispack.com DESCRIPCIÓN: ACHEMA is the world forum of the process industry and the trend-setting technology summit for chemical engineering, environmental protection and biotechnology. ORGANIZADORES: Dechema LUGAR DE CELEBRACIÓN: Frankfurt am Main, Alemania FECHA DE CELEBRACIÓN: 15 al 19 de junio de 2015 MÁS INFORMACIÓN: http://www.achema.de SIL 2015 DESCRIPCIÓN: 15º Salón Internacional de la Logística y la Manutención ORGANIZADORES: El Consorci Barcelona LUGAR DE CELEBRACIÓN: Fira de Barcelona (Barcelona) FECHA DE CELEBRACIÓN: 9 al 11 de julio de 2015 MÁS INFORMACIÓN: www.silbcn.com/es PHARMA PROCESS 2015 BIOTECHNICA 2015 DESCRIPCIÓN: Evento nº 1 en Europa en Biotecnología, Ciencias de la vida y tecnología de laboratorio. ORGANIZADORES: Deutsche Messe LUGAR DE CELEBRACIÓN: Hannover FECHA DE CELEBRACIÓN: 6 al 8 de octubre de 2015 MÁS INFORMACIÓN: www.biotechnica.de CPHI WORLDWIDE MADRID DESCRIPCIÓN: Feria internacional de la industria farmacéutica ORGANIZADORES: UBM LUGAR DE CELEBRACIÓN: Feria de Madrid FECHA DE CELEBRACIÓN: 13 al 15 de octubre de 2015 MÁS INFORMACIÓN: www.cphi.com DESCRIPCIÓN: Foro de innovación mundial de la industria farmacéutica, biofarmacéutica y de las ciencias de la vida ORGANIZADORES: Fira Barcelona LUGAR DE CELEBRACIÓN: Fira de Barcelona (Barcelona) FECHA DE CELEBRACIÓN: 27 y 28 de octubre de 2015 MÁS INFORMACIÓN: www.firabcn.es EMPACK 2015 BIOLATAM DESCRIPCIÓN: BIOLATAM es una plataforma de desarrollo de negocio en el sector biotech y usuarios (farma, diagnóstico, alimentación, energía) con foco en América Latina ORGANIZADORES: Asebio, Chile Biotech LUGAR DE CELEBRACIÓN: Santiago de Chile FECHA DE CELEBRACIÓN: 16 y 17 de noviembre de 2015 MÁS INFORMACIÓN: www.asebio.com 4 NOVIEMBRE/DICIEMBRE14 DESCRIPCIÓN: punto de encuentro de los profesionales del envase y embalaje. Se celebra de forma conjunta con LOGISTICS, la plataforma comercial del almacenaje, manutención y logística, y PACKAGING INNOVATIONS. ORGANIZADORES: easyFairs LUGAR DE CELEBRACIÓN: Feria de Madrid FECHA DE CELEBRACIÓN: 18 y 19 de noviembre de 2015 MÁS INFORMACIÓN: www.easyfairs.com TECHNOPHARM DESCRIPCIÓN: International Trade Fair for Life Science Process Technologies ORGANIZADORES: Nürnberg Messe LUGAR DE CELEBRACIÓN: Exhibition Centre Nürnberg, Alemania FECHA DE CELEBRACIÓN: 19 al 21 de abril de 2016 MÁS INFORMACIÓN: www.technopharm.de FARMESPAÑA INDUSTRIAL NOTICIASACTUALIDAD Transmisor de humedad y punto de rocío Michell Instruments LTD, representada en España por Anisol, presenta su nueva generación de higrómetros por impedancia EASIDEW para la medida de punto de rocío, aplicaciones en laboratorio y salas de ambiente controlado. Las principales ventajas del EASIDEW son: Amplio rango de medida para bajos puntos de rocío -100ºC a +20ºC. Tecnología por impedancia con sensor cerámico que asegura estabilidad en la medida. Versión para punto de rocío más altos -40ºC a + 60ºC. Posibilidad de roscarlo directamente al conducto de medida o conectarlo a través de un bloque de muestreo en bypass. Multitud de opciones de toma de muestra para proceso que per- mite su trabajo a presiones de hasta 400barg para aplicaciones en la industria petroquímica o del gas. Posibilidad de conectar el sensor externamente a un display con alarmas que permitirá mostrar los valores en ppm(s). Simplicidad de uso y mantenimiento. Las aplicaciones del EASIDEW son muy amplias y cubren cualquier industria donde se necesite un sistema portátil, robusto y fiable como en la industria petroquímica (gases de reciclo), del gas (producción, transporte y almacenamiento de gas natural, gases del aire y CO2), farmacéutica (procesos de secado), siderúrgica y centrales térmicas. Los accesorios del EASIDEW incluyen filtros, display remoto, bloque de muestreo y varios dispositivos de acondicionamiento de muestra. Grifols Engineering desarrolla una máquina para automatizar el tapado de tubos de análisis Esta compañía de Grifols especializada en ingeniería biofarmacéutica, ha desarrollado una nueva máquina cuya función principal es la automatización del tapado de los tubos de análisis de muestras. El desarrollo atiende la necesidad del área de análisis de plasma para la elaboración de medicamentos derivados del plasma de Grifols, que llega a analizar diariamente 15.000 muestras mediante instrumentación automática de diagnóstico por diversas técnicas. La nueva máquina automatiza el taponado. Así se elimina el problema ergonómico de llevar a cabo manualmente el tapado de los tubos, además de optimizar el tiempo dedicado por el personal a esta función. Otro aspecto a resaltar es la mejora de la seguridad del proceso ya que reduce el riesgo de contaminación cruzada por vertido de plasma. Funcionamiento de la máquina El rack de 32 tubos se introduce por la apertura de la parte frontal. Unas guías fijan el rack y un sistema de arrastre lo lleva al interior tras su detección por el sensor de presencia, colocándolo debajo de una estrella. Colocado el rack, una tolva dispensa los tapones que son presionados por un dispositivo sobre cada tubo a medida que avanza por las guías. Completado el proceso, el rack con los tubos tapados sale por la misma apertura de entrada. El sistema de control está basado en un PLC de pantalla táctil para operar y supervisar el proceso. Diversos sensores y detectores verifican la correcta colocación de los tapones y posición de los tubos. La capacidad de producción de la nueva taponadora es de 32 segundos por rack, es decir, poco más de un segundo por tubo. Grifols Engineering ofrece soluciones de ingeniería aplicada y desarrollo de maquinaria para la producción industrial farmacéutica, siendo un socio estratégico de empresas del sector Biotec y del diagnóstico. Nuevo subdirector general de Grupo Cofares Recientemente se ha incorporado al Grupo Cofares José Luis Sanz Otero, para ocupar el puesto de Subdirector General, de nueva creación en la empresa. José Luis Sanz ha desarrollado su vida profesional en el campo de la gran distribución 6 mayorista, ocupando diversos puestos en Makro S.A., tanto en las áreas de operaciones como en marketing. Posteriormente se incorporó a la compañía catalana Miquel Alimentació, primer operador de capital español en el mercado nacional de la distri- NOVIEMBRE/DICIEMBRE14 bución mayorista, en calidad de director regional de la enseña GM para Cataluña. Los últimos cuatro años ha desempeñado la labor de Director General de la empresa de cash&carry, CashDiplo, tercer operador nacional del mismo sector. FARMESPAÑA INDUSTRIAL Storopack se extiende al mercado japonés Storopack, especialista en embalaje de protección, ha adquirido la empresa japonesa EJ Co., Ltd. (Ebina/ Kanagawa-Ken) con efecto a partir del 15 de octubre de 2014. EJ Co., Ltd. es desde hace ya varios años socio comercial de Storopack en Japón para la venta de almohadillados de papel PAPERplus® y de cojines de aire AIRplus®. Ahora EJ pasa a formar parte de Storopack. EJ Co., Ltd. es líder del mercado de Loose Fill (chips de embalaje) en Japón y suministra este embalaje de protección a clientes en todo el país bajo las marcas ECOsoft®, ECOtouch® y HItouch®. En el marco de su integración en el grupo Storopack, EJ Co. Ltd. cambiará de razón social a lo largo de los próximos meses para convertirse en Storopack Japan Co. Ltd. La empresa tiene centros de producción propios en Ebina/KanagawaKen, donde también se en- cuentra su sede central, así como en Nagoya. Youichi Isawa, gerente de EJ Co., Ltd. desde hace más de 6 años, seguirá asumiendo la responsabilidad de las actividades de la empresa cuya plantilla actual es de 33 trabajadores. Durante el proceso de integración en el grupo empre- sarial Storopack, Youichi Isawa cuenta con el apoyo de Frank Imkamp que en calidad de presidente de Asia-Pacífico es responsable del desarrollo de mercado de la división de Packaging en esta región. “Esta adquisición es un paso estratégico de suma importancia para nosotros”, explica Frank Imkamp. El hecho de contar con una presencia directa nos permite estar cerca del cliente y entender mejor el mercado japonés. El objetivo es ampliar la oferta de productos de manera progresiva y de acuerdo con las necesidades. Además, esta adquisición refuerza la posición de Storopack como líder del mercado internacional en el ámbito de Loose Fill. +(* ("#"0*"&%.' (" % ")*/&+( (," $$" # !% )* ("#"0"1%$ "%* $ !$& $# ! ! $ /' FARMESPAÑA INDUSTRIAL NOVIEMBRE/DICIEMBRE14 ---"&%")&) )!%& "%!&"&%")&) ) 7 NOTICIASACTUALIDAD Inscripciones abiertas. El Paperless Lab Academy 2015 tendrá lugar en España, Barcelona, los días 14 y 15 de Abril Después del éxito de la última edición en Mayo 2014, Peter Boogaard, CEO de Industrial Lab Automation, se asocia con NL42 Consulting para llevar la quinta edición al sur de Europa. Gracias a los sponsors, el registro al evento es gratis* para los visitantes. La dinámica interactiva del congreso le proporcionará múltiples ideas para volver a su rutina diaria con otra perspectiva. La academia incluye 16 seminario-taller, numerosas presentaciones prácticas de usuarios finales, encuentro de networking, cena de congreso y más de 15 demostraciones en directo de los principales proveedores internacionales. El tema central de este año tratara sobre cómo ORGANIZAR EL PROCESOS DE CAMBIO en un laboratorio de I+D o de QAQC en organizaciones del mundo farmacéutico, biotecnológico, bienes de consumo e industrias químicas. Organizar los procesos de cambio en un laboratorio requie- – re una visión integrada e intersectorial y estos serán los temas principales de la edición 2015: Adoptando nuevas formas de pensar para enfrentarse al cambio – Adoptando procesos de captura de datos automáticos – “Sin papel” vs. “menos papel” – Estrategias que fomenten la colaboración multifuncional entre investigación, desarrollo, garantía de calidad y producción. El ciclo de producción completo (PLM) Optimización de procesos internos y externos – Nuevo conceptos para integrar calidad y efi- Valtria firma nuevos contratos en el sector APIs Valtria reafirma su presencia como ingeniería e instaladora experta en el desarrollo de instalaciones en el sector APIs. Tras el contrato firmado con Ercros para la ampliación del edificio de purificación de fosfomicina en su centro de producción de Aranjuez, Valtria firma contratos para ampliar las áreas de producción de APIs de Crystal Pharma en Valladolid y Malta; para la ampliación del área de producción de Bemiparina de Rovi en Granada; la ampliación del edificio 15 de Hovione en Portugal y para Uquifa y Gentec en el área de Barcelona. Esta última fue objeto de una presentación en el encuentro anual de socios de ISPE sobre soluciones de contención (“Categorización 8 de Productos Activos”), a cargo de Mario Nahra de la Ingeniería IPB y Joan Amor, Director Técnico Pharmanoid del grupo GENTEC, S.A. Los nuevos contratos firmados por Valtria, demuestran el Know-how adquirido en sus más de 25 años de trayectoria, tanto en construcción de salas limpias, como en instalaciones críticas (aguas, vapor, CIP/SIP) para la fabricación de APIs y la manipulación de productos tóxicos o explosivos. NOVIEMBRE/DICIEMBRE14 ciencia con los colaboradores externos. – Procesos Reglamentarios (FDA, EMA, REACH, normativas nacionales) Consolidación de sistemas – Simplificando y consolidando los sistemas existentes – Estrategias de trazabilidad del ciclo de vida de los sistemas – Actualizando los potenciales de los productos LIMS, ELN, LES, SDMS, ERP/QM & PLM Adoptando nuevas tecnologías – Uso de la nube en procesos científicos. Gestión de Riesgo. – Uso de las tablets y dispositivos móviles. Análisis estadística científica “ in-silico” y estrategias de visualización – Saas/cloud vs. Tradicional. Impacto tecnológico y operacional ( privacidad, OPEX vs CAPEX) – Impacto sobre el panorama legal debido al “ Big Data” Programas Cursos de Formación Pre-congreso, día 13 de Abril 2014: (En español) NL42 Consulting: Cómo cumplir con las regulaciones de la tecnología de información en un laboratorio (En inglés) Mass Informatics Ltd: Advanced Paperless Compliance Training (En inglés) Industrial Lab Automation: Introduction course to European Data Protection for scientific data Esperamos verle pronto en la próxima edición 2015 del Paperless Lab Academy. Contacto: [email protected] / www.paperlesslabacademy.com Válvula de muestreo aséptica con instrumentación ASEPCO lanza una combinación de válvula aséptica de muestreo para aplicaciones bio-farmacéuticas con hasta tres sensores incorporados. Dispone de versiones para conexión clamp y para soldar directamente a la pared del reactor/fermentador. Está diseñada para ahorrar espacio, ya que cuenta con una válvula de muestreo y hasta 3 sondas de instrumentación en un conector ASEPCO de 4”. Es apto para sondas de 12mm, con varias longitudes. Dispone de camisas para el montaje fácil de sondas, toma SIP para limpieza detrás del asiento de la válvula cerrada. El cambio de diafragma puede realizarse de forma sencilla en 60 segundos, sin herramientas. Existen versiones especiales según especificación del cliente. Para más información: www.asepco-spain.com FARMESPAÑA INDUSTRIAL PDE Valores toxicológicos de límites de exposición Análisis de riesgos para el uso compartido o no de equipamiento e instalaciones en áreas multiproducto Cumplimiento nueva normativa EMA (20 Noviembre 2014): Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities. Instrumentos Testo lleva a Matelec 2014 sus novedades en cámaras termográficas Instrumentos Testo S.A. presentó en la pasada edición de Matelec todas las novedades de su gama de cámaras termográficas, que se centran principalmente en un rango de cámaras para una gran variedad de aplicaciones y la serie profesional para las aplicaciones más exigentes. La serie de inicio a la termografía es la testo 870; las novedades en Matelec fueron los nuevos Sets con los que los profesionales de instalaciones y el mantenimiento preventivo se beneficiarán de importantes ventajas. En los nuevos Sets, aparte de una notable reducción en el precio, se incluye la funda de transporte y la tecnología SuperResolution con la que se cuadruplican los píxeles de la cámara: mediante esta tecnología exclusiva Testo, las cámaras de la serie testo 870 con detector de 160x120 pasan a tener una resolución de 320x240. La cámaras que componen la gama inicial que se presentó en Matelec se complementan con la serie testo 875i, unas cámaras con unas prestaciones superiores a la serie testo 870, especialmente indicadas para el mantenimiento preventivo industrial y eléctrico así como para el sector de las auditorías energéticas. Como parte de la FARMESPAÑA INDUSTRIAL promoción en feria, y para facilitar el acceso a la termografía y hacer más fácil una posible decisión de compra, ahora y hasta final de año el cliente que lo solicite podrá probar sin compromiso durante una semana una unidad de esta serie, rellenando un sencillo formulario que encontrará en nuestra página web (adjuntar el link). Además, la serie testo 875i también incluye la tecnología SuperResolution mencionada anteriormente. La serie profesional también presentó en Matelec unos nuevos Sets con la inclusión de múltiples accesorios y una importante reducción de precio respecto a los Sets estándar. Esta serie de cámaras termográficas están dirigidas a los profesionales de la inspección fotovoltaica y eléctrica, así como a inspectores y auditores energéticos o departamentos de I+D. Estas promociones en cámaras termográficas también incluyen, para todas las unidades de cualquier serie adquiridas desde Matelec y hasta fin de año, un curso gratuito de termografía de 12 horas, impartido por termógrafos profesionales acreditados. Con esta iniciativa, Testo pretende facilitar a sus clientes y usuarios el aprovechamiento al máximo de sus productos. Cumplimiento normativa y guidelines EMA, FDA ICH, ISPE y PDA. Datos avalados y firmados por expertos toxicólogos y médicos relativos a la caracterización de dosis-respuesta (NOAEL, LD 50, LC50, ED50, LOAEL) de los productos farmacéuticos que servirán para el cálculo de otros parámetros claves como: PDE/ADE (exposición diaria aceptable) ADI (ingesta diaria segura) MRLs (nivel de riesgo mínimo) RfD (dosis de referencias) y RfC (concentraciones de referencias). Definición del potencial genotóxico y carcinogénico, factores de seguridad, vía de administración, biodisponibilidad y tipología de pacientes por producto para el cálculo de la PDE/ADE (exposición diaria aceptable). Realización de diagrama de proceso, definición de equipos y elementos compartidos entre los productos y cálculo de superficie compartida entre los productos objetos del estudio. Cálculo científico del ARL (Nivel aceptable de residuos), MAC (Máximo remanente admisible), SAL (Limite por superficie) a partir de datos toxicológicos. Estudio de las distintas estrategias factibles y recálculo de los límites de limpieza e informe final que justifique el uso compartido o no de equipos e instalaciones. Realización de análisis de riesgos para el uso compartido o no de equipamiento e instalaciones entre productos en un área de fabricación. Formación en la metodología empleada. [email protected] Tlfo. 912 771 076 NOVIEMBRE/DICIEMBRE14 9 NOTICIASACTUALIDAD Dara Pharmaceutical Packaging presenta su equipo SX-210-PP. Monobloc compacto, para la alimentación, llenado y cerrado automático de inyectables La máquina que le presentamos es una combinación de llenadora y/o cerradora compacta, para trabajar viales cilíndricos en vidrio, plástico o bien metal, para el acondicionado automático de todo tipo de productos líquidos, semilíquidos o en polvo, en zona estéril o sala limpia. Volúmenes de dosificación desde 0,1 hasta 250 ml. Para producciones de hasta 6.000 uph. Su diseño ha sido realizado conforme a las normas cGMP y de la US FDA en especial correspondencia con las exigencias de la industria farmacéutica, biotecnológica, cosmética, química e industrias afines. La estación de llenado está equipada con bombas peristálticas SpeedFill para productos líquidos o con dosificadores volumétricos para productos semi-líquidos o en polvo. Los programas de llenado son almacenados y pueden ser llamados de nuevo desde el panel de mandos. Parámetros como el volumen a dosificar o la cinemática del proceso de llenado son almacenados en la memoria del sistema. El usuario puede crear nuevos programas de llenado de forma rápida y sencilla. La estación de pick & place coloca el obturador a su justa altura sobre el vial, según sea obturador para inyectable o para liofilizado. La manipulación de los obturadores se realiza por vacío. La estación de cerrado está equipada con un cabezal de crimpado neumático, con el cual se consigue la mínima generación de partículas durante el proceso de cerrado. Todos los cierres de uso corriente pueden ser tratados sin problemas, tales como: Obturadores de goma para inyectables o liofilizados. Cápsulas de aluminio / Flipoff. Para productos que se deban liofilizar, existe la posibilidad de dividir en dos máquinas independientes los procesos de: Llenado y pre-colocación de obturadores de goma para liofilizado. Colocación de cápsulas de aluminio y cerrado de viales. Para trabajar en zona estéril disponemos de diferentes unidades de flujo laminar o RABs adaptadas a cada tamaño de máquina. El aire estéril que se Global Quality Management El mes de febrero de 2015 es la fecha indicada para el evento interactivo de Labware “Ciclo completo de un proceso productivo”. El evento tendrá lugar en el Hotel Barceló Sants (Plaza Països Catalans, sn, Barcelona). Labware presentará en vivo y de forma interactiva el Ciclo completo de un proceso productivo para la producción de un pastel de chocolate. En el Global Quality Management, a los visitantes les espera una multitud de nove- 10 NOVIEMBRE/DICIEMBRE14 dades Labware, como el IPKB (Integrated Knowledge on IT Applications), un módulo cuyo objetivo es la explotación eficiente de la información disponible en distintos sistemas de las diferentes áreas relacionadas con el proceso de producción. De esta manera se centraliza el conocimiento y se reducen los tiempos en el análisis y gestión de la información. Podremos ver en acción la nueva Plataforma Ip6, concebida para dar soluciones a necesidades presentes y futuras brindando tanto eficiencia y rentabilidad como trazabilidad integrada de recursos, operaciones y producto desde la entrada de materiales hasta la preparación de pe- genera asegura una extracción continua y eficaz de partículas y microorganismos fuera del área de trabajo. Con la utilización de un aislador, la zona estéril se limita al área de trabajo de la máquina llenadora - cerradora, permitiendo trabajar fuera de zona estéril, respetando los estándares de la industria farmacéutica. La máquina es adecuada tanto para lotes de ensayos clínicos como para extensos lotes de producción. Equipamientos opcionales: IPC para el control de la dosis por peso. Gaseado antes, durante o después del proceso de llenado. Cabezal de cerrado con rulina tangencial. Expulsión de viales con defectos de proceso. Software para la documentación de todos los datos relevantes de la producción. Flujo laminar / RABs / Aislador. Validación IQ / OQ. Impresión / Codificación. Para más información www. dara-pharma.com didos y expediciones pasando por controles de calidad, pesadas y producción. Habrá presentaciones y demostraciones multimedia, así como ponencias a cargo de expertos sobre logística, producción, control de calidad y retorno de la inversión. El Global Quality Management promete ser un evento con soluciones para la integración de la información y la implantación de la mejora continua. FARMESPAÑA INDUSTRIAL Miebach Consulting: rediseño de los procesos operacionales de Picking Farma Miebach Consulting ha colaborado con Picking Farma en el rediseño de los procesos operacionales de su centro de distribución de Santa Perpètua de Mogoda. El proyecto ha consistido en la definición e implementación de un paquete de mejoras concretas que han permitido alcanzar un nivel de calidad de servicio de referencia en el sector, y a su vez incrementar de forma muy significativa la eficiencia del personal y la capacidad operacional del centro de distribución. Picking Farma es un operador logístico especializado que actúa como cabecera de distribución de diversas y reconocidas compañías del sector farmacéutico. La compañía realiza la preparación de pedidos, tanto de reposición a los distribuidores del sector como de entregas directas a farmacias y hospitales, desde sus instalaciones de almacenamiento a temperatura controlada ubicadas en Santa Perpètua de Mogoda (Barcelona). El portafolio de servicios de Picking Farma también incluye todos los servicios adicionales requeridos por la distribución comercial en el sector farmacéutico: gestión de material promocional para la red farmacéutica, gestión de muestroteca, gestión de devoluciones, reacondicionamiento de productos (reetiquetado, reestuchado y cambios de formato de presentación) y preparación de agrupaciones promocionales. El proyecto desarrollado con la colaboración de Miebach Consulting se FARMESPAÑA INDUSTRIAL inició con un diagnóstico preliminar de los procesos operacionales existentes, realizado en el año 2012, que permitió diagnosticar las debilidades y cuellos de botella del centro de distribución, así como identificar las posibles vías de mejora, a partir de la utilizacion de tecnología ya contrastada, incorporando algunas soluciones aplicadas en otros sectores. Una vez identificada la mejor solución global y validada su rentabilidad y su potencial de crecimiento, en el transcurso de 2013 se acometió su implantación. La operación incluyó dos elementos clave. Por una parte, un cambio radical de la estrategia de verificación de pedidos, que ha permitido mejorar significativamente la calidad de servicio. Esto asegura a su vez un flujo continuo en el proceso de preparación y una reducción de los recursos asociados, permitiendo también realizar una verificación selectiva a la carta con arreglo a los requerimientos específicos de cada cliente. Por otra parte, una instalación automática que gestiona la reposición inmediata de producto a los puestos de preparación, en los que se realiza el picking y la verificación mediante un sistema combinado de pesaje y Put-to-Light. Como resultado, Picking Farma ha incrementado sustancialmente la capacidad operacional de su centro de distribución de Santa Perpètua de Mogoda, asegurando la mejor calidad de servicio a sus clientes. NOVIEMBRE/DICIEMBRE14 11 NOTICIASACTUALIDAD Las puertas súper rápidas de apertura horizontal Novosprint cumplen 30 años Hace treinta años, Arnold Butzbach tuvo la idea revolucionaria de construir una puerta de alta velocidad en la que la lona estaba dividida en dos mitades que se enrollaban a los lados, en vez de una sola lona que subiera. Esta filosofía que permite que la altura de paso esté disponible en todo momento y en la mitad del tiempo, se ha mantenido igual desde los comienzos de la serie. Por contra, la tecnología se ha desarrollado de forma continua para mantener la puerta siempre actualizada con los últimos avances. Uno de los más importantes que se ha realizado recientemente ha sido substituir el sistema hidráulico por un accionamiento eléctrico, lo que ha aumentado aún más la velocidad de apertura y ha hecho que el modelo Novosprint sea todavía más eficiente. Angel Mir - Portes Bisbal SL es el distribuidor oficial de Butzbach en el Estado español y Portugal. Cuenta con más de 45 años de experiencia, siendo la mayor empresa española de fabricación y montaje de puertas industriales, residenciales y equipamiento logístico. Está presente en más de 50 países de Latinoamérica, Europa y África. Es el fabricante que dispone de la gama más amplia de productos y soluciones para los cerramientos y la logística. Apertura Horizontal: El principio de apertura horizontal de las Novosprint proporciona seguridad, tanto para el usuario como para los bienes transportados y la propia puerta. Toda la altura de paso está disponible simultáneamente en ambos lados. 12 Última Tecnología: La tecnología de estas puertas está más actualizada que nunca y ofrece muchos beneficios para el usuario. Con el cambio del sistema hidráulico por el accionamiento eléctrico, las Novosprint son las puertas industriales más rápidas del mundo, con una velocidad de apertura de hasta 5 metros por segundo. El paso queda completamente libre en cuestión de segundos, evitando que se formen colas y retrasos. Los tiempos de cierre, que también son muy cortos, reducen el tiempo de apertura efectiva al mínimo y minimizan las molestias ocasionados a los empleados en cuanto a ruido, corrientes de aire y frío. ADAPTABLES Las Novosprint son adaptables a las necesidades de empresas de todos los sectores, con modelos con doble lona para zonas de mayor aislamiento térmico y acústico para equipamientos con un alto nivel de ruido, o puertas con elementos que proveen una mayor higiene para las industrias alimentaria y farmacéutica, por citar sólo algunos ejemplos. También existen opciones para aumentar la seguridad de los empleados, como el uso de las Novosprint como puerta anti-pánico en rutas de salidas de emergencia, ya que se abren automáticamente en caso de fallo de alimentación eléctrica. Las puertas de la serie HS5E pueden llegar a dimensiones de luz de paso ancho x alto de 4.750 mm. x 4.500 mm. Las de la serie HS3E hasta 9.000 mm. x 6.000 mm. NOVIEMBRE/DICIEMBRE14 Jungheinrich: Líder en innovación para soluciones intralogísticas En la próxima LogiMAT 2015 de Stuttgart, la compañía con sede en Hamburgo desplegará sus inteligentes e innovadoras soluciones de manutención para aplicaciones intralogísticas. En consonancia con el eslogan del evento “Intelligent Networking: Mastering Complexity”, Jungheinrich demostrará su expertise en la 13ª feria internacional para la distribución, los equipos de manutención y los flujos de información LogiMAT 2015 (1012 de febrero). En su stand de más de 500 metros cuadrados de espacio de exposición en el “Hall de apiladoras” o Hall 6 (stand 6B03/6C03), la compañía con sede en Hamburgo expondrá su amplia gama de carretillas y soluciones intralogísticas. “Como proveedores líderes en soluciones intralogísticas, aprovecharemos nuestra presencia en esta plataforma ferial para presentar nuestra competencia en soluciones completas ante una audiencia internacional,” explicó Marcus Karst, Director de Marketing en Jungheinrich. Y añadió: “Aquí demostraremos a los visitantes profesionales nuestras soluciones intralogísticas inteligentes, innovadoras y sostenibles, desde carretillas hasta soluciones de automatización y digitalización. ¡Y vamos a presentar todo ello de forma tangible para los visitantes de la feria!” Una novedad especial en el stand de Jungheinrich de esta próxima edición es la presencia del ganador en la máxima categoría de los premios IFOY 2014, la carretilla contrapesada eléctrica EFG S40s. otros productos destacados incluyen el Auto Pallet Mover (APM) ERC 215a, las tractoras de la serie 5, el sistema de almacenaje LRK y la carretilla trilateral EKX 410 para pasillo estrecho y equipada con navegación de almacén. Los visitantes de todo el mundo tendrán la oportunidad de probarla en vivo, en un espacio del stand dedicado a ello. En la feria de Stuttgart Jungheinrich mostrará también un sistema completo de tractoras de la serie 5 con sus respectivos remolques. La presencia de Jungheinrich en el Hall 6 estará rodeada por la presentación del protfolio completo de la empresa, por ejemplo el sistema de gestión de flotas ISM Online, servicios financieros, equipos de ocasión y alquiler, el negocio ProfiShop y el área de selección de RRHH. Jungheinrich también expondrá en el “Software Hall” (Hall 7, stand 7D/221), con stand propio. En un espacio de exposición de cerca de 100 metros cuadrados, el integrador logístico desplegará su expertise en materia de software y hardware IT para aplicaciones logísticas, que incluirán el sistema de gestión de almacenes ‘Jungheinrich WMS’ y los terminales de radiofrecuencia. Los visitantes podrán ser testigos de demostraciones en vivo de soluciones integradas y holísticas para la logística interna, tanto para almacenes manuales como para almacenes totalmente automatizados, y comprobar cómo proyectos integrales pueden implementarse con éxito en tan sólo ocho semanas. FARMESPAÑA INDUSTRIAL Telstar desarrolla dos SAS-pasamateriales que utilizan procesos integrados de biodescontaminación ionHP AEPIMIFA confirma su participación activa en Farmaforum durante la asamblea general de socios AEPIMIFA, asociación española de profesionales de la industria farmacéutica, alimentaria, cosmética y afines reunió en Madrid a una parte importante de sus asociados en la asamblea general que se celebró en el hotel Silken Puerta de América. Dentro de las actividades programadas para el año 2015, José Cristóbal, Presidente de AEPIMIFA, confirmó la partici- FARMESPAÑA INDUSTRIAL pación de la asociación en la segunda edición Farmaforum tanto dentro del programa de conferencias como en las jornadas y talleres que se celebrarán en paralelo y que suponen una de las principales novedades de esta edición. La organización de Farmaforum considera que el apoyo y la presencia de AEPIMIFA contribuirá, sin duda alguna, al éxito de esta segunda edición. Telstar ha diseñado y fabricado dos SAS-pasamateriales con volúmenes superiores a 5 m³, cada uno con su propio sistema integrado de biodescontaminación ionHP (Peróxido de Hidrógeno ionizado), que serán instalados entre dos zonas con niveles distintos en una instalación de fabricación aséptica de Virbac. Telstar, que cuenta con una sólida experiencia en la instalación de sistemas de biodescontaminación en aisladores, ha adaptado con éxito este tipo de sistema de biodescontaminación ionHP en un SAS para transferir materiales en instalaciones asépticas. Las unidades de SAS, que han sido desarrolladas por el centro tecnológico especializado en sistemas de contención de Telstar ubicado en Dewsbury (Reino Unido), están diseñadas para esterilizar materiales que no son aptos para la esterilización mediante otros métodos y son capaces de biodescontaminar una unidad completamente cargada en menos de 2 horas, al mismo tiempo que consiguen una reducción de 6 log en la biocarga, validado mediante un Bio-indicador (BI) Challenge. La duración de 2 horas comprende una aireación hasta <1 ppm de H2O2 mientras trabajan independientemente con el sistema de instalaciones de climatización. NOVIEMBRE/DICIEMBRE14 13 NOTICIASACTUALIDAD Calzados Robusta diseña una colección para Salas Blancas en la Industria Farmacéutica Calzados Robusta ha puesto siempre especial interés en la industria farmacéutica. Para ello ha diseñado una colección específica para salas blancas, salas de producción donde el producto ya está envasado y no se puede tocar directamente. Son modelos con puntera de protección de aluminio resistente hasta 200 J y suela de PU antideslizante. Esta colección incluye 7 modelos, todos ellos blancos con suela verde y con distintos diseños y sistemas de cierre: cordones, velcro, elástico, tipo sandalia… Gracias al estudio de las necesidades de este sector, Robusta ofrece un calzado laboral antideslizante, antiestático, con modelos lavables, preparado para combatir la fatiga y proporcionar confort al usuario gracias a su ligereza. Además, dispone de una web con un apartado específico que incluye estos modelos diseñados para la industria farmacéutica. Robusta® ha diseñado esta línea de calzado para el sector farmacéutico teniendo en cuenta los estándares de calidad que indican las GMP´s (Good Manufacturing Practices) o buenas prácticas de manufactura. Este sistema, ideado para minimizar errores en la manufactura de productos farmacéuticos, abarca todos los aspectos de la fa- bricación, incluyendo el calzado más adecuado para cada puesto de trabajo y las diferentes salas de producción. Además, todos los modelos están confeccionados según la normativa europea para el calzado de seguridad, protección y trabajo de uso profesional. El calzado profesional dirigido a la industria farmacéutica debe reunir características específicas de seguridad, comodidad y prevención de riesgos. Por eso Robusta® ha creado esta colección especializada de zapatos, deportivos y mocasines diseñados para estos entornos. La colección Feeling Robusta® es el resultado de un estudio cuidadoso de las necesidades de los puestos de trabajo en industrias de farmacia, así como de una constante experimentación biomecánica para aplicar los avances científicos al calzado profesional. Cada modelo se desarrolla en el laboratorio de biomecánica de Robusta® e incorpora todos los requisitos necesarios para una total seguridad y confort. De cada modelo se ofrece en la web una completa información sobre sus características técnicas y, también, varias fotografías generales y de detalle. En la web también se pueden encontrar diferentes accesorios, como calcetines, medias o plantillas. El calzado de Robusta para la industria farmacéutica cuenta con unos argumentos técnicos que lo avalan. El diseño está avalado por el Laboratorio de Biomecánica de Robusta de tal forma que su diseño interior está determinado por una horma que proporciona confort y previene la fatiga: un calzado desarrollado para personas que trabajan de pie y que caminan varios kilómetros al día, tanto en señora como en caballero. Y un diseño exterior determinado por los diferentes usos y pues- tos de trabajo en los que se deben prevenir los riesgos de accidentes. Feeling Robusta®, se caracteriza por la extensión y variedad de diseños y modelos, todos ellos testados en el ámbito laboral. Componentes: a) Pieles: Muy transpirables, para eliminar sudor, con tacto muy suave. b) Suelas: Creadas fundamentalmente para proporcionar el máximo confort, aportando el mínimo peso, la densidad del material adecuada, y diseño y colocación de sus pastillas para evitar el resbalamiento. c) Plantillas: Es el elemento fundamental para maximizar las bondades del diseño, pieles y suelas. Las plantillas de Robusta están diseñadas íntegramente por su Laboratorio de Biomecánica, para proporcionar confort y descanso, y ausencia de humedad en el interior del calzado. La colección de industria farmacéutica tiene 21 modelos e incluye calzado de seguridad para personal de laboratorio, auxiliares, etc. de la industria farmacéutica. Están diseñados en función de las tres áreas de trabajo de un laboratorio farmacéutico: además de salas blancas, salas verdes (salas de fabricación, donde el producto se puede tocar) y salas grises, de almacenaje. Comindex se consolida en Portugal ampliando su equipo de asesores técnico-comerciales José Luís Barroso se ha incorporado para liderar la expansión de Comindex en el país luso. Barroso es licenciado en Ingeniería Biológica en la especialidad de Procesos Químicos, Tecnológicos y Alimentarios, estudios que completó 14 NOVIEMBRE/DICIEMBRE14 posteriormente con un postgrado en Gestión Empresarial. A nivel profesional empezó trabajando en un laboratorio, luego fue Responsable de Producción y más adelante Responsable de I+D. Su última posición antes de incor- porarse a Comindex fue como Responsable de Compras y Logística. José Luís Barroso proporcionará una excelente asistencia técnica y comercial, gracias a su formación y a su experiencia en empresas de pinturas y plásticos. FARMESPAÑA INDUSTRIAL Burdinola se adjudica los nuevos laboratorios de Fresenius Kabi en EEUU Burdinola, especialista en soluciones integrales de laboratorios, ha resultado adjudicataria de los nuevos laboratorios que la compañía farmacéutica Fresenius Kabi dispondrá en Grand Island, en el estado de Nueva York. Fresenius Kabi es la división farmacéutica de la multinacional alemana Fresenius y constituye un importante referente mundial en productos farmacéuticos y dispositivos médicos para el cuidado de la salud. El proyecto incluye la reforma del laboratorio químico y microbiológico en diferentes fases de trabajo y comienza en su primera etapa por la renovación del laboratorio de control de calidad. El equipo de Burdinola llevará a cabo el lab-planning y el equipamien- to de los laboratorios en un proyecto de gran envergadura a todos los niveles, que arranca con un amplio estudio de necesidades. En la fase de diseño y planificación, se analizará la actividad a realizar en el laboratorio –flujos de trabajo, ocupación, riesgos asociados, etc– y las características constructivas para ir definiendo en detalle los diferentes espacios, así como la identificación de los puestos de trabajo, dando respuesta a todas las necesidades. De acuerdo a este análisis se realizarán posteriormente las propuestas de diseño que se materializará en los planos de distribución con todos los elementos integrados, teniendo en cuenta la aplicación de la normativa vigente en todos los equipos e instalaciones incluidas. Burdinola se encargará también del equipamiento integral de los laboratorios a partir de su amplia gama de sistemas modulares que combinan criterios estéticos, funcionales y medioambientales. La versatilidad, la capacidad de personalización y la resistencia son características comunes de todos los elementos que integran las distintas series fabricadas por la firma. En su conjunto, Burdinola aportará al proyecto de Fresenius Kabi los últimos avances tecnológicos en materia de seguridad, materiales y equipamiento, abordándolo desde la sostenibilidad, el ahorro energético y el respecto con el medio ambiente. Fresenius Kabi tiene su sede en Alemania donde se fundó hace más de 100 años. En la actualidad, emplea a más de 30.000 personas en todo el mundo y cuenta con una red de 16 centros de I+D y más de 70 centros de producción. Está presente en más de 65 países y sus ventas anuales se cifran en más de 5 mil millones de dólares. Desde el 2008, la compañía ha ampliado su presencia en los Estados Unidos con la adquisición de distintas empresas especializadas. Constantia Flexibles Pharma Constantia Flexibles desarrolla, fabrica y suministra soluciones de embalaje flexible para el mercado farmacéutico, cuidado personal y alimentación. Somos un proveedor líder de envases con aluminio para medicamentos aplicaciones médicas y cosméticas, incluyendo comprimidos, cápsulas, polvo y productos médico-técnicos. Constantia Flexibles Farma ofrece ・Una producción a nivel global y una red de servicios que suministra desde pequeños a grandes lotes ・Tecnologías de proceso líderes en el mercado: · laqueado - laminación de aluminio · laminación, extrusión - laminación y coating · fabricación de cilindros · impresión - gofrado · troquelados, corte y bobinado ・Los más altos estándares de producción en sala limpia ・Una gran especialización y compromiso con los sistemas y procesos de calidad ・Materiales de embalaje registrados en el fichero Drug Master (US Food and Drug Administration). Contact Madrid: FARMESPAÑA INDUSTRIAL Constantia Tobepal, S.L (Sociedad Unipersonal) Avda. de América 45, 1º B E-28002 Madrid T +34 915 631 120 / +34 915631122 Contact Barcelona: Constantia Tobepal, S.L (Sociedad Unipersonal) Via Augusta 15-25, Arroba San Cugat Edificio B1, Planta 4ª, Despacho 10 08174 Sant Cugat del Vallés T +34 934 152 545 NOVIEMBRE/DICIEMBRE14 www.cflex.com 15 NOTICIASACTUALIDAD Reduce 95% de falsos rechazos: sin efecto de producto con Profile Advantage Los fabricantes y las empresas de procesamiento de los sectores de carne y aves, lácteos, panadería y comida preparada, así como los fabricantes de productos envasados en láminas metalizadas, pueden alcanzar niveles de sensibilidad del detector que hasta ahora solo se lograban en la inspección de productos secos. Durante años, el ‘efecto de producto’ (la señal eléctrica detectada en algunos alimentos con un contenido muy elevado de humedad o sal, o envasados en láminas metalizadas) ha reducido considerablemente la sensibilidad del detector por debajo de los niveles logrados en la inspección de alimentos secos no conductivos. El detector de metales Profile Advantage recién presentado garantiza la eliminación del fenómeno del ‘efecto de producto’ del proceso, gracias al uso de un algoritmo de inspección sofisticado. Esto mejora en un 50 % los niveles de sensibilidad del detector independientemente del material de empaquetado, garantizando que el sistema Profile Advantage detecta más contaminación metálica que los sistemas tradicionales en aplicaciones exigentes como alimentos húmedos, calientes o refrigerados. Además, la solución es capaz de reducir rigurosamente el número de falsos rechazos. Por ejemplo, los fabricantes de carne y productos avícolas pueden reducir habitualmente en hasta un 95 % las tasas de falsos rechazos asociados al ‘efecto de producto’ cuando intentan detectar la contaminación metálica más pequeña. El detector Profile Advantage emplea la tecnología pionera de frecuencia multisimultánea (MSF) para modificar el rendimiento. Debido a los cambios recientes en los códigos de conducta de los comerciantes y niveles 16 más rigurosos aplicados en la industria alimentaria por la iniciativa mundial de seguridad alimentaria (GFSI, por sus siglas en inglés), los fabricantes están buscando métodos para garantizar la eliminación de contaminación metálica con una eficiencia elevada de la línea de producción y ahorros en el balance final. Profile Advantage permite lograr un ahorro notable en los costes gracias a la reducción de rechazos de productos y un menor tiempo de inactividad operativo para investigar el problema. Para los fabricantes de alimentos que desean controlar la eficacia global del equipo (OEE, por sus siglas en inglés), presenta un coste total de propiedad (TCO, por sus siglas en inglés) atractivo para la inspección de metales en comparación con otras tecnologías. “Durante un tiempo, la superación del fenómeno del efecto de producto ha sido un problema vital para nuestra industria,” comentó Jonathan Richards, responsable del departamento de marketing de Mettler-Toledo Safeline Metal Detection. “Nuestro sistema Profile Advantage es un avance tecnológico fundamental para la detección de metales, ya que ayuda a que los fabricantes aumenten la protección de la marca, reduzcan los costes y mejoren la productividad. Como consecuencia, el nuevo sistema apoya a las empresas alimentarias a fabricar únicamente productos sin contaminantes en la instalación de producción. Al combinar este rendimiento fiable con un cálculo global del coste total de propiedad bajo, la detección de metales se convierte en la tecnología preferida”. El algoritmo ‘3S’ y la función de supresión de la señal de producto son claves para la NOVIEMBRE/DICIEMBRE14 tecnología de frecuencia multisimultánea del detector Profile Advantage. A diferencia de los detectores de metales convencionales que solo capturan y almacenan la señal del producto activo, el sistema Profile Advantage modifica la señal durante la configuración, de forma que el producto alimentario se presenta como si fuera un producto seco sin el efecto de producto. Una vez que comienza la producción, el algoritmo de detección se aplica a todos los productos que pasan por el detector. Por lo tanto, se logran niveles muy superiores de sensibilidad del detector, ya que la señal del producto activo se percibe como insignificante y el envolvente de detección de la contaminación se optimiza. La función de supresión de la señal de producto no solo puede hacer frente a aplicaciones exigentes de productos húmedos o con un contenido elevado de sal o los que están en evolución de refrigeración o descongelación, sino también a los productos envasados en lámina metalizada. Este tipo de empaquetado ha complicado hasta ahora que los detectores compitan con otros tipos de tecnología en cuanto al rendimiento. No obstante, con la tecnología MSF, el sistema Profile Advantage satisface los niveles de inspección de producción de la mayoría de tipos de metales y ofrece un rendimiento considerablemente superior cuando se detecta contaminación de aluminio. La gama Profile Advantage combina un detector muy sensible con una analítica predictiva e innovadora del rendimiento de detección que realiza el software del sistema usando una función de supervisión de estado. Así se puede reducir el número de pruebas diarias de rendimiento que requieren mucho tiempo. La máquina informa a los operarios acerca de si es preciso realizar cualquier medida preventiva con el objeto de mantener el nivel de la fábrica de la sensibilidad del detector. La reducción de las pruebas del rendimiento necesarias cada día mejora la productividad y maximiza el tiempo de funcionamiento y la efectividad global de equipos. Para los fabricantes de productos con requisitos rigurosos de higiene, el detector Profile Advantage incorpora un diseño robusto e higiénico conforme con los niveles del Grupo Europeo de Ingeniería y Diseño Higiénico (EHEDG, por sus siglas en inglés). Con una cubierta fabricada con acero inoxidable, el sistema puede resistir incluso los procesos de lavado más fuertes, mientras que los bordes y las esquinas curvos eliminan los puntos de acumulación de suciedad, minimizando el riesgo de contaminación biológica de los alimentos. Además, el nuevo detector de metales está también disponible con transportadores personalizados opcionales, que permiten a los fabricantes adaptar el sistema a sus necesidades de producción específicas. FARMESPAÑA INDUSTRIAL Instrumentación y sistemas para la industria farmacéutica Validación de procesos térmicos IBERFLUID junto a su representada ELLAB se posicionan como un referente en las validaciones de procesos térmicos para la industria farmacéutica. Dataloggers Tracksense Pro Comunicación inalámbrica en tiempo real. Temperaturas de trabajo: -80ºC a 400ºC Esterilización con vapor. Liofilización. Despirogenización. Óxido de etileno y H2O2. Cámaras de estabilidad. Ultracongeladores. Sede Central Barcelona +34 933 333 600 iberfluid@iberfluid.com Delegación Centro Alcobendas (Madrid) +34 916 611 717 madrid@iberfluid.com Delegación Norte Basauri (Vizcaya) +34 946 715 012 nor@iberfluid.com www.iberfluid.com Delegación Sur Sevilla +34 955 452 780 sevilla@iberfluid.com Delegación Portugal Lisboa +351 210 993 616 portugal@iberfluid.com AEPIMIFA Éxito de participación, un año más, en la asamblea general de socios de AEPIMIFA A EPIMIFA, asociación española de profesionales de la industria farmacéutica, alimentaria, cosmética y afines reunió el pasado 20 de noviembre en Madrid a una parte importante de sus asociados en la vigésimo cuarta Asamblea General, que se celebró en el hotel Silken Puerta de América. El evento contó con el patrocinio de Roda & Plast S.L., Ingeclima y Sika. Siguiendo el formato de asambleas anteriores, el evento comenzó con un análisis del estado de la asociación y una breve mención a las actividades desarrolladas en el 2014, destacando las siguientes. Colaboración Elur- Farmaclúster: 2ª Jornada tecnológica destinada a departamentos de ingeniería, de mantenimiento, de producción y de calidad de la industria farmacéutica, química, cosmética y afines. Colaboración Farmaforum-AEPIMIFA: en el evento, celebrado el pasado mes de marzo de 2014, participaron Yves Billiet-Prades, que colaboró en una de las mesas redondas, y José Cristóbal que impartió la ponencia “Aplicación de Normativa (USA/UE) antifalsificación (serialización) desde el punto de vista de Fabricante a Terceros”. Colaboración con Sivart: curso de formación y visita al Competence Centre de GS1 España AECOC. 18 NOVIEMBRE/DICIEMBRE14 DURANTE LA ASAMBLEA SE ESTABLECIÓ LA MISIÓN DE SER EL MEJOR Y MÁS PRÓXIMO SOPORTE, SIN ÁNIMO DE LUCRO, PARA EL DESARROLLO INTEGRAL DE LOS PROFESIONALES EN EL ÁMBITO DE LAS OPERACIONES EN LOS SECTORES FARMACÉUTICO Y AFINES, A NIVEL NACIONAL Colaboración con la asociación AICA: prevención de riesgos laborales sector químico y farmacéutico. Cursos de formación: curso sobre negociación avanzada y un curso sobre cómo desarrollar competencias interculturales. Jornadas organizadas por socios: Tecnoferia 2014 de la mano de Festo y Roda & Plast y la jornada organizada por Sika “Refuerzo sobre estructuras de hormigón” Creación de un newsletter. Preparación del plan estratégico. En relación a las actividades previstas para el 2015, se destacó: La participación de la asociación en la segunda edición de Farmaforum, tanto en el programa del congreso (en el que colaborarán con la conferencia “Tendencias en llenado aséptico”), como a través de la organización de una jornada para los socios (mesas redondas en la sala Aepimifa). La visita prevista a la planta de Esteve, Curso sobre Quality Risk Management Asamblea de Socios y las Elecciones a la Junta Directiva. Uno de los momentos más destacados de la jornada fue la propuesta de cambio de estatutos, que contó con el apoyo de los socios en una votación realizada durante el evento. La propuesta de modificación de estatutos tiene como objetivo facilitar la renovación de la Junta a través de un proceso electoral y adaptar la asociación a los nuevos tiempos, en los que la accesibilidad telemática y la agilidad en la toma de decisiones se han convertido en elementos decisivos para el buen funcionamiento de la organización. Los puntos más destacados de la propuesta son los siguientes: Diferenciar socio numerario de protector. Modificación del quorum y convocatorias. Junta Directiva: forma de elección, presentación candidaturas y votación telemática. Asambleas: votaciones de puntos del orden del día. Posibilidad contratar los servicios de un Administrador que realizará las funFARMESPAÑA INDUSTRIAL ciones que la Junta Directiva considere que deba delegar. A continuación, se presentó el plan estratégico de la asociación, apoyado en tres ítems: MISIÓN, VISIÓN y OBJETIVOS ESTRATÉGICOS. Misión La asociación establece, como propósito general, ser el mejor y más próximo soporte, sin ánimo de lucro, para el desarrollo integral de los profesionales en el ámbito de las operaciones en los sectores farmacéutico y afines, a nivel nacional. Visión Bajo este epígrafe se indicó el enfoque con el que se pretende desarrollar la actividad de la asociación. AEPIMIFA quiere: Ser un referente para los profesionales de su área de influencia. Ser una organización en la que todos sus miembros se sientan protagonistas, beneficiados y orgullosos de formar parte de ella. Potenciar la acción social desde la Asociación Destacar por contribuir al crecimiento de los sectores implicados Fomentar la colaboración entre sus asociados y otros profesionales Facilitar a los asociados la comunicación , interpretación y acceso a las fuentes informativas FARMESPAÑA INDUSTRIAL Objetivos estratégicos Durante la asamblea, se establecieron tres líneas de actuación, básicas y prioritarias para la asociación y sus asociados: Incrementar los servicios y calidad a los socios, mediante las siguientes actividades: – Visitas a plantas/entidades – Mesas redondas – Cursos de formación – Participación en eventos – Difusión de eventos – Bolsa de trabajo Aumentar la participación/involucración de los socios: – Promover la difusión de actividades/posibilidades – Ampliar los canales de comunicación – Conocer qué quieren los asociados (protectores y numerarios) – Provocar encuentros con/entre los asociados Lanzamiento hacia sectores afines, sectores hasta ahora no presentes con una presencia minoritaria en la asociación. La parte formativa y lúdica del evento llegó de la mano del Mago More con la ponencia “El poder positivo del cambio”. More (José Luis Izquierdo Martín) es un líder en presentación de eventos y experto comunicador, en el que han confiado algunas de las más grandes empresas naciona- les e internacionales (PwC, Banco de Santander, Cepsa, Telefónica, BBVA, Morgan Stanley, Renault, Chrysler, ING, El Corte Inglés, BMW, Endesa, Ferrovial o Microsoft). Su enfoque innovador, junto a su sólida formación empresarial, le ha permitido especializarse en el diseño de productos exclusivos orientados a organizaciones y ejecutivos que buscan mejorar su comunicación. Para la Asamblea General de Socios de AEPIMIFA preparó una impactante conferencia con el claro objetivo de enseñar a introducir imaginación, innovación y creatividad (conceptos no siempre suficientemente valorados en el ámbito industrial y/o empresarial) en los procesos de producción, con el fin de contribuir a mejorar en competitividad. La ponencia tocó los siguientes puntos: O cambias o te cambian. Cambios en el entorno, la distribución y los productos. Nuevos clientes, nuevos retos. Convivir con la tecnología. ¿Cuándo innovar? Excusas para no cambiar. Por dónde empezar. Fórmula de la innovación. Crear el camino. La Asamblea anual finalizó con una cena de confraternización en la que participaron la mayoría de los asistentes y que sirvió para estrechar los lazos entre los miembros de AEPIMIFA NOVIEMBRE/DICIEMBRE14 19 En la ya clásica jornada ISPE de Gestión de plantas farmacéuticas, este año ISPE ha innovado trayendo a un profesor del IESE, quien ha desarrollado un ejercicio práctico de servicio a domicilio de medicamento directamente desde el fabricante. ISPE celebra el VI encuentro de gestión de plantas farmacéuticas E l 27 de Noviembre, ISPE España celebró en el hotel AC Madrid Feria su jornada sobre gestión de plantas farmacéuticas, que contó con IESE como partner académico y que se inició con la bienvenida a todos los asistentes y ponentes por parte del presidente de ISPE España, Carles Cirera, y se explicaron brevemente las actividades que desde ISPE España se llevan a cabo, así como el calendario de actos previsto para el 2015. La primera ponencia, “Quality efficiency by design” fue impartida por Olivier Depardieu de Oxo Pharma and Amina Belabbes, uno de los consultores de Altran que trabajó en el proyecto de vacunas de GSK. Su exposición se centró en el coste que supone la documentación actual para una empresa farmacéutica (PNT’s, protocolos, guías de fabricación, etc.) y las fuentes de errores que una documentación compleja puede acarrear. Ilustró su ponencia con 3 casos prácticos de proyectos llevados a cabo con 3 empresas farmacéuticas, haciendo ver a la audiencia las ventajas de tener una documentación simple y concisa en lugar de largos y complejos procedimientos. La segunda presentación, titulada “Nuevos servicios en la cadena de valor del medicamento: ¿Es posible convertir una compañía farmacéutica en un proveedor de Salud?”, fue llevada a cabo por el profesor 22 NOVIEMBRE/DICIEMBRE14 CALENDARIO DE EVENTOS 2015 Título Fecha Lugar Contención Enero Barcelona/Madrid Encuentro Anual Socios ISPE Marzo Madrid, Farmaforum Calidad Abril Barcelona/Madrid Excelencia Operacional Mayo Barcelona/Madrid Mantenimiento Junio Barcelona/Madrid HVAC Julio Barcelona/Madrid Octubre Barcelona/Madrid Automatización y GAMP Gestión de Plantas Farmacéuticas Noviembre Barcelona Fabricación Estéril Diciembre Barcelona/Madrid del IESE Eduard Calvo, Production, Tech. & Operations Management, que desarrolló un ejercicio práctico con la participación de la audiencia. Seguidamente, el Dr. Heiko Rengel, de Boehringer Ingelheim, con su ponencia “Sharing experiences and learning on how to pass a Chinese FDA inspection”, compartió sus experiencias obtenidas tras pasar una auditoría de las autoridades FDA chinas en su planta de Sant Cugat, haciendo énfasis en el altísimo nivel de exigencia y profesionalidad que éstas mostraron en todo momento. En la segunda parte de la jornada, tuvimos el privilegio de contar con la presencia de uno de los máximos expertees europeos en temas regulatorios, Bryan Wright, quien dio una ponencia sobre desabastecimiento de medicamentos en el mercado y la proble- mática que ello conlleva (“ISPE Drug Shortages Prevention Plan- Global Initiative”). Finalmente, John P. Manning, jefe de Technology Strategy, de Novartis Basilea, nos explicó en su ponencia titulada “Moving from decentralized approach for technology to global centralized strategy. Standardization benefits for manufacturing sites”, las ventajas de los criterios de estandarización aplicados en las plantas de Novartis a nivel mundial, así como el ahorro de costes y mejora de la gestión alcanzado con esta decisión. Para cerrar la jornada hubo un sorteo de una tablet entre los asistentes al acto y, seguidamente, se ofreció una excelente comida buffet servida en el mismo hotel, donde los asistentes departieron y conversaron entre ellos y con los ponentes FARMESPAÑA INDUSTRIAL Inspired by nature, driven by innovation Efficient processes are key to preserving natural product quality in the biopharmaceutical industry. We provide innovative answers to achieve this. Ingeniería. Ingeniería de procesos. Gestión de proyectos. Sistema de aguas. IV Solutions plants. Consultoría Biofarmacéutica. Análisis de requerimientos regulatorios en instalaciones (EMA, FDA). Grifols Engineering We know how. Escalado de procesos biotecnológicos. Sistemas de contención. Desarrollo de Maquinaria. CIP, SIP. Purificaciones biológicas. Dosificación estéril (GSF®). Dosificación aséptica de bolsas. Grifols Engineering, S.A. Can Guasch, 2, 08150 Parets del Vallès, Barcelona - SPAIN Tel. [34] 935 710 868 Fax [34] 935 710 393 www.grifolsengineering.com Avda de Fuentemar, 31 28823 Coslada (Madrid) - SPAIN Tel: [34] 917 479 466 5555-Valley Boulevard, Los Angeles California 90032 - USA Tel. (323) 227 7016 Fax (323) 441 7928 CLIMATIZACIÓN Miguel Ruiz, Responsable HVAC en INGELYT Con una correcta instalación un recuperador de placas puede ser un elemento de ahorro energético válido en las instalaciones farmacéuticas y hospitalarias. En este artículo se desmonta la leyenda negra que rodea a estos intercambiadores. ¿Quién teme a los recuperadores de placas? L a recuperación de energía en las extracciones en los sistemas de tratamiento de aire es un punto importante en un sistema de eficiencia energética. Dentro de los sistemas de tratamiento de aire, los sistemas que trabajan en “todo aire exterior” son los más sensibles a la recuperación energética. En la industria farmacéutica, biotecnológica, laboratorios de investigación, incluso en el ámbito hospitalario, los sistemas tipo “todo aire exterior” son muy comunes y en muchos casos de uso obligatorio. En un sistema 100% aire exterior la corriente de aire de extracción contiene una gran cantidad de energía térmica, por lo que la recuperación de esa energía es muy importante en el balance energético de la instalación. La recuperación de energía en el aire de extracción es un tema bien conocido en el mundo de la climatización convencional; dentro de los diversos sistemas de recuperación destacan los recuperadores de flujos cruzados, también conocidos como recuperadores de placas o recuperadores estáticos. Los recuperadores de placas representan un sistema muy económico de recuperación, con un mantenimiento nulo y ratios de recuperación energética supe- riores al 50%. Estas características les hacen ser muy apreciados en la recuperación energética del aire de extracción en los sistemas de climatización convencional. Sin embargo en algunos ámbitos de la industria farmacéutica, laboratorios de contención y, sobre todo, en el ámbito hospitalario los recuperadores de placas no están bien vistos y en algunos casos están expresamente prohibidos. ¿A qué se debe que un elemento sencillo, barato y eficiente sea rechazado en ámbitos en los que el ahorro energético puede ser crucial? La Leyenda Negra Básicamente, el rechazo a los recuperadores de placas viene del hecho de que los fabricantes han “confesado” que no son totalmente estancos y pueden tener una tasa de fugas del orden de 0,1%, con diferenciales de presión de 400 Pa. Hay fabricantes de elementos de arquitectura y cerramientos (también usados en los hospitales) que a tasas de fuga muy superiores a ese orden lo denominan “estanqueidad total”. Sin embargo, en algunos ámbitos farmacéuticos y hospitalarios esta tasa de fugas, aplicada al caso particular de los recuperadores, se declara “inadmisible” y, como consecuencia, los recuperadores de placas son vetados sin ninguna opción de defensa. EN UN SISTEMA 100% AIRE EXTERIOR LA CORRIENTE DE AIRE DE EXTRACCIÓN CONTIENE UNA GRAN CANTIDAD DE ENERGÍA TÉRMICA, POR LO QUE LA RECUPERACIÓN DE ESA ENERGÍA ES MUY IMPORTANTE EN EL BALANCE ENERGÉTICO DE LA INSTALACIÓN 24 NOVIEMBRE/DICIEMBRE14 De arriba a abajo: recuperador, detalle del intercambiador y detalle de las placas. Desmontando el Mito Sin entrar a valorar si 0,1 % es mucho o poco ¿nadie se ha preguntado hacia dónde va esa fuga o hacia dónde se puede conducir esa fuga? Porque, ¿qué pasa si es el aire “nuevo” el que se mezcla en una proporción de 0,1% con el aire de extracción que se expulsa al exterior? ¿Por qué se presupone que es el “aire contaminado” el que se mezcla con el “aire nuevo” y vuelve a entrar al recinto tratado? FARMESPAÑA INDUSTRIAL CLIMATIZACIÓN Figura 1. Figura 2. Lo que ocurre en el interior de un recuperador de placas no es nada que no obedezca a las leyes de la física: si hay una fuga, ésta irá desde la zona de mayor presión hacia la zona de menor presión, por tanto, si se puede garantizar que el aire contaminado se mantiene a presiones inferiores a las del aire “nuevo”, no habría ninguna posibilidad de contaminación, siempre que el diferencial de presiones entre ambos lados mantenga una amplitud suficiente para contrarrestar posibles efectos venturi. Hay que recordar que, en el interior de las zonas de contención, presiones diferenciales de 10 y 15Pa entre salas “contaminadas” y “no contaminadas” separadas por una simple puerta (cuya tasa de fuga es desde luego bastante mayor de 0,1%) se consideran suficientes para garantizar el nivel de contención. Un recuperador de placas está formado por la superposición de placas paralelas que forman canales por los que pasan los 26 NOVIEMBRE/DICIEMBRE14 flujos de aire. Cada placa separa la corriente de impulsión de la corriente de extracción y, mediante la transmisión térmica a través de la superficie de placa, ambas corrientes intercambian su energía sin mezclarse. Para que el intercambio en el recuperador sea eficiente es necesario un alto nivel de turbulencia en la corriente de aire y esta turbulencia se traduce en perdida de carga. Cuanto mayor es la perdida de carga, mayor es la turbulencia y por tanto mayor la recuperación; de hecho perdidas de carga inferiores a 100Pa hacen que un recuperador sea prácticamente ineficiente. Las presiones operativas de un recuperador oscilan entre los 150 y 250Pa. Por debajo de 150Pa el recuperador es ineficiente y por encima de 250-300Pa el consumo energético de ventilador puede empezar a penalizar el balance total de energía del sistema. Por tanto, un valor del orden de 175200Pa se puede considerar adecuado. Los flujos de aire de extracción e impulsión no se mezclan, ya que están separados por las placas, pero en los bordes, donde las placas se cierran mediante pliegues, pueden producirse fugas de una corriente a otra. En recuperadores de buena calidad esta corriente de fuga se ha cuantificado en un 0,1% para diferencias de presión de 400Pa. En una primera aproximación podría pensarse que simplemente el mantenimiento de la corriente de extracción a presiones menores que la corriente de impulsión sería suficiente para asegurar que la fuga se produce en el sentido de la seguridad, es decir de la corriente de aire “limpio” hacia la de aire “sucio”. En los recuperadores de placas los flujos de aire circulan “cruzados”, sus direcciones están giradas 90º por lo que hay zonas donde una corriente está entrando al recuperador y la otra corriente está saliendo, con lo cual una corriente está en su máxima presión para entrar al recuperador, mientras que la otra corriente ya ha consumido su presión y está saliendo del recuperador. Debido a las altas presiones en el interior del recuperador, pueden existir puntos en los que las presiones internas compensen e incluso inviertan el régimen de presiones del exterior. En la figura 1 puede verse que, aunque en líneas generales la corriente de extracción (rojo) está a menor presión que la corriente de impulsión (azul), hay zonas del recuperador (marcadas en azul) en las que el aire de extracción está a mayor presión que el aire de impulsión y la contaminación de la corriente de aire limpio con el aire de extracción sería posible. Por tanto, es muy importante la selección de la perdida de carga del recuperador en relación con las pérdidas de carga de extracción e impulsión, para evitar la aparición de zonas del recuperador con un régimen de presiones inverso. En la fig. 2 se representa el esquema más usual en la extracción de una zona de contención. En el lado de extracción hay filtración HEPA y prefiltración (al menos a nivel F8), además de toda la red de conductos de extracción. En el lado de la impulsión de “aire nuevo” solo estaría el prefiltro de protección del recuperador, normalmente a nivel F8. Por tanto está claro que la red de extracción estará a una presión bastante menor (más negativa) que la red de aire de impulsión. Transponiendo estos valores sobre un esquema típico de extracción (figura 3) se puede ver que dentro del recuperador el mínimo valor de presión entre ambas corrientes es de 200Pa, siempre favorable al aire de extracción. FARMESPAÑA INDUSTRIAL CON UNA CORRECTA INSTALACIÓN, UN RECUPERADOR DE PLACAS PUEDE MANTENER FÁCILMENTE UNA PRESIÓN DIFERENCIAL DE, AL MENOS, 75-100PA ENTRE LA CORRIENTE DE AIRE “LIMPIO” Y AIRE “CONTAMINADO”; ESTO ES 10 VECES MÁS QUE EL RÉGIMEN DE PRESIONES QUE SE CONSIDERA Figura 3. Figura 4. Como regla general la perdida de carga del recuperador debe ser inferior al diferencial de pérdida de carga entre impulsión y extracción. En el caso de la figura 1, este diferencial es de 100Pa, mientras que el recuperador tiene 200Pa, y esto da lugar a la existencia de puntos internos del recuperador con inversión de presiones. En el caso de la figura 2, el diferencial es de 400Pa por lo que los 200Pa del recuperador no pueden producir inversión de presiones. La colmatación de filtros puede alterar el valor de la diferencia entre extracción e impulsión, sobre todo por el hecho de que el filtro F8 en la entrada de aire exterior se colmatará más rápido y más frecuentemente que los filtros de extracción, aumentado su pérdida de carga y disminuyendo el diferencial de presión entre ambas corrientes de aire. Un control de la presión diferencial entre los dos puntos FARMESPAÑA INDUSTRIAL críticos del recuperador (manómetro “P” en la figura 3) sería recomendable para asegurar y certificar que en ningún momento hay una inversión de presiones en el interior del recuperador. Otra forma de asegurar que en todo momento se mantiene el régimen de presiones es instalar la extracción en “aspiración” con presión negativa pura e instalar la impulsión en presión positiva pura, situando el ventilador de impulsión antes del recuperador. Esta disposición asegura un régimen correcto de presiones en todo momento, independientemente del grado de colmatación de filtros: aire contaminado en presión negativa pura y aire nuevo en presión positiva pura; pero somete a las placas del recuperador a un diferencial de presiones muy alto que puede llegar a provocar deformaciones permanentes. Como puede verse en la figura 4, el ventilador de “SEGURO” impulsión no sólo tiene que vencer la perdida de carga del recuperador, sino de los filtros, baterías y red de conductos, por lo que al colocar el ventilador de impulsión antes del recuperador la presión en este sería la de todo el sistema, no solo su propia perdida de carga. En el ejemplo de la figura 4 se han considerado presiones muy moderadas; seguramente una instalación de este tipo requeriría incluso presiones mayores en su régimen de funcionamiento. Aun así, puede verse que las diferencias de presión en el interior del recuperador llegan a alcanzar los 1.300Pa. En general, los fabricantes recomiendan que no haya diferencias mayores de 700800Pa entre las placas en recuperadores pequeños y 1800 Pa en recuperadores grandes, aunque hay recuperadores especiales que pueden soportar diferencias de 6.000 Pa e incluso, con placas de acero inoxidable, alcanzan los 8.000Pa. Por tanto, una composición en presiones positivas/negativas puras como la indicada en la figura 4 es técnicamente posible, siempre que se seleccione el recuperador correcto. Conclusión Con una correcta instalación, un recuperador de placas puede mantener fácilmente una presión diferencial de, al menos, 75100Pa entre la corriente de aire “limpio” y aire “contaminado”; esto es 10 veces más que el régimen de presiones que se considera “seguro” dentro de las propias zonas de contención. Por otra parte, la corriente de aire “contaminado” ha sido filtrada por filtro HEPA, por lo que es un 99,999% más limpia que el ambiente interior de la zona en contención. La tasa de fugas del recuperador es de solo un 0,1% y, por último, es el sistema de recuperación más económico de instalar y mantener, con tasas de recuperación superiores al 50%. Por tanto: ¿Quién teme a los recuperadores de placas? NOVIEMBRE/DICIEMBRE14 27 TRATAMIENTO DE AGUAS Ana Lázaro Martín, Directora Ingeniería TCI, S.L.L. Se denomina rouging al proceso de corrosión general o uniforme que se produce en los aceros inoxidables, cuya coloración característica se debe a la presencia de óxidos de hierro. Este fenómeno cobra importancia en sistemas de agua de alta pureza y /o vapor. En el siguiente artículo se repasan las causas y factores que contribuyen a la aparición del rouging, así como los métodos de detección, eliminación y la posición de las distintas normativas de la industria. Aceros inoxidables y rouging 1. Aceros inoxidables Los aceros inoxidables son aquellos aceros en los que el porcentaje de Cromo es superior al 11,5%. Se llaman inoxidables porque a partir de ese porcentaje de Cromo, la velocidad de corrosión del acero disminuye drásticamente, lo que no significa que desaparezca. Tasa de corrosión de aleaciones Hierro-Cromo en pulverización intermitente de agua a temperatura ambiente. Fuente: W. Whitman and E. Chappel, Industrial and Engineering Chemestry Fundamentals, vol 13 (1926) p. 533. El uso de los aceros inoxidables está muy extendido porque, además de la ya elevada resistencia mecánica de los aceros en general, la presencia de Cromo mejora su resistencia a la corrosión, su resistencia a la oxidación y su aspecto exterior. Los tipos principales de aceros inoxidables son: 1. Ferríticos: aquellos en los que el porcentaje de Cormo está entre un 12% y un 30%. Este tipo de aceros inoxidables apenas contiene Carbono, su presencia es inferior al 0,1% en peso. 2. Martensíticos: aquellos en los que el porcentaje en Cromo está entre un 12% y un 27% pero con una presencia 28 NOVIEMBRE/DICIEMBRE14 de Carbono entre el 0,1% y el 1,0% en peso. 3. Austeníticos: aquellos en los que el porcentaje de Cromo está entre un 17% y un 25% y con una presencia de Níquel entre el 8% y el 20%. Este tipo de aceros presentan una estructura de estado sólido tipo FCC (cúbica centrada en las caras), lo que les otorga una resistencia a la corrosión muy alta y buenas propiedades de conformación. 4. Dúplex: aquellos que son una mezcla entre austeníticos y ferríticos, con un porcentaje de Cromo entre un 23 y un 30% y un porcentaje de Níquel entre un 2,5 y un 7%. Por sus buenas propiedades mecánicas y químicas, los aceros inoxidables más empleados en la construcción de instalaciones y equipos para fabricación de medicamentos o cosméticos son los aceros inoxidables de tipo austenítico, cuyo diagrama de fases Fe-Cr-Ni es el siguiente: Diagrama Fe-Cr-Ni a temperatura ambiente (Wever y Jellinghaus) Donde: _ o Ferrita es la fase del acero con estructura en estado sólido tipo BCC (cúbica centrada en el cuerpo), estable desde temperatura ambiente hasta 910ºC y a partir de 1400ºC hasta los 1510ºC, donde el acero pasa a estado líquido. En su composición el contenido de Carbono debe ser inferior al 0,09% en peso. ¯ o Austenita es la fase del acero con estructura en estado sólido tipo FCC (cúbica centrada en las caras), estable a temperaturas entre los 910ºC y los 1400ºC, para contenidos en Carbono inferiores al 2% en peso. m o Martensita es la fase del acero con estructura en estado sólido tipo BCC (cúbica centrada en el cuerpo), formada a partir de acero austenítico templado a muy alta velocidad y, por tanto, con la misma composición química que éste. El porcentaje en Carbono debe mantenerse siempre muy bajo, preferiblemente por debajo del 0,03% para evitar que retire el Cromo, formando con él carburos (Cr23C6), que entre otros inconvenientes, favorecen la corrosión intergranular del acero inoxidable. Dentro del grupo de los aceros austeníticos están los correspondientes a la serie 300 según la American Iron and Steel Institute (AISI). Las propiedades que presentan estos aceros derivan de su naturaleza de la siguiente manera: Por tener estructura FCC: son paramagnéticos, tenaces a bajas temperaturas y fácilmente conformables. Por ser monofásicos: son fácilmente soldables y endurecen cuando son trabajados en frío. FARMESPAÑA INDUSTRIAL 2. Rouging Como ya hemos visto, los aceros inoxidables austeníticos son los más resistentes a la corrosión, pero no son inmunes. La corrosión puede dividirse en dos grandes grupos: corrosión general o uniforme y corrosión localizada: La corrosión general es aquella en la que la disolución del metal es uniforme en la superficie del acero en contacto con el medio corrosivo. La corrosión localizada es aquella en la que la disolución del acero se produce sólo en una pequeña zona, pero el grado de ataque es relativamente alto. En el caso de la corrosión general o uniforme, el grado de corrosión se mide en mm/año o micras/año. En los aceros inoxidables, la corrosión general o uniforme también se llama “ROUGING”, por su color rojizo. En estas reacciones, el FeO(OH) u óxido ferroso hidratado es el óxido responsable del color anaranjado y se trata de la forma correspondiente al mineral conocido como Limonita. El Fe2O3 u óxido férrico es el óxido responsable del color azulado y el Fe3O4, conocido como magnetita es el óxido responsable del color negro. La presencia de uno de estos óxidos o de varios de ellos explica las distintas coloraciones del fenómeno rouging. Rouging en una tubería de agua WFI a 80ºC. Proliferación del Rouging en el interior de un reactor de fabricación 2.1. Responsables de la coloración La inspección de muestras de aceros inoxidables con distintos tipos de rouging, mediante las técnicas de Microscopía Electrónica (SEM), espectroscopía de energía dispersiva (EDS) y espectroscopía de RX (XPS) ha demostrado que las distintas coloraciones del rouge se deben, en todos los casos, a la presencia de óxidos de Fe. El resto de óxidos metálicos que pueden detectarse, no aportan coloración. Esta capa de oxidación comienza como una película estable de la que no se desprenden partículas. A medida que el espesor de rouging crece, la superficie de contacto va cambiando de color, desde el dorado, pasando por el azul hasta llegar a distintos matices de negro. Las relaciones estequiométricas que explican la formación de los óxidos de hierro responsables de la coloración del rouging son las siguientes: 2Fe + 4H2O A 2FeO(OH) + 3H2(g) 2FeO(OH) AFe2O3 + H2O 2Fe + 3H2O A Fe2O3 + 3H2(g) FeO + Fe2O3 A Fe3O4 FARMESPAÑA INDUSTRIAL 2.2. Formación del rouging La teoría más extendida para explicar la formación del rouging se basa en la avidez de las aguas de alta pureza y el vapor puro por el dióxido de carbono del aire. Cuando el agua y el aire entran en contacto, parte del CO2 se disuelve en forma de ácido carbónico (H2CO3). El ácido carbónico disuelto en el agua crea un medio reductor que ataca la capa superficial de los aceros inoxidables. El ataque de esta capa permite al Fe quedar expuesto y ser oxidado, formando el rouging. Los fluidos más puros como el agua calidad inyectable a alta temperatura o el vapor puro son muy agresivos y atacan a los aceros, así como a toda una variedad de metales pesados. Estas interacciones entre el oxígeno y el dióxido de carbono y los iones metálicos dan como resultado óxidos y carbonatos metálicos que componen los depósitos coloreados conocidos como rouging. Rouging en el interior de una válvula de membrana de un lazo de WFI a 80ºC. Esta hipótesis parece que sugiere que si se extinguiera la fuente de oxígeno y dióxido de carbono, el fenómeno de ataque se detendría. Sin embargo, en el caso de los sistemas de aguas altamente purificadas o de vapor puro, el agua y el vapor se están consumiendo y generando constantemente, por lo que el fenómeno no se detiene en ningún momento. 2.3. Factores que contribuyen a la formación del rouging Después de analizar los potenciales contribuidores a la formación del rouging se han identificado los siguientes aspectos: 1. Composición química de los materiales y acabado superficial. 2. Métodos de fabricación e instalación. 3. Entorno del proceso: temperatura, velocidad del fluido y presencia de gases. 2.3.1. Composición de los materiales Influencia de la composición química Como ya hemos visto anteriormente, en la industria farmacéutica y biotecnológica, los aceros más utilizados para entrar en contacto directo con el producto son los aceros inoxidables austeníticos. Dentro de esta gran familia de aceros, los más utilizados son el acero inoxidable AISI 316 y el AISI 316L. En desuso han quedado ya los aceros AISI 304 y AISI 304L. La “L” de “bajo contenido en carbono” ayuda a minimizar la cantidad de carburos que se forman durante el proceso de soldadura, aumentando así su resistencia a la corrosión por fatiga (véase tabla 1). Sin embargo, con esta denominación, AISI 316L, en función del estándar que se utilice, se pueden encontrar diferentes composiciones químicas. Para entenderlo mejor, presentamos la tabla comparativa 2. Las propiedades mecánicas y ante la corrosión del AISI 316L mejoran cuando, dentro de los rangos establecidos para este tipo de acero: Se minimiza el contenido en Carbono, Fósforo y Azufre. Se maximiza el contenido en Cromo, Manganeso, Molibdeno, Nitrógeno, Níquel y Silicio. Influencia del acabado superficial. El grado de rouging depende también del tipo de acabado que tenga la superficie del acero inoxidable, si es una superficie no pulida, si está pulida mecánicamente o si está electropulida. La selección de un tipo de acabado u otro no es sencilla. Por un lado se sabe que NOVIEMBRE/DICIEMBRE14 29 TRATAMIENTO DE AGUAS TIPOAISI ϯϬϰ ϯϬϰ> ϯϭϲ ϯϭϲ> %Cr ϭϴͲϮϬ ϭϴͲϮϬ ϭϲͲϭϴ ϭϲͲϭϴ %Ni ϴͲϭϮ ϴͲϭϮ ϭϬͲϭϰ ϭϬͲϭϰ %C(máx.) Ϭ͕Ϭϴ Ϭ͕Ϭϯ Ϭ͕Ϭϴ Ϭ͕Ϭϯ %Fe ϲϱͲϳϭ ϲϱͲϳϭ ϲϮͲϲϵ ϲϮͲϲϵ %Mo Ϭ Ϭ ϮͲϯ ϮͲϯ biofilm suponen un riesgo para la capa de pasivación y, por tanto, un acicate para la aparición de rouging. Tabla 1. ELEMENTO ƌ DŶ DŽ Eŝ W ^ŝ ^ E ASTMA270 Ϭ͕ϬϯϱŵĂdž͘ ϭϲ͕ϬͲϮϬ͕Ϭ Ϯ͕ϬŵĂdž͘ Ϯ͕ϬͲϯ͕Ϭ ϭϬ͕ϬͲϭϰ͕Ϭ Ϭ͕ϬϰϱŵĂdž ϭ͕ϬŵĂdž Ϭ͕ϬϬϱͲϬ͕Ϭϭϳ DIN17457 Ϭ͕ϬϯŵĂdž͘ ϭϲ͕ϱͲϭϴ͕Ϭ Ϯ͕ϬŵĂdž͘ Ϯ͕ϱͲϯ͕Ϭ ϭϮ͕ϱͲϭϱ͕Ϭ Ϭ͕ϬϰŵĂdž Ϭ͕ϳϱŵĂdž Ϭ͕ϬϯŵĂdž͘ Ϭ BS316S12 Ϭ͕ϬϯŵĂdž͘ ϭϲ͕ϱͲϭϴ͕Ϭ Ϭ͕ϱͲϮ͕ϬŵĂdž Ϯ͕ϮϱͲϯ͕ϬϬ ϭϭ͕ϬͲϭϰ͕Ϭ Ϭ͕ϬϰϱŵĂdž ϭ͕ϮͲϭ͕ϬŵĂdž Ϭ͕ϬϯŵĂdž͘ Ϭ ENDIN1.4404 Ϭ͕ϬϯŵĂdž͘ ϭϲ͕ϱͲϭϴ͕Ϭ Ϯ͕ϬŵĂdž͘ Ϯ͕ϬͲϮ͕ϱ ϭϬ͕ϬͲϭϯ͕Ϭ Ϭ͕ϬϰϱŵĂdž ϭ͕ϬŵĂdž Ϭ͕ϬϭϱŵĂdž Ϭ Ϭ͕ϭϭŵĂdž ENDIN1.4435 Ϭ͕ϬϯŵĂdž͘ ϭϳ͕ϬͲϭϵ͕Ϭ Ϯ͕ϬŵĂdž͘ Ϯ͕ϱͲϯ͕Ϭ ϭϮ͕ϱͲϭϱ͕Ϭ Ϭ͕ϬϰϱŵĂdž ϭ͕ϬŵĂdž Ϭ͕ϬϭϱŵĂdž Ϭ͕ϭϭŵĂdž Tabla 2. la formación del rouging es más lenta en el caso de que la superficie del acero inoxidable haya sido electropulida antes de ponerla en contacto con el medio, pero, por otro lado, la eliminación del rouging que se forme, por medios mecánicos o químicos puede dañar significativamente el acabado electropulido además de reducir la vida efectiva de los componentes en cuestión. Imagen de SEM de tubería antes y después de ser electropulida. 2.3.2. Métodos de fabricación Para la construcción de los equipos y sistemas de tuberías empleados en la industria farmacéutica y biosanitaria se utiliza la soldadura TIG, de manera manual o bien automática, con máquina orbital. Este tipo de soldadura, en atmósfera inerte de gas, garantiza que durante el proceso de calentamiento del acero para soldarlo no sea posible la presencia de oxígeno y, por tanto, la formación de óxidos metálicos, que quedarían incrustados en el cordón de soldadura. La presencia de estos óxidos permitiría la proliferación de la corrosión por picadura, la fragilización de la unión y la proliferación del rouging. 30 NOVIEMBRE/DICIEMBRE14 Proliferación del Rouging alrededor de una soldadura no TIG. 2.3.3. Entorno del proceso Influencia de la temperatura La influencia de la temperatura es el efecto cuya repercusión en la aparición del rouging mejor se conoce. No es posible establecer un valor de temperatura a partir del cual la evolución del rouging sea distinta, sino que se trata de algo gradual. En sistemas de agua a baja temperatura, es decir, entre 4 y 10ºC, y hasta temperatura ambiente el rouging normalmente no aparece. Sin embargo, en sistemas a partir de 65ºC y en las redes de vapor puro, el rouging es habitual. También es habitual la presencia de rouging en los sistemas a temperatura ambiente que son sanitizados a alta temperatura periódicamente. Influencia de la velocidad Los sistemas de aguas purificadas, altamente puras e inyectables suelen diseñarse con velocidades mínimas de 1,5m/s (5f/s) para evitar que pueda producirse adhesión bacteriana a las superficies metálicas, que creen biofilm. Las capas de Rouging en el interior de una bomba de un lazo de WFI a 80ºC. Por otro lado, una velocidad de circulación de agua excesiva puede provocar burbujeos o cavitación, sobre todo en la parte central del impulsor de la bomba. Estas burbujas impactan a gran velocidad sobre la bomba y la tubería de impulsión y pueden ocasionar daños en la capa superficial del acero. Como sucede con la formación de biofilm, si la capa superficial resulta dañada, la aparición de rouging será muy probable. Influencia de la presencia de gases Se ha observado que la presencia de oxígeno disuelto en el agua disminuye el desarrollo del rouging. De ahí que éste sea más frecuente en los sistemas a alta temperatura, donde los gases son menos solubles. También se ha observado cierta relación entre la presencia de CO2 gas y la formación de rouging. De ahí que en los sistemas de almacenamiento y distribución de aguas altamente purificadas o en los depósitos de fabricación, en los que hay renovación constante del aire en contacto con el agua a través del filtro de venteo de los tanques, la presencia de rouging en las zonas del depósito donde impacta el agua rociada por las bolas de limpieza, es una realidad. Rouging en la zona de aspersión de la bola de limpieza de un tanque de almacenamiento de agua purificada a 20ºC. FARMESPAÑA INDUSTRIAL TRATAMIENTO DE AGUAS Sin embargo, aún no ha conseguido demostrarse que la inertización del depósito mediante introducción contralada de Nitrógeno consiga eliminar la presencia de este fenómeno. Este proceso, llamado blanketing, debe seleccionarse si con él se consigue proteger la calidad del producto, pero no como método para evitar el rouging. 2.4. Métodos de detección del rouging La detección de la presencia de rouging en los sistemas de aguas altamente purificadas y vapor puro fabricados en acero inoxidable AISI 316L y expuestos a altas temperaturas de manera continua o intermitente, no es posible mediante mediciones de temperatura, caudal, presión, conductividad o TOC. La presencia de rouging puede detectarse mediante análisis del fluido de proceso (métodos no invasivos) o bien mediante análisis de la composición de las capas superficiales del acero (métodos invasivos). Los métodos de análisis de muestras del fluido de proceso son métodos en los que se puede identificar la composición química del rouging y cuantificarla, sin embargo, requieren de muestreos continuos y periódicos en diferentes puntos del sistema, lo que incrementa considerablemente la carga de trabajo. En el caso de los métodos invasivos de análisis de la superficie del material, lo que se suele hacer es colocar testigos en diferentes puntos de la instalación, que periódicamente se van cambiando, para poder ser estudiados con métodos destructivos en laboratorio. Rouging en una junta clamp de PTFE de un lazo de WFI a 80ºC. Actualmente, existen métodos de monitorización del rouge. Para ello utilizan equipos de detección visual mediante la medida del grado de reflexión de la superficie del acero inoxidable. Estos equipos proporcionan alarmas en el caso de que el grado de reflexión cambie. El inconveniente de estos métodos es que el equipo no distingue si el 32 NOVIEMBRE/DICIEMBRE14 cambio en el grado de reflexión es debido a la presencia de rouging o en realidad se debe a cualquier otro motivo. En cualquier caso, ninguno de los métodos que se emplean actualmente para la detección y/o medición del rouging aporta información alguna sobre las medidas a implantar para evitar su aparición o sobre el impacto que tiene en la calidad del fluido de proceso en el sistema en cuestión. 2.5. Métodos de eliminación del rouging Los sistemas de aguas altamente purificadas y de vapor puro, fabricados en acero inoxidable AISI 316L y expuestos a altas temperaturas de manera continua o intermitente, deben disponer de un nivel máximo de aceptación de cantidad de partículas y óxidos metálicos, basado en un análisis de riesgos que incluya daños potenciales en el producto. Junto con estos datos, debe disponerse de métodos para la eliminación del rouging basados en la observación y la cuantificación del aumento tanto del nivel de partículas, como de la acumulación de óxidos en la superficie metálica. Hasta el momento no ha podido demostrarse que la presencia de rouge en el sistema altere la calidad del fluido. Sin embargo, se trata de partículas de óxidos metálicos que no deberían permanecer en el sistema y que, después del correspondiente análisis de riesgos una vez detectadas, deben implementarse los métodos para su eliminación. Para la eliminación del rouging, existen métodos físicos y métodos químicos, sin embargo, ninguno de ellos garantiza que el rouging no vuelva a aparecer. Se trata de procedimientos que deberán repetirse con la frecuencia que determine el análisis de riesgos disponible. Los métodos físicos, como el pulido, pueden ser utilizados en aquellos casos en los que todas las superficies afectadas por el rouging sean accesibles. Sin embargo, estos métodos no están muy extendidos porque el pulido implica la disminución del espesor de la pared del acero y debe hacerse con sumo cuidado. Además, estos métodos no son válidos en los lazos de agua y las redes de distribución de vapor puro, porque las superficies a limpiar ya no son accesibles a la herramienta del pulidor. Por tanto, los métodos más empleados para eliminar el rouging son métodos químicos. Entre los compuestos químicos utilizados para la eliminación del rouge en los sistemas de aguas altamente purificadas y vapor puro, se encuentran las disoluciones de ácido nítrico (HNO3), de ácido cítrico (C6H8O7), de ácido fluorhídrico (HF) y de fluoruro de amonio (NH4HF2) o mezclas de ellas. En el caso de este tipo de rouging, la efectividad del ataque químico dependerá también del tipo de acabado superficial del acero inoxidable. Cuanto menor sea la rugosidad del acero, mejor será el resultado del ataque. En estos casos, los ácidos orgánicos más débiles precisarán de altas temperaturas (de 60ºC a 80ºC) y largos tiempos de exposición (de 8 horas a 48 horas o más), las disoluciones fluoradas de ácido nítrico de temperaturas medias (de ambiente a 40ºC) y las disoluciones fluoradas de ácido cítrico de altas temperaturas y cortos períodos de exposición (de 2 a 24 horas). Antes de proceder a la ejecución de la limpieza química, deberán retirarse del sistema todos aquellos componentes que puedan verse dañados. Asimismo, dependiendo del producto químico empleado y de la naturaleza de los elastómeros presentes en el sistema, es posible que tras la limpieza, sea necesaria la sustitución de todos los elastómeros que hayan estado en contacto con el producto de limpieza. Reactor antes y después de eliminar el rouging. Una vez realizado el ataque químico con ácido, el producto deberá neutralizarse antes de ser vertido a la red de drenajes. El sistema objeto de la limpieza deberá ser aclarado con abundante agua, de la misma calidad que la del producto final, y deberá verificarse de manera analítica, mediante medida de conductividad o medida de pH, que se ha eliminado por completo del sistema. Es recomendable proceder a la pasivación del sistema una vez que se ha eliminado el rouging. Esto ayudará a disminuir la frecuencia con la que es necesario proceder a la eliminación de este fenómeno en dicho sistema. 2.6. Posicion de las normativas FDA Actualmente, la FDA no dispone de posición documentada sobre el rouging, su existencia, o presencia en sistemas de agua ultra pura, vapor puro o proceso farmacéutico. La 21 CFR (Code of Federal Regulations) Capítulo 1, parte 211, subparte FARMESPAÑA INDUSTRIAL D – Equipment, Sección 211.65(a) afirma que los equipos y sistemas deberán construirse de forma que la superficie de contacto de sus componentes no deben ser reactivos, aditivos o absortivos de manera que no puedan alterar la seguridad, la naturaleza, la actividad, la calidad o la pureza del producto más allá de los requerimientos establecidos. En relación con la limpieza y mantenimiento de los equipos e instalaciones, la 21 CFR (Code of Federal Regulations) capítulo 1, parte 211, subparte D – Equipment, sección 211.67(a) dice que los equipos y utensilios deben ser limpiados, mantenidos y sanitizados a intervalos apropiados para prevenir fallos y contaminaciones que pudieran alterar la seguridad, naturaleza, calidad o pureza del producto, más allá de los requerimientos establecidos. USP La USP ni identifica el rouging como un contaminante, ni propone límites de alerta / acción o métodos para la detección. El ámbito de la USP alcanza la calidad del agua que se usa, pero el sistema que la proporciona y el rouging es un asunto relacionado con los materiales seleccionados en el sistema. La USP requiere muestreos representativos; por tanto, la calidad del sistema se deduce de las muestras y de la calidad de éstas durante los períodos de muestreo. Además, los criterios de diseño van dirigidos a asegurar la calidad del agua y a mantener el sistema bajo control durante largos períodos de tiempo. El usuario deberá comprobar si la calidad del agua obtenida de un sistema que muestra presencia de rouging está de acuerdo con la USP así como con sus requerimientos de usuario. No existen guías o regulaciones específicas para eliminar el rouging o para establecer unas frecuencias de eliminación. Hasta ahora no hay casos documentados que demuestren que la presencia de rouging en sistemas de agua ultrapura o vapor puro haya provocado que éstos dejen de cumplir los requerimientos de las actuales farmacopeas. EP La Farmacopea Europea no aborda el tema del rouging ni da ninguna pauta a este respecto Bibliografía – – – – – – – – – – ISPE Baseline Guide, 2011, “Water and Steam Systems”, Chapter 10. Michelle Gonzalez, September/October 2001, “Stainless Steel Tubing in the Biotechnology Industry”, Pharmaceutical Engineering Journal, Vol.21, No. 5. Joseph J. Manfredi, March 2006, “Materials”, Ultrapure Water. John C. Tverberg and James A. Ledden, 1999, “Rouging of Stainless Steel in WFI and High Purity Water Systems”, Institute for International Research, “Preparing for Changing paradigms in High Purity Water”. Troels Mathiesen and Jan Elkjaer Frantsen, 2007, “Rouging of stainless steel in WFI systems – Examples and present understanding”. NACE Corrosion, Paper 07193. John J. Kilkeary, Daryl L. Roll and Tim Sowell, 2000, “New Developments in Passivation technolgy”, Interphex. ASME BPE, The American Society of Mechanical Engineers, Bioprocessing Equipment, An American National Standrad. ASME B31.3, Teh American Society of Mechanical Engineers, process, Piping, An American National Standard. W.Whitman and E. Chappel, Industrial and Engineering Chemistry Fundamentals, vol.13, p.533. E.E. Thum, “Book of Stainless Steels”, 2ª. Ed., American Society for Metals. Somos una empresa de servicios externa e independiente que ofrecermos los servicios que a continuación detallamos: tREALIZAMOS TODO LO RELACIONADO EN EL ÁMBITO DE SEGURIDAD E HIGIENE. CONTEO DE PARTÍCULAS ESTADO DE FILTROS ETC.. MEDICIONES AMBIENTALES. MEDICIONES DE CAMPOS ELECTROMAGNÉTICOS. tAUDITORIAS/ VALIDACIÓN DE ARMARIOS, VITRINAS, CENTRIFUGAS CABINAS SEGURIDA CLASE III, CAMPANAS, LABORATORIOS FARMCEUTICOS, ESTERILIZADORES,QUIROFANOS, SALAS BLANCAS, SALAS- AREAS LIMPIAS, LABORATORIOS ETC. tNUESTRA EMPRESA DISPONE DE TODOS LOS REGISTROS DE METROLOGIA 16 V / 006 / 98 IRI ENT.CONSULTORIA 108183 tREPARACIONES/ MANTENIMIENTO DE EQUIPOS ELECTROMEDICOS. CON CERTIFICADO DE CALIBRACION POR LABORATORIO ACREDITADO EN METROLOGIA. 16 I/154R.REGISTRO INDUSTRIAL 28/101183, CUMPLIMOS CIRCULAR 3/2012 DEL MINISTERIO DE SANIDAD SERVICIOS SOCIALES E IGUALDAD. tMEDICIONES DE AGENTES QUÍMICOS REAL DECRETO 374/ 2001. BOE 104. tCertificados Láser de diagnostico, Yag, Argón, Rubí, Alejandrita, Diodo Etc. Equipos de Estética Ultrasonidos, Microondas, Magnoterapia etc. t REVISIONES SEGURIDAD ELÉCTRICAS. EMPRESA AUTORIZADA EN BAJA / ALTA TENSION. t Preparación y Montaje de Cuadros Eléctricos. t Acometidas Eléctricas. t Tendido de Redes Eléctricas y de Comunicaciones. t Certificación de Instalaciones de Baja Tensión según REBT aprobado según Real Decreto 842//2002, de 2 de agosto. C/ Velázquez, Nº119 - 3ºF 28006 Madrid Tel. / Fax: 91 564 86 31 Urgencias: 626 70 76 46 Email: [email protected] FARMESPAÑA INDUSTRIAL www.picweb.es t Actualización de las instalaciones a la Normativa actual. t Proyectos de obra nueva en Edificios y Locales de publica concurrencia. t Esterilización: Áreas de riesgo y equipamiento. Desinfección de ámbito Clínico. t Equipamiento, accesorios. EQUIPOS DE PROTECCIÓN ( EPIS )-PURIFICADORES DE AIRE. PRODUCTOS DESECHABLES (Guantes Látex etc. Gorros, Batas, Mascarillas etc.)33 NOVIEMBRE/DICIEMBRE14 PRECIOS DIRECTAMENTE DE FÁBRICA. TRATAMIENTO DE AGUAS Carmen Carrillo, Directora de I, D&D. Antonio Matachana, S.A. [email protected] El tratamiento de efluentes contaminados biológicamente es indispensable en las plantas es instalaciones con áreas de biocontención. En muchos casos, dado el volumen de residuos, no es posible la esterilización manual. El tratamiento por inactivación térmica es una opción ideal en la mayoría de los casos. Inactivación térmica de efluentes biocontaminados: tipología y funcionamiento E n los últimos años, el número de áreas de biocontención ha ido aumentando de forma progresiva en toda Europa. De existir apenas unos pocos laboratorios, hemos pasado a un número muy superior de centros. Y no sólo ha crecido su número sino también su tamaño. Donde antes encontrábamos una zona de escasos metros cuadrados, en la que se generaba pocos litros de residuos biocontaminados que se recogían en un recipiente y se procesaban en el esterilizador, hoy encontramos centros que generan efluentes líquidos biocontaminados en tal volumen que la práctica de la recogida manual y su posterior procesado en el esterilizador ya no es viable. Es absolutamente indispensable procesar de forma apropiada estos efluentes antes de su vertido a la red de desagüe por varios motivos, que veremos a continuación: pueden ser los condensados provenientes de los equipos ubicados dentro de la zona de biocontención (por ejemplo, los drenajes de las cabinas de seguridad biológica). La inactivación se realizará por medios físicos o químicos de eficacia probada. Esto es, el sistema ha de poder ser validado en su ubicación final y debemos disponer de sistemas que nos permitan conocer si el proceso de inactivación ha sido correcto. El riesgo biológico La legislación española es clara en este aspecto, ya que disponemos de un Real Decreto, el 664/1997, que establece la protección de los trabajadores contra los riesgos relacionados con la exposición a agentes biológicos durante el trabajo. Esta regulación nos indica que se deben inactivar, antes de su vertido, los fluidos que puedan contener microorganismos viables. Especialmente, debemos considerar que: Los efluentes de fregaderos y duchas deberán recogerse e inactivarse antes de su liberación (facultativo en NCB3 y obligatorio en NCB4). Obviamente, ampliaremos esta inactivación a todos aquellos efluentes que se puedan considerar de riesgo, como Figura 1. Tratamiento de efluentes industriales de muy alta capacidad 34 NOVIEMBRE/DICIEMBRE14 El impacto medioambiental Cualquier vertido de agua a la red pública de desagües debe cumplir con la legislación local sobre esta materia. Están sujetos a autorización previa todos los vertidos de aguas residuales (domésticos o sanitarios o industriales) que puedan contaminar el dominio público hidráulico. Ello supone que hemos de tener en cuenta no solamente el riesgo biológico que ya hemos mencionado, sino también las posibles alteraciones de pH, presencia de contaminantes y otros elementos que puedan alterar el ciclo del agua. Por este motivo, el tratamiento de efluentes debe incorporar, en los casos que sean necesarios, un sistema adicional para la corrección del pH y de aquellos otros posibles parámetros que se encuentren fuera del requerimiento legal. Por ejemplo, y aunque estos valores pueden variar de un municipio a otro, es habitual encontrar restricciones en cuanto a la presencia de líquidos orgánicos inmiscibles en agua, elementos potencialmente corrosivos, productos que puedan generar gases nocivos… En especial, prestaremos atención específica a las restricciones en cuanto a temperatura, pH y metales tóxicos. Los tratamientos de efluentes por calor Como hemos mencionado, los efluentes líquidos de la zona de biocontención pueden ser procesados mediante tratamientos químicos o físicos. En este artículo nos centraremos únicamente en la inactivación de efluentes biocontaminados mediante el proceso físico más habitual: el tratamiento térmico. La inactivación térmica Cuando queremos inactivar un líquido biocontaminado con un tratamiento térmico, aplicaremos los principios de la esterilización por calor húmedo. Esto es, tomaremos como referencia para nuestro proceso (para temperaturas, tiempos y otros parámetros como por ejemplo valores de F0 del proceso, elección del microrganismo de reto para validar el proceso…) los mismos valores que están aceptados para la esterilización por vapor saturado. Podemos recurrir a las normas europeas que cubren la esterilización por calor húmedo para producto sanitario que, aunque desarrolladas para otro ámbito específico, son FARMESPAÑA INDUSTRIAL HAY QUE TENER EN CUENTA NO SOLAMENTE EL RIESGO BIOLÓGICO QUE YA HEMOS MENCIONADO, SINO TAMBIÉN LAS POSIBLES ALTERACIONES DE PH, PRESENCIA DE CONTAMINANTES Y OTROS ELEMENTOS QUE PUEDAN ALTERAR EL CICLO DEL AGUA perfectamente aplicables para el proceso que nos ocupa. Dentro de los tratamientos térmicos de efluentes encontraremos, además, dos grandes grupos: los tratamientos de procesado por lotes y los tratamientos de procesado en continuo. Veremos a continuación las diferencias entre un sistema y otro y las aplicaciones concretas de cada uno de ellos. El tratamiento térmico de efluentes por lotes En el procesado por lotes dispondremos, normalmente, de un primer recipiente, que puede ser de acero inoxidable, que hace las funciones de depósito acumulador de los efluentes. Este depósito enviará el líquido biocontaminado a un segundo recipiente, generalmente de igual o menor tamaño, hasta su llenado total. Una vez lleno, en este segundo recipiente (el digestor o ‘kill tank’) se calentará el fluido contaminado hasta alcanzar temperaturas de esterilización. Habitualmente, esto supone llevar el líquido hasta 134ºC y mantener esta temperatura un mínimo de 20 minutos. Cuando el líquido ya ha sido esterilizado, se procede a su enfriamiento para su posterior vertido a la red de desagües. Es importante recordar que el objeto de todo tratamiento de efluentes, sea del tipo que sea, no sólo es desactivar el fluido biocontaminado, sino verterlo a la red de desagües en condiciones seguras, como ya se ha mencionado anteriormente. Ello implica que la temperatura final del vertido, su pH y otros posibles residuos disueltos o en suspensión han de estar dentro de lo que marquen las normativas locales sobre este punto. Por ello, no sólo se debe enfriar el efluente hasta temperaturas aceptables de vertido, sino que, en algunos casos, será necesario añadir como mínimo algún tratamiento adicional de pH para regular el mismo. FARMESPAÑA INDUSTRIAL Figura 2, pie: Figura 2. Tratamiento de efluentes biocontaminados por lotes: vista general esterilización es la espora del Geobacillus Stearotermophilus y que debemos tener, al menos, una reducción de la población inicial de 1/1.000.000 para validar esterilidad), realizar un ciclo de esterilización completo y tomar muestras del fluido final, ya estéril. El cultivo de esta muestra debe indicar la reducción esperada del microorganismo de referencia. Los resultados correctos en tres ensayos repetitivos nos garantizan que nuestro tratamiento funciona de manera adecuada. Adicionalmente, el control de rutina acostumbra a ser muy simple, ya que los parámetros de proceso (temperatura y tiempo) son de fácil supervisión por parte del usuario. El tratamiento térmico de efluentes en continuo En el procesado por lotes, como hemos mencionado, tendremos unos requerimientos de suministro energético y de espacios relativamente elevados. El espacio es realmente un bien escaso, por lo que se impone la necesidad de buscar equipos de menor tamaño que, además, presenten menores consumos. Figura 3, pie: Figura 3. Detalle de doble kill tank Los tratamientos por lotes suelen tener altos requerimientos energéticos (necesarios para calentar el volumen total de efluentes hasta la temperatura de esterilización requerida) y acostumbran a ser más voluminosos que los tratamientos en continuo. Por el contrario, estos equipos son fácilmente escalables, ya que se pueden diseñar para tratar desde unos pocos litros por lote hasta varios miles de litros de efluentes manteniendo el mismo principio funcional. La gran ventaja de los equipos de tratamiento por lotes es que son sistemas fácilmente validables: se realiza la validación termométrica y/o microbiológica del lote, que es la unidad de referencia del proceso. La validación termométrica supondrá tomar lectura de la temperatura dentro del ‘kill tank’, en diferentes puntos del fluido, y comprobar que estas lecturas se ajustan al proceso estipulado; esto es, que se alcanzan, por ejemplo, 134ºC/20 minutos en todos los puntos del lote. Si deseamos realizar una validación microbiológica, únicamente debemos preparar un lote con carga microbiológica conocida (recordemos que en este tipo de proceso el organismo de referencia para comprobar la Figura 4, pie: Figura 4. Tratamiento de efluentes en continuo Esta necesidad queda perfectamente cubierta por los tratamientos de efluentes en continuo. Existen en el mercado sistemas de pequeño tamaño, coste reducido y, normalmente, con discreta capacidad de procesado, que son los equipos óptimos para las áreas de biocontención de tamaNOVIEMBRE/DICIEMBRE14 35 TRATAMIENTO DE AGUAS ño pequeño o medio no generen grandes volúmenes de efluentes biocontaminados. Igual que en el caso anterior, el efluente biocontaminado se recoge en un depósito previo. A partir de aquí, el líquido pasa a través de unos tubos calefactores que subirán la temperatura del líquido, en los casos óptimos, hasta más de 160ºC, lo que permite acortar de forma notable el tiempo de exposición, esto es, el tiempo que debemos mantener el fluido a la temperatura fijada. En este caso, sin embargo, el fluido no permanece dentro de un recipiente durante el tiempo de exposición, sino que continúa moviéndose a través del sistema. Figura 5, pie: Figura 5. Detalle de los tubos calentadores Por este motivo, el diseño del equipamiento es más crítico que en el caso anterior, ya que debemos asegurar que la velocidad de paso del líquido a través de los elementos calefactores es lo suficientemente lenta como para garantizar, con el margen de seguridad suficiente, el tiempo de exposición previsto. Y, al igual que en el caso anterior, debemos prever un sistema de enfriamiento del efluente y de compensación del pH, antes de su vertido a la red de desagües. La validación de los sistemas de efluentes en continuo presenta mayor complejidad que en los procesos por lotes. La ausencia del ‘lote’ hace que debamos definir un procedimiento de validación específico, que 36 NOVIEMBRE/DICIEMBRE14 ES IMPORTANTE RECORDAR QUE EL OBJETO DE TODO TRATAMIENTO DE EFLUENTES, mente de haber realizado adecuadamente los mantenimientos preventivos que indican los diferentes fabricantes. SEA DEL TIPO QUE SEA, NO SÓLO ES DESACTIVAR EL FLUIDO BIOCONTAMINADO, SINO VERTERLO A LA RED DE DESAGÜES EN CONDICIONES SEGURAS evalúe no sólo la temperatura alcanzada durante el proceso, sino la estimación del tiempo de exposición, que va a depender de la velocidad del fluido a través del sistema. Si optamos por la validación microbiológica, debemos establecer un proceso de muestreado por intervalos de tiempo que nos permita evaluar el funcionamiento ‘en continuo’ de nuestro equipo. El control de rutina de los parámetros físicos también será algo más complicado que en el caso anterior, puesto que, además de verificar temperaturas y tiempo, debemos asegurar que la velocidad de paso del fluido por los tubos calefactores es la correcta. Si la velocidad de paso fuese excesivamente alta, no el efluente no permanecería a la temperatura de proceso el tiempo suficiente y la esterilización del líquido no sería correcta. Es responsabilidad del fabricante del equipo aportar el procedimiento de control y validación que aplica a su diseño. Con esta información el usuario debe ser capaz de realizar el control de rutina y, si procede, la validación del equipo, con la misma facilidad que en los sistemas de tratamiento por lotes. Otras consideraciones En la elección de cualquier equipamiento debemos tener en cuenta los aspectos generales de funcionamiento del centro y también, muy especialmente, los requerimientos a nivel de usuario. En equipos críticos, como es este caso, es necesario evaluar otros puntos, tales como la claridad en la utilización (el usuario ha de poder manejar el sistema de forma ergonómica e intuitiva; los mensajes, especialmente los de alarmas, ha de ser claros y entendibles…), la posibilidad de implementar sistemas auxiliares como tratamientos de pH o equipos CIP (Clean-In-Place) si se requiere, y la gestión eficaz del mantenimiento del equipo. No debemos olvidar que estos sistemas trabajan en condiciones de altas presiones y temperaturas, por lo que su correcto funcionamiento depende fuerte- Figura 6, pie: Figura 6. Interface de usuario: simple y ergonómico ¿Qué sistema debemos elegir? El tratamiento en continuo y el tratamiento por lotes tienen características de diseño, funcionamiento y validación diferentes, que hacen que podamos elegir uno u otro en función de nuestros requerimientos específicos. Además, no podemos olvidar los otros factores, que también han de ser cuidadosamente evaluados en la elección de cualquier equipamiento: la ergonomía en uso, la facilidad de limpieza y la simplicidad en el mantenimiento son también importantes elementos a considerar. La correcta evaluación de nuestro entorno (humano, técnico, normativo y, por supuesto, presupuestario) nos llevará, sin duda alguna, a la elección del sistema más adecuado para cada una de nuestras instalaciones Información adicional y referencias – – – – Guía Técnica para la evaluación y prevención de los riesgos relacionados con la exposición a agentes biológicos http://www.insht.es/InshtWeb/Contenidos/ Normativa/GuiasTecnicas/Ficheros/agen_bio.pdf OMS, Manual de bioseguridad en el Laboratorio, Tercera edición, http://www.who.int/csr/resources/publications/ biosafety/CDS_CSR_LYO_2004_11SP.pdf Norma UNE-EN 17665-1 Esterilización de productos sanitarios. Calor húmedo. Parte 1: Requisitos para el desarrollo, validación y control de rutina de un proceso de esterilización para productos sanitarios Norma UNE-EN 556-1 Esterilización de productos sanitarios. Requisitos de los productos sanitarios para ser designados “ESTÉRIL”. Parte 1: Requisitos de los productos sanitarios esterilizados en su estado terminal. FARMESPAÑA INDUSTRIAL Cleaner than clean. Stericlean. www.staubli.es Aplicaciones farmacéuticas, médicas y ciencias de la vida. Diseñados para proteger al producto y a la plantilla, los robots Stericlean, completamente resistentes al H2O2, automatizan los procesos tanto en entornos aislados como en sala blanca. Los Stericlean le permiten optimizar y proteger su producción con altos estándares de calidad, exactitud, flexibilidad y fiabilidad. Stäubli Robotics es su socio para una automatización aséptica. Stäubli Española, S.A., +34 93 720 54 08 Staubli es una marca de Stäubli International AG, registrada en Suiza y otros países. © Stäubli, 2014 Bioquell: Probado por etamente pl m co t bo ro H O2 resistente al 2 FILTRACIÓN Equipo Técnico de ICT FILTRACIÓN www.ictfiltracion.com Los sectores industriales Farmacéutico y Químico (API) son dos referentes de calidad y seguridad en procesos productivos y producto final por su alto nivel de exigencia normativa. Debido a esto, y alineándose en el mismo objetivo de calidad y seguridad, los proveedores de sistemas y elementos de filtración de estos sectores industriales deben autoexigirse el mismo nivel de cumplimiento normativo. La filtración como aliado del rendimiento y la calidad P ara la industria farmacéutica y de química fina existe el objetivo prioritario de garantizar mezclas con un bajo coeficiente de variación, pero no por prioritario es éste su único objetivo. Por eso, los elementos filtrantes suministrados por los proveedores especializados en filtración de alta exigencia también deben cumplir otros tres objetivos básicos, como son optimizar los tiempos de los procesos productivos, facilitar un producto final puro exento de contaminantes y controlar la emisión y pérdida de producto durante el proceso de fabricación. Sólo productos diseñados y fabricados para cumplir estos objetivos pueden considerarse soluciones filtrantes de alto rendimiento para sectores de alta exigencia, como el farmacéutico y el químico. Conseguir alinearse en esta batería de objetivos es fruto de una estrecha colaboración entre el proveedor de elementos filtrantes y los laboratorios y los fabricantes de maquinaria de proceso. Cuando esta convergencia ocurre, el proveedor de elementos filtrantes se convierte en un socio estratégico de primer nivel para la industria farmacéutica y química, una posición de privilegio que permite fabricar los consumibles para los filtros de la 38 NOVIEMBRE/DICIEMBRE14 mayoría de fabricantes, diseñar y producir soluciones a medida de los laboratorios, proponer mejoras que aumentarán el rendimiento de los procesos y los sistemas filtrantes y ofrecer servicios especializados exclusivos de asesoramiento y asistencia técnica. Tejido, confección y acabado Sólo disponiendo de capacidad para fabricar soluciones personalizadas y a medida, incluso artesanales, es posible confeccionar elementos filtrantes perfectamente adaptados a la instalación para que ésta funcione de forma óptima. Acabados eficientes, ajuste dimensional preciso o adaptación a especificaciones concretas de diseño o técnicas, entre otras, aportan al sistema un rendimiento mayor, máxima eficiencia energética, ciclos de vida más largos, menores costes de mantenimiento y reposición y máximas garantías de funcionamiento. La investigación sobre innovadores materiales y materias primas y su posterior aplicación proporcionan al mercado elementos filtrantes de una alta eficiencia económica y ecológica. La elección correcta entre los diferentes tejidos existentes será una clave fundamental para el correcto y óptimo fun- cionamiento del sistema de filtración. Así, la composición y las especificaciones técnicas del tejido referidas a la filtración de partículas (materia, permeabilidad, retención de partículas, ligamento, espesor, urdimbre, trama, número de hilos en sentido urdimbre y trama, resistencia a la rotura y acabados del tejido), junto a una cuidada confección y perfecto acabado, determinarán la eco-eficiencia de todo el sistema. Una de las características principales del tejido de filtración es la permeabilidad al aire. Esta propiedad va muy vinculada al rendimiento del filtro, es decir, la superficie filtrante de una tela o manga de filtración tiene que tener la suficiente permeabilidad para filtrar todo el fluido que el proceso genera. La otra característica importante es la capacidad de retención del tejido. Se requiere que el tejido retenga la mayor cantidad de partículas, ya que en la mayoría de casos estos sólidos tienen un alto valor económico. En especial en las industrias químicas y farmacéutica. Se utilizan diferentes métodos para determinar la capacidad de retención de un tejido. Algunos de ellos tienen una validez muy relativa, y otros tienen un coste muy elevado. El método de ensayo TEXMAX-60255 de FARMESPAÑA INDUSTRIAL FILTRACIÓN Texfiltra, utilizado por ICT FILTRACIÓN, da una respuesta práctica y rigurosa a esta necesidad. Este método de ensayo describe el proceso con el cual un gas (aire) contaminado con micro-esferas de cristal es fluidificado y forzado a través de un tejido (la muestra a estudiar). Las partículas no retenidas por el tejido, es decir, las que lo han atravesado, son recogidas en un filtro de membrana absoluto para ser medidas, y así determinar la capacidad de retención del tejido. El método tiene en cuenta el tamaño de las microesferas de cristal a utilizar, dependiendo de la permeabilidad del tejido. Determinado el tejido, la relevancia recae en los procesos y las técnicas constructivas, de confección, fabricación y acabado del elemento filtrante. Aquí el objetivo se centra en aumentar el rendimiento y el ciclo de vida de los productos, reducir las posibilidades de fugas, mejorar los costes de producción y presentar soluciones más competitivas. En esta fase del proceso la capacidad industrial del proveedor de elementos filtrantes resulta fundamental para superar los objetivos normativos y legislativos de calidad de producto y seguridad medioambiental y personal. Esta capacidad industrial se compone de laboratorio de análisis de muestras, sistemas de corte láser, sistemas de ultrasonidos y termosoldadura para el acabado y cierre de mangas o tubulares y, decisivo para producir soluciones personalizadas y a medida de alta calidad, maquinaria de corte manual, máquinas de coser en todas sus variantes, ejecución de procesos artesanales y equipos semiautomáticos para acabado de piezas, inclusión de presillas, ollaos, etcétera. FDA, ATEX y GMP Todos los materiales utilizados en la confección de elementos filtrantes para la industria farmacéutica y química deben cumplir la normativa FDA. En el caso de ICT FILTRACIÓN, el cumplimiento de la normativa viene certificada por los proveedores, facilitando la trazabilidad del producto. Asimismo, son materiales antiestáticos (conductivos) para garantizar la certificación ATEX. En todos los casos, la fabricación del elemento filtrante debe garantizarse de acuerdo a las normas de correcta fabricación, calidad y seguridad de la industria farmacéutica (Good Manufacturing Practices - GMP’s), mediante procedimientos normalizados de trabajo (PNT’s). Existen casos particulares donde a requerimiento del cliente los elementos filtrantes llevan incorporada una codificación perso40 NOVIEMBRE/DICIEMBRE14 nalizada (etiqueta o bordado) para facilitar su trazabilidad. Análisis y ensayo en laboratorio Cuando el proveedor de elementos filtrantes tiene la capacidad técnica de analizar y ensayar en laboratorio propio se multiplica también su capacidad de adecuar el elemento filtrante a las necesidades reales del cliente y ajustar los costes a la solución óptima, sin sobredimensionamientos de ningún tipo. Las muestras enviadas por el laboratorio farmacéutico o químico al laboratorio de análisis y ensayos del permitirán fabricante del elemento filtrante permitirán a éste detectar la causa de determinados problemas presentes en elementos filtrantes del cliente y determinar cuál es la mejor solución. Además, es el laboratorio de análisis y ensayos quien pone en evidencia las capacidades reales de los tejidos técnicos y, por tanto, las posibilidades de mejora o alternativas a proponer para lograr una mayor eficiencia económica y ecológica. Detalles que marcan grandes diferencias Más allá de las exigentes normativas y legislaciones que rigen a los sectores farmacéutico, químico o alimentario, los procesos de fabricación y acabado seguidos por algunos fabricantes de elementos filtrantes pueden suponer un plus de beneficios, especialmente cuando se trata de productos singulares, personalizados y a medida, o producción de pocas unidades, que en ocasiones significa efectuar un trabajo de gran habilidad artesanal imposible sin la habilidad y pericia de personal experto. Algunos detalles que marcan grandes diferencias entre productos aparentemente similares en su función son, entre otros, la incorporación de sistemas de cierre por cremallera, la posibilidad de in- corporar sistemas de identificación según criterios de trazabilidad personalizados por el cliente, la confección de mangas a medida para conexión, trasvase y descarga con tejidos que cumplen las especificaciones FDA y ATEX, el etiquetado o bordado permanente de información especificada por el cliente para su correcta identificación, el envasado y empaquetado siguiendo directrices del cliente, el sistema de cierre reforzado por cuerda, el sistema de fabricación robusto con doble tapa, cordón interior y costuras reforzadas, los tejidos antiestáticos con acabados mecánicos para facilitar el desprendimiento del polvo. Mención especial tienen la disponibilidad de fichas técnicas y los sistemas de unión por termosoldaura. La entrega de fichas técnicas completas con información sobre procedencia del tejido (fabricante, país de origen…), composición y especificaciones técnicas (permeabilidad, resistencias, tolerancias...), las certificaciones y las homologaciones que avalan dichas informaciones, así como las prestaciones que aportarán durante el uso, facilitan las garantías necesarias sobre la materia prima, además de la trazabilidad. Por su parte, siempre que sea posible su aplicación durante la confección, la unión de las partes del tejido por termosoldadura en sustitución de tradicional cosido garantiza máxima eficiencia de filtración y mínimo riesgo de contaminación cruzada por desprendimiento de fibras. Conclusión La especialización, la experiencia y el conocimiento son tres cualidades que debe a portar a la relación el proveedor de elementos filtrantes en sectores de alta exigencia. Esto se traducirá en beneficios porque se garantizarán productos fabricados de forma uniforme y controlada, de acuerdo con las exigentes normas de calidad y seguridad de la industria farmacéutica (GMP’s); porque sus procesos de fabricación estarán perfectamente adaptados gracias a la aplicación de Procedimientos Normalizados de Trabajo (PNT’s) y formación continua de sus empleados; porque el proveedor facilitará la adaptación de los procesos productivos del laboratorio a la legislación medioambiental internacional, porque ofrecerá soluciones competitivas de alto rendimiento y máxima eficiencia medioambiental que garantizará mezclas con un bajo coeficiente de variación, optimizará los tiempos de los procesos productivos, facilitará un producto final puro exento de contaminantes y controlará la emisión y pérdida de producto durante el proceso de fabricación FARMESPAÑA INDUSTRIAL Nuestro compromiso es garantizar su éxito Dibujando el futuro de la automatización a nivel mundial, SU EFICIENCIA logística ha sido y seguirá siendo siempre nuestro objetivo. SSI Schaefer C/Can Pi, 17, Pol. Ind. Gran Via Sur, 08908 L’Hospitalet de Ll., Barcelona Tel. 902 109 669, [email protected], www.ssi-schaefer.es ESTERILIZACIÓN Vittorio Mascherpa, Consultor Senior I+D, Fedegari Autoclavi Este artículo compara los conceptos de esterilización, desinfección y descontaminación con referencia a los distintos enfoques de cada uno. Por otra parte, se valora la descontaminación por vapor de peróxido de hidrógeno y su uso en aisladores para ensayos de esterilidad. También se presenta un nuevo método tecnológico para la vaporización del peróxido de hidrógeno y su dosificación. Descontaminación por peróxido de hidrógeno: usos y desarrollos técnicos Esterilización, desinfección y descontaminación: una revisión terminológica Durante mucho tiempo la palabra esterilización ha sido utilizada para describir aquellos procesos en la industria de la alimentación, farmacéutica u hospitalaria que buscan la inactivación completa de especies microbianas viables en la superficie o en la mayor parte de un objeto, incluyendo esporas bacterianas. Según esta definición comúnmente aceptada, un objeto es estéril cuando está completamente libre de contaminación por organismos viables. Por tanto, la esterilidad puede ser vista como un concepto absoluto: el objeto es o no estéril. De todas maneras, si nos movemos desde el campo abstracto de las definiciones hacia las prácticas diarias, la complejidad aumenta: ¿cómo podemos constatar objetivamente que un objeto es estéril? Una característica común de todos los métodos de ensayos de condiciones de esterilidad es la destrucción de la misma sobre el objeto de ensayo, por lo que ya no es viable para el alcance de ensayo: en otras palabras, no hay métodos de ensayo de esterilidad de un objeto dado que respeten íntegramente su integridad sin afectar sus propiedades y dañar, en mayor o menor medida, la condición de estéril. En el caso de productos estériles envasados se puede llegar a afirmar que no existen métodos no destructivos. Esta paradoja puede resumirse en que la esterilización es un proceso especial en que sus resultados (eficacia de la esterilización) no pueden verificarse por la consiguiente inspección y ensayo del producto esterilizado (Publimed, US National Library of Medicine, PMID: 10402868) 42 NOVIEMBRE/DICIEMBRE14 Figura 1: Vista interior de un autoclave de esterilización. Por lo tanto, una verificación estadística de un proceso de esterilización satisfactorio es el único método alternativo y económicamente sostenible que implica la necesidad de lograr una alta probabilidad de que un lote esterilizado no contiene productos no estériles. Por ese motivo, la esterilización industrial es definida como un proceso utilizado para eliminar organismos viables de un producto con una probabilidad específica (PDA, TR#1 revisado en 2007) Desinfección El significado de esta palabra no es inequívoco. En la mayoría de casos describe un proceso que elimina los patógenos de objetos inertes, excepto de esporas bacterianas, a un nivel de seguridad específico. Pero, ¿a qué nivel? La FDA define como un nivel de desinfección elevado una reducción a 6-Log para un conjunto de especies micro bacterianas en un tiempo de contacto reducido. La Guía para Desinfección y Esterilización en Instalaciones Sanitarias especifica que, en un tratamiento de desinfección, los objetos o equipos para cuidados de pacientes que no invadan tejidos estériles o el sistema vascu- lar, pero en contacto con membranas mucosas o piel lesionada, deberían estar libres de microorganismos, aunque un número bajo de esporas bacterianas es permisible. Este criterio remarca la diferencia entre objetos a esterilizar previamente a invadir el cuerpo humano (elementos críticos) y aquellos que requieren solamente desinfección (elementos semi-críticos); la distinción está esencialmente relacionada con prácticas clínicas y se refiere a las diversas capacidades de diferentes partes del cuerpo humano, externas o internas, de resistir la presencia de un número bajo de esporas bacterianas. Todo lo anterior difiere de la definición de los Estándares Europeos EN-ISO 15883, relevante para equipos lavadores-desinfectadores. Según este estándar, la desinfección es la reducción del número de microorganismos viables en un producto a un nivel previamente especificado como adecuado para el uso o manejo de dicho producto. La desinfección es un proceso especial, como la esterilización, por lo que también debe ser validado. Tanto la esterilización como la desinfección pueden obtenerse por procesos termales o químicos, o incluso de otras maneras (filtración, irradiación, etc.). La farmacopea recomienda como mejor práctica, siempre que sea posible, el tratamiento por calor húmedo, que inactiva a los micro organismos viables y las esporas bacterianas por contracto con vapor condensado y/o agua súper calentada durante un tiempo de exposición específico y en condiciones estrictamente controladas. La enorme variedad de productos a esterilizar y su envasado han dado lugar al desarrollo de autoclaves diferenciadas, sofisticadas y flexibles (ver Figura 1) Un autoclave-lavadora puede diseñarse siguiendo dos enfoques: la función de desFARMESPAÑA INDUSTRIAL ESTERILIZACIÓN Izquierda, figura 2: Vista interior de un autoclave-lavadora (incluyendo función de esterilización). Derecha, figura 3: Imagen de un Aislador de Ensayo de Esterilidad. infección puede integrarse en el aparato de lavado o la función de lavado en un esterilizador. Teniendo en cuenta que los materiales a lavar y desinfectar suelen estar bien especificados, el esterilizador es, en general, un sistema más sencillo (ver Figura 2). La elección de diseño de Fedegari para sus autoclaves-lavadoras es el segundo enfoque, ya que mantiene la flexibilidad de un esterilizador por vapor tradicional también desde el punto de vista de la reducción del nivel de microorganismos. El contacto entre vapor y los elementos, requerido por la esterilización/desinfección por calor, ha demostrado ser muy útil para la función de lavado. De acuerdo con la definición de la Universidad de Columbia, la descontaminación es una actividad que reduce la carga microbiana para prevenir de infección o contaminación inadvertida. La idoneidad de un procedimiento de descontaminación depende de la situación dada. Por ejemplo, la instrumentación quirúrgica debe ser estéril, pero dicho nivel no es necesario para superficies ambientales como suelos y paredes. La palabra descontaminación puede referirse tanto a esterilización y lavado y a desinfección. Descontaminación por agente químico: un ejemplo La típica aplicación de descontaminación por agente químico son los Aisladores para Ensayo de Esterilidad y los Aisladores para Ensayo de Proceso Aséptico/Esterilidad. Según la definición del PIC de 2007 (Convención de Inspección Farmacéutica por sus siglas en inglés), un aislador es una disposición de barreras físicas en la que el aislador puede estar sellado para llevar a cabo ensayos de fugas basados en presión para cumplir con los requisitos de los lími44 NOVIEMBRE/DICIEMBRE14 tes específicos. Internamente dispone de un espacio de trabajo que está separado del ambiente que lo rodea. Las manipulaciones pueden realizarse en un espacio sin comprometer su integridad. Obviamente, un ensayo de fugas no es un enfoque en sí mismo, sino el método para verificar que la zona de trabajo está separada del resto. Los aisladores industriales para proceso aséptico son aisladores donde el espacio interior y las superficies expuestas están controladas a nivel microbiológico. Uno de los métodos para obtener este control microbiológico es el llamado proceso esporicidal, es decir, un tratamiento gaseoso, líquido o por vapor es aplicado en las superficies utilizando un agente reconocido como capaz de matar esporas bacterianas o fúngicas. Este proceso suele validarse utilizando indicadores biológicos que contienen esporas bacterianas. El número log de reducción de esporas no está especificado en la definición, pero un objetivo de 6-Log es aplicado habitualmente. En otras palabras, un Aislador para Ensayos de Esterilidad (ver Figura 3) es un espacio a prueba de fugas en que un operador puede manipular un objeto a través de los guantes: ni las manos ni el ambiente contactan con los elementos a manipular. El tratamiento de la zona interior del aislador previene a las muestras bajo ensayo de contaminación bacteriológica. Entre los agentes más fiables el vapor de peróxido de hidrógeno (H2O2) es el más popular: sus propiedades antibacterianas, antimicóticas y antivirales son conocidas y están científicamente documentadas. La efectividad del tratamiento depende de la concentración y distribución homogénea del vapor dentro del aislador. Para vaporizar el peróxido de hidrógeno antes de su dispersión en la zona de trabajo del aislador, una cantidad controlada de peróxido líquido es rociada en una placa calentada sobre la que un flujo de aire fluye. En el caso de los aisladores Fedegari, se instalan uno o más ventiladores de circulación en la parte superior de la zona de trabajo para garantizar la distribución homogénea del agente esterilizante y esporoizante. Mejoras tecnológicas En los aisladores tradicionales, la concentración de vapor se controla por la “receta”. Esto significa que una cantidad total prefijada de peróxido de hidrógeno líquido es rociada sobre la placa de vaporización. Este método implica un control de concentración indirecto, pero durante el propio tratamiento de descontaminación (durante el periodo de exposición), no hay control posible. El grupo Fedegari ha diseñado y desarrollado un método nuevo basado en la experiencia de más de 50 años en el control de esterilización. Este método se basa en la medición continua de la concentración de peróxido de hidrógeno en la superficie de trabajo durante todas las fases del tratamiento. La diferencia entre la concentración fijada y la obtenida es constantemente controlada y determina el flujo del peróxido de hidrógeno líquido liberado en la placa de vaporización. Este control es realizado tanto durante el periodo de preparación como de exposición para evitar la caída progresiva en la concentración del agente esterilizante y esporoizante por consumo. Este método ofrece una respuesta en tiempo real sobre el control de la concentración del peróxido de hidrógeno durante el proceso y garantiza la estabilidad del mismo durante la fase crítica del tratamiento FARMESPAÑA INDUSTRIAL PUBLIRREPORTAJE Esysa: sistemas de tratamientos, filtración y depuración de Aguas para uso farmacéutico E l agua, en la industria farmacéutica, se utiliza tanto en lo que hace referencia a la preparación de formas farmacéuticas estériles y no estériles, como al lavado de equipos e instrumentos utilizados para la preparación de éstas. La gran diversidad de aplicaciones del agua de uso farmacéutico hace que, para cada una de ellas, la calidad del agua requerida no sea necesariamente la misma, sin olvidar la importancia de la filtración de sus aguas residuales. De ahí la necesidad de buscar la alternativa que mejor se adecúe al uso al que se vaya a destinar. Nuestra empresa, ESYSA Sostenibilidad y desarrollo S.L., es especialista en buscar este tipo de soluciones a través de la distribución de diferentes sistemas de filtraciones insertos en elementos modulares, fijos o transportables, para la producción de agua adecuada a los diferentes usos farmacéuticos o industriales y tratamiento de aguas residuales. En ESYSA contamos con un equipo humano comprometido con nuestros objetivos y con nuestros clientes. Nuestra misión consiste en dar a nuestros clientes la solución más adecuada a sus necesidades, a través de un servicio profesional, asesoramiento especializado, propuestas innovadoras y soluciones integrales de proyectos a cualquier escala. El suministro de agua a bajos costes de producción y gasto energético cumple, en todo momento, con las Normas de Calidad de Agua Potable de la Organización Mundial de la Salud (OMS-WHO) y normativas del sector industrial residual a nivel mundial. A continuación se recogen las principales normativas de aplicación que regulan las diferentes calidades de agua en la Industria Farmacéutica: 46 NOVIEMBRE/DICIEMBRE14 Farmacopea europea Farmacopea americana. Usp 21 Cfr- current Fed. Regulations (US) FDA. Guide to Inspections of High PurityWater Systems Normas de correcta fabricación (CGMP) En Esysa, dedicamos nuestra actividad al asesoramiento y solución de problemas de Ingeniería de Tratamientos y Depuración del Agua en general, especializándonos en Tecnologías donde suministramos sistemas y desarrollos de equipos a medida que se completan con la oferta de las mejores tecnologías del mercado. El sistema de agua de los laboratorios farmacéuticos es siempre uno de los principales puntos críticos de cualquier tipo de inspección. El diseño, construcción, cualificación y control en rutina -monitorización- de un sistema de producción de agua para uso farmacéutico es fundamental para asegurar que el producto final mantendrá unos estándares de calidad adecuados y reproducibles en el tiempo. Por ello es importante establecer un plan exhaustivo y lógico cuando se aborda la validación de un sistema de producción de agua para uso farmacéutico. A diferencia de otras validaciones, la de los sistemas de producción de agua, plantea diferentes cuestiones a los profesionales del sector: ¿cómo definir los estándares de ca lidad adecuados?, ¿cuál es el ámbito de la validación?, ¿cómo establecer el plan de monitorización en rutina?, ¿qué impacto tienen? Para lograr una solución óptima no basta con seleccionar el componente adecuado, como una bomba dosificadora, un sensor o un aparato de regulación. Solo la adecuación correcta y la selección de un equipo a medida y su conexión en red garantizan el mejor resultado posible. Las aplicaciones pueden ser muy complejas. La superioridad de una aplicación y, por tanto, su eficiencia, están determinadas por una gran cantidad de posibles interacciones entre los componentes necesarios, como equipos de filtración, sensores, control y regulación y, especialmente, su diseño hidráulico especifico. Para obtener un resultado óptimo en una aplicación no basta con seleccionar los componentes adecuados correspondientes a cada requisito, como contrapresión, capacidad de transporte, temperatura, alcance de medición o resistencia química. El mejor resultado posible no se consigue hasta lograr la perfecta adecuación de todos los componentes, la correcta conexión entre estos y, dado el caso, con un mando superior, así como la consideración de las posibles perturbaciones procedentes del entorno. Por lo tanto, para solucionar su problema de aplicación nuestros especialistas aportan, además de una vasta experiencia con nuestros productos en el sector farmacéutico, los conocimientos necesarios sobre la química del agua, la hidráulica, la técnica de medición y regulación, la microbiología y otros campos. El agua ultrapura se emplea principalmente en la industria de los semiconductores y en la industria farmacéutica. Debido a que en la industria de los semiconductores cada vez se trabaja a escala más pequeña, las especificaciones se vuelven mucho más estrictas. FARMESPAÑA INDUSTRIAL De izquierda a derecha: Intercambio iónico; Termosol y Electrodesionización. Por definición el agua ultrapura sólo contiene H2O e iones H+ y OH- en equilibrio. Por lo que la conductividad de la misma es aproximadamente de 0,054 uS/cm a 25oC, o de 18,3 MOhm expresada en términos de resistencia. Normalmente la producción de agua ultrapura se realiza en dos etapas. Por ejemplo, partiendo de agua del grifo o de agua subterránea, primero se desmineraliza mediante procesos ecológicos de filtración, membranas o mediante un sistema de intercambio iónico para alcanzar una conductividad de 10 uS/cm. El agua desmineralizada se procesa después a través de un Lecho Mixto o mediante Electrodesionización. Los procesos más utilizados en la filtración del agua para uso farmacéutico dependen de las características de la misma y suelen ser: sistema de intercambio iónico, osmosis inversa y ultrafiltración. Nosotros ofrecemos además un novedoso proceso ecológico de filtración. El intercambio iónico es una operación de separación basada en la transferencia de materia fluido-sólido, que implica la transferencia de uno o más iones de la fase fluida al sólido por intercambio o desplazamiento de iones de la misma carga, que se encuentran unidos por fuerzas electrostáticas a grupos funcionales superficiales. La eficacia del proceso depende del equilibrio sólido-fluido y de la velocidad de transferencia de materia. Los sólidos suelen ser de tipo polimérico, siendo los más habituales los basados en resinas sintéticas. La osmosis inversa es un procedimiento que garantiza el tratamiento desalinizador físico, químico y bacteriológico del agua. Funciona mediante membranas de poliamida semipermeables, enrolladas en espiral, que actúan de filtro, reteniendo y eliminando la mayor parte de las sales disueltas al tiempo que impiden el paso de las bacterias y los virus, obteniéndose un agua pura y esterilizada. FARMESPAÑA INDUSTRIAL Aguas con un elevado contenido de sales como, sodio, calcio, boro, hierro, cloruros, sulfatos, nitratos y bicarbonatos, pueden ser tratados con la osmosis inversa hasta alcanzar los límites considerados como agua aceptable para su utilización. Las membranas semipermeables son la clave y responsables de separar las sales del agua. Dichas membranas pueden considerarse como filtros moleculares. El tamaño de los poros de estos filtros membranas es extremadamente reducido, por lo que se requiere una presión considerable para hacer pasar cantidades de agua a través de ellas. La elección del modelo de membrana más apropiado es según el agua a tratar y su empleo posterior, determinando el tipo de instalación más idónea. Las suciedades que quedan en las membranas son posteriormente arrastradas y lavadas por la misma corriente de agua. De esta forma el sistema realiza una autolimpieza constante. Esta corriente de agua de desperdicio necesaria, está en relación directa con el tipo de membrana que se utiliza y sus exigencias. La ultrafiltración es un proceso físico mecánico a través del cual partículas y moléculas son retenidas por tener un tamaño superior al poro de la media o membrana. Dentro de esta clasificación se incluyen aquellas membranas o cartuchos cuya retención nominal va desde 0.1 micras hasta 0.005 micras. La tecnología de ultrafiltración ha empezado a ganar terreno en la primera década de este siglo. Se han desarrollado membranas más resistentes y tecnologías de aplicación que permiten mejorar la calidad del agua en muchos procesos de manera muy segura. El proceso ecológico de filtración, es un proceso de bio-filtración basado en procesos naturales desarrollados por microorganismos que modifican y/o metabolizan elementos y compuestos químicos que para ellos constituyen nutrientes indispensables. Los microorganismos ya se encuentran de forma natural en las aguas a tratar y lo único que debemos hacer es potenciar su actividad para su desarrollo. Un ambiente adecuado incluye: temperatura, oxígeno, alimentos y alojamiento (soporte biológico BIO). Los microorganismos utilizan para su metabolismo una serie de iones que son oxidados mediante reacciones exotérmicas y se transforman liberándose energía en el proceso de oxidación. Los microorganismos consiguen una oxidación eficiente y más rentable en comparación a procesos físico y químicos consiguiendo la precipitación y/o bio asimilación de los metales. Desde ESYSA, sostenibilidad y desarrollo S.L., ofrecemos soluciones a través de sistemas y desarrollos de equipos, a medida de las necesidades de nuestros clientes, respaldados por las mejores tecnologías del mercado: sistemas de intercambio Iónico, osmosis inversas, ultrafiltración, desalación, reutilización, potabilización y todo lo relacionado con aplicación y proceso industrial del agua, con las certificaciones correspondientes en cualquier parte del mundo. ESYSA es una empresa que se encuentra en plena expansión internacional teniendo como objetivo ser una referencia en el sector de la sostenibilidad, tanto en el tratamiento de aguas, como en eficiencia energética, energías renovables y construcción modular, gracias al trabajo conjunto con fabricantes, proveedores y partners. Nuestra misión consiste en dar a nuestros clientes la solución más adecuada a sus necesidades, a través de un servicio profesional, asesoramiento especializado, propuestas innovadoras y soluciones integrales de proyectos a cualquier escala Para más información: [email protected] Tel. 617 708 514 NOVIEMBRE/DICIEMBRE14 47 CASO PRÁCTICO SSI Schäfer SSI Schäfer ha realizado en la capital eslovena, Ljubljana, uno de los centros de distribución más modernos en Europa Central para el mayorista farmacéutico Salus. Se ha creado una instalación a partir de una concepción integral que combina y vincula entre sí diferentes zonas de almacén y de picking. Solución modélica para el mayorista farmacéutico Salus L a estructura de productos y pedidos diferenciados requiere la más alta flexibilidad del sistema de gestión del almacén junto con una combinación inteligente de las estrategias de picking. Teniendo en cuenta estos requisitos, SSI Schäfer ha realizado para Salus, el mayorista líder de productos farmacéuticos y médicos en el mercado esloveno, uno de los centros de distribución más modernos en Europa Central. “En la instalación antigua habíamos llegado al límite de nuestra capacidad”, explica el Jefe de Proyecto de Salus, Andrej Hocevar. “Además, no estaba bien comunicada; por lo tanto, la decisión de construir un nuevo centro de distribución fue coherente y lógica. Ofrece más espacio y un mayor grado de eficacia debido a su equi48 NOVIEMBRE/DICIEMBRE14 FARMESPAÑA INDUSTRIAL pamiento moderno y a su configuración de los procesos. De esta forma, nuestros clientes reciben un servicio mucho mejor.” El encargo fue adjudicado a SSI Schäfer, Giebelstadt, como contratista general para la intralogística. La instalación realizada puede definirse como una solución sectorial modélica con múltiples particularidades que cubren los requerimientos del comercio farmacéutico. En el nuevo centro de distribución en Ljubljana están concentrados unos 13.000 artículos diferentes con un stock medio que asciende a aproximadamente 6 millones de unidades. El almacén abarca 30 zonas con cuatro áreas de temperaturas distintas. De esta manera agrupan pedidos para la entrega a farmacias u hospitales teniendo en cuenta un picking por órdenes de varias FARMESPAÑA INDUSTRIAL etapas según las diferentes estrategias de picking. El almacén de gran altura es totalmente automático con 6 pasillos y 2 transelevadores con cambio de pasillo por curva, con más de 5.100 ubicaciones para el almacenamiento a profundidad simple de palets con un peso de 1.000 kg cada uno. Se ha instalado también una zona de despacho de mercancías separada para el crossdocking de palets completos. El almacenamiento y el picking de artículos individuales se realizan mediante un almacén miniload automático y un Schäfer Carousel System (SCS). Además, el centro de distribución dispone de dos zonas de almacén manuales: una especial para el almacenamiento de artículos a temperatura controlada, y otra dedicada al picking para grandes cantidades. Todas las zonas de almacén y de picking y sus correspondientes procesos de manipulación en Salus son controlados por el sistema de gestión de almacenes WAMAS® de SSI Schäfer. “Con la vinculación y el sistema de control y mando IT integral de estos sistemas y de estas estrategias de tratamiento diferenciadas hemos realizado uno de los proyectos más complejos de los últimos años, tanto a nivel de sistemas como de control de procesos mediante las tecnologías de información”, opina Peter Diener, jefe de proyectos de SSI Schäfer. “La optimización de los procesos conseguida comporta para Salus unos tiempos de ciclo extremadamente rápidos, un incremento notable del rendimiento y una alta seguridad de los procesos a pesar de trabajar con una gama de artículos muy diferenciada y heterogénea.” “Gracias a la combinación única de los sistemas, la automatización de los procesos y a su capacidad de rendimiento podemos agrupar y entregar diariamente hasta un total de 10.000 a 12.000 líneas de pedido agrupadas en unos 300 pedidos”, comenta el Sr. Hocevar. Esto es posible por tener unos flujos inteligentes de las mercancías, flujos que ya habían sido comprobados por SSI Schäfer en la fase de concepción y planificación mediante simulaciones en el diseño del almacén. El Schäfer Carousel System permite un picking de unidades altamente dinámico con hasta 1.000 picks por hora y operario. Requisitos del proyecto Los principales objetivos/requisitos del proyecto: Gran flexibilidad para adaptarse a los cambios en las dimensiones garantizando el uso eficiente del espacio Integración de sistemas automáticos con las dimensiones optimizadas para garantizar la eficiencia en el uso del espacio Diseño vanguardista e innovador del picking para gestionar la amplia gama de productos Mayor eficiencia y calidad del picking Gestión de más de 13.000 referencias Diseño de las zonas de almacenaje para adaptarse a las 4 diferentes condiciones de temperatura requeridas Base de la colaboración con SSI Schaefer Salus seleccionó a SSI Schaefer como contratista general de intralogística tras una oferta de licitación internacional. Gracias a su experiencia en la industria farmacéutica con muchas instalaciones a nivel mundial y al ratio coste/productividad, Salus asignó el contrato a SSI Schaefer. El sistema que se ha implantado puede considerarse como una solución ejemplar por contener la estructura modular necesaria para ajustarse a los requisitos específicos de la industria farmacéutica. Solución Para ayudar a Salus a cumplir los requisitos de rendimiento y mejorar la calidad de servicio, SSI Schaefer diseñó e implantó una solución que permite gestionar los pedidos considerando los diversos procesos de picking multinivel creando diferentes zonas en el almacén de gran altura (HBW) totalmente automático según las estrategias de picking. El nuevo almacén de gran altura actúa como almacén central y da servicio a todas las zonas de almacenaje y áreas de picking del centro de distribución. De acuerdo con las especificaciones, se incluyó una estación de picking para realizar la preparación manual de los pedidos a granel de los clientes. Desde el almacén también se suministran algunos productos a las estaciones de embalaje, productos que se despaletizan y se ponen en bandejas adecuadas para almacenarse en el miniload. Asimismo se creó un área separada para el cross-docking de pallets enteros. El almacenaje y picking de productos individuales se hace vía un sistema de almacenaje de piezas pequeñas (Miniload) y un carrusel (Schaefer Carousel System, SCS). NOVIEMBRE/DICIEMBRE14 49 CASO PRÁCTICO Además el centro de distribución de Ljubljana tiene 2 áreas de almacenaje que se operan manualmente: una especial para productos a temperatura controlada y otra para el picking de grandes cantidades. Y finalmente 3 áreas de cuarentena para gestionar las devoluciones, los productos dañados y productos que tienen que destruirse. A pesar de la amplia gama de productos, los procesos se han optimizado respetando los procesos de seguridad. Los resultados obtenidos han sido gracias a un diseño inteligente de los flujos de materiales realizado por SSI Schaefer, diseño que se define tras varias simulaciones de flujos en tiempo de proyecto hasta encontrar el óptimo. en tiempo real las ubicaciones y el stock disponible en todas las zonas del almacén. Schaefer Miniload Crane & Pick-by-Light Los productos, reempaquetados en bandejas, se transportan en cintas por los 3 pasillos del miniload con 28.840 ubicaciones para bandejas. Hay 3 transelevadores con 72 ciclos dobles por hora que almacenan las bandejas; estas últimas se rellenan con productos hasta un máximo de 25 kg y los transelevadores las retiran para llevarlas a las estaciones de picking que son alimentadas directamente por el miniload. En las estaciones de picking, las bandejas se llenan de forma óptima tanto con los productos de alta rotación como con los C. Para optimizar la eficiencia de los operarios, SSI Schaefer implementó pantallas que los guiaban y les indicaban Sistemas SSI Schaefer implantados y su impacto en las instalaciones de Salus HBW – Almacén de gran altura totalmente automático Los pallets se almacenan en un almacén de gran altura completamente automático, con 6 pasillos y 2 transelevadores con cambio de curva. Tiene una capacidad para 5100 ubicaciones de pallet con más de 1000 kg por pallet y cada transelevador puede mover 35 pallets por hora. El software logístico de SSI Schaefer gestiona 50 NOVIEMBRE/DICIEMBRE14 FARMESPAÑA INDUSTRIAL el canal de picking para el siguiente pedido. En total existen 2.150 canales en los 4 niveles. Guiado por sistemas integrados Pick-by-Light, los operarios cogen respectivamente sus pedidos y los introducen en las bandejas de expedición gracias a pantallas iluminadas que le indican de qué bandeja y qué cantidad tienen que coger para cada pedido. La ubicación de las bandejas con los pedidos preparados FARMESPAÑA INDUSTRIAL en las estaciones de picking está previamente planificada por el software de SSI Schaefer de forma que los pedidos puedan completarse eficientemente y sin errores en las estaciones. Con los pertinentes algoritmos, los productos de alta rotación se depositan en estaciones de picking estáticas mientras que los productos C se depositan en posiciones dinámicas de picking temporales. Schaefer Carousel System (SCS) Para el picking de los productos C, Salus usa el método mercancía al hombre. Con este método las bandejas se llevan a uno de los 3 carruseles para almacenarlas de acuerdo con las órdenes especificadas por el SGA, desde las estaciones de reempaquetado o desde el miniload vía la línea de cintas a través de las cintas. Entonces las bandejas se descuentan del stock y se llevan a una estación de picking moderna en la que, una vez realizados los procesos de picking, vuelven a figurar como stock disponible. Así, por un lado, se vuelve a realizar el picking para ordenar las bandejas en el carrusel de acuerdo con las especificaciones del software. Por otro lado, el sistema Pick-by-light va guiando a los operarios para hacer el picking de pedidos; las bandejas con los pedidos preparados salen por la cinta de expedición. El SCS es un sistema muy dinámico que permite un rendimiento de 250 ciclos/hora. Con medidas optimizadas y diseño modular, es una de las soluciones más innovadoras y eficientes para procesos de picking y de consolidación para almacenes NOVIEMBRE/DICIEMBRE14 51 LIMPIEZA DE EQUIPOS Abimael Cumplido, Jefe de Ingeniería y Servicio Técnico en Farma Alimenta La versatilidad de los equipos y de las producciones multiproducto de hoy en día han convertido la limpieza en uno de los pasos más importantes dentro de los procesos de producción. A esta relevancia se suma la búsqueda de la eficiencia máxima de los costes, inclinándose hacia la automatización de los procesos de limpieza y su especificación, reproducción y validación dependiendo de cada producto. CIP, CLEAN IN PLACE Métodos de limpieza automáticos T anto las compañías farmacéuticas como las autoridades sanitarias están prestando cada vez más más atención tanto al proceso de limpieza como a su validación y monitorización, por varios motivos: Por motivos de disminución del riesgo de contaminación cruzada, no solo por la variedad de productos, sino por el diseño y los materiales que se emplean en la construcción de los equipos, cada vez de mejor calidad, aunque muchas veces no son fácilmente desmontables para limpieza. Se pone especial precaución al trabajar con determinados tipos de producto como son los hormonales o citostáticos, atendiendo a las condiciones de seguridad tanto del operario como del ambiente. En esos casos, incluso los procesos de limpieza se realizan bajo total contención. Existen otras razones, como la económica ya citada, puesto que la limpieza CIP reduce tiempos muertos o tareas exigidas por la actividad manual que, además, eleva los costes. Son motivos que decantan más aún la balanza para la automatización de procesos y que, además, permiten, por ejemplo, utilizar métodos y recetas de limpieza más agresivos (detergentes, temperaturas, presiones, caudales, etc.) que en procesos manuales. A la reproductividad de procesos y la monitorización y registro de parámetros más importantes de la limpieza, hay que sumar como consecuencia, la facilidad para la validación de procesos. 52 NOVIEMBRE/DICIEMBRE14 En la fabricación de formas sólidas, los equipos de fabricación se pueden limpiar de manera manual, de forma semiautomática o totalmente automática. A estos efectos, cabe aclarar las diferentes terminologías: «WIP», Washing In Place, es un proceso de limpieza semiautomático o completamente manual con resultados no definidos o resultados de limpieza no acordes a los requerimientos GMP (Good Manufacturing Practices). «CIP», Cleaning In Place, proceso completamente automatizado con un resultado de limpieza acorde a requerimientos GMP, incluyendo todos los factores que tienen influencia en el resultado de la limpieza. Ello incluye un criterio de aceptación de limpieza que pueda alcanzarse para ser validable. A la hora de diseñar un proceso de limpieza automático, no solo hay que tener en cuenta el CIP como tal, sino las características del equipo a limpiar. Hay una serie de componentes que aun siendo estándares y comúnmente utilizados en muchos equipos, exigen de un análisis previo que garantice que su correcto uso ayudará a la limpieza. Estos son: juntas, sensores, uniones, mirillas, etc. En el diseño de juntas, hay que tener en cuenta, independientemente del componente o instrumento que soporten, si son susceptibles de almacenar producto, y no solo sólido, sino también soluciones o suspensiones que se generen de la limpieza húmeda. En estos casos, hay propuestas de juntas pulidas hinchables o incluso mecánicamente expansivas, que en contacto con el producto, garantizan un flujo de entrada y salida del producto de limpieza durante el CIP, lo que igualmente favorece el proceso de secado. En cuanto a componentes como mirillas, la mejor opción es que estén soldadas de forma sólida al cuerpo del equipo, sin juntas. La soldadura garantiza una suave transición y una superficie plana. En cuanto a los sensores, requieren especial atención aquellos que se utilizan para medir presiones diferenciales, puesto que exigen de la instalación de un tubo flexible con un extremo abierto para la toma. En estos casos, se recomienda cambiar de principio de medición y, por tanto, de instrumento, por ejemplo, utilizando una medición de presión absoluta tras un disco cerámico instalado a haces del cuerpo del equipo a través de una junta. Los filtros, altamente recomendados para la limpieza, son los de acero inoxidable, preferiblemente circulares, que garantizan la limpieza y estabilidad a largo plazo en el caso de que sea necesario un soplado de aire previo al proceso de limpieza. El contenedor o cuerpo del equipo debe estar dispuesto de tal manera que sea totalmente drenable por gravedad. En el caso de que en el fondo del contenedor se haya instalado un componente o instrumento, es recomendable situarlos en una base que disponga de cierta pendiente, a fin de que cualquier lado o superficie del FARMESPAÑA INDUSTRIAL Tabla 1. Tabla 2. S= Swab samples; R = Rinse samples. a)= Para el cálculo de la cantidad de residuo, se utilizaron el límite de detección. b) La cantidad se calcula a partir de las cantidades teóricas. La concentración real puede estar en realidad muy por debajo del número mostrado. componente esté bajo pendiente y no se pueda acumular líquido. Un componente fundamental en el diseño del equipo a limpiar son las boquillas o nozzles de limpieza. Por un lado, en cuanto a su ubicación, deben estar dispuestas de tal manera que el producto de limpieza alcance toda la superficie del equipo, especialmente aquella que deba tener un impacto directo. Y por otro lado, en cuanto al tipo de nozzle a emplear. Hoy en día existen nozzles retráctiles que se expanden durante el proceso de limpieza y, posteriormente, con aire comprimido, se secan y vuelven a su posición inicial, enrasados con la superficie del equipo. El proceso de limpieza de un equipo viene determinado por una serie de parámetros, como son: Tipos de limpieza: – Inundación. Normalmente con prelavado y aclarado final. En este caso, el agua introducida es descargada directamente fuera del equipo después de que haya tenido contacto con la superficie a limpiar. Esto garantiza que con el prelavado la suciedad o los restos del producto más grueso son eliminados, y con el aclarado final, desaparecen también residuos tensoactivos o, incluso, la cal del agua. – A presión, mediante nozzles o boquillas de limpieza. Existe la posibilidad de diseñar estos sistemas con recirculación o sin recirculación. Para decidirse por uno de estos dos FARMESPAÑA INDUSTRIAL Tabla 3. a) Medidas por debajo del límite de detección. sistemas hay que tener en cuenta el tiempo necesario del agente de limpieza con las superficies a lavar, y los ahorros o bien en agua y detergente que ofrece el sistema con recirculación, o bien el ahorro de energía que puede suponer el segundo caso sin recirculación. – Recirculación. Este es el proceso real de limpieza, favorecido por el tiempo de contacto del producto residual con el agente de limpieza. Tipo y concentración de detergentes. Los productos usados incluyen bases, ácidos, emulsificantes, solventes y agentes oxidantes en concentraciones apropiadas. Temperatura, presión y flujo del producto del fluido de limpieza. Duración de la limpieza. Secuencia de la limpieza, tanto del fluido como de los elementos utilizados para limpiar, como los nozzles. La correcta evaluación y combinación de todos estos parámetros permite desarrollar un ciclo de limpieza óptimo para cada producto y equipo lo que, a su vez, facilita su validación. WIP vs. CIP Existen estudios donde se puede comprobar la optimización de los procesos de limpieza automáticos en comparación a los manuales. Hacemos referencia a uno realizado en la Universidad de Basilea (Suiza), en el Centro Farmacéutico, por A. Xchiffmann como parte de la disertación de su tesis. En él, se compara la limpieza de un lecho fluido convencional con una limpieza en el mismo lecho fluido pero incorporando un CIP. Conventional GPCG 15 frente a CIP GPCG 15 SC. Tras realizar un proceso de granulación en una mezcla con un 33 por ciento de ingrediente activo (cetaminophen), se procedió a la limpieza. En el caso de un lecho fluido convencional, se inició la limpieza por el operador, siguiendo su procedimiento habitual. Los filtros textiles se limpiaron fuera de la unidad en una lavadora. En el caso del lecho fluido con CIP, todo el equipo se ha limpiado automáticamente sin intervención del operario. El procedimiento de muestreo se compuso combinando muestreo con torundas y con la inmersión de ciertas partes del sistema en la cantidad definida del correspondiente solvente. La receta del proceso de limpieza se muestra en la tabla 1. Con un consumo de agua de red de 0,84m3 y de agua desmineralizada de 0,28m3. La combinación de detergentes fue: m2 a. 1% p3 cosa cip 95 (alcalino) b. 1.5% p3 cosas cip 92 (alcalino + tensoactivo) El procedimiento de análisis del muestreo fue conforme al método descrito en la USP 23 (Acetaminophen Capsules) para HPLC (Detección UV) y los resultados obtenidos tras repetir en tres ocasiones el ciclo de limpieza, en cuanto a residuos fueron los expuestos en la tabla 2. NOVIEMBRE/DICIEMBRE14 53 LIMPIEZA DE EQUIPOS Con una desviación estándar relativa en cada elemento mostrada en la tabla 3. De este estudio, analizando los datos, se puede concluir que: En un lecho fluido con CIP hay menos zonas contaminadas analíticamente demostrables. La cantidad de residuos en las muestras es superior en el lecho fluido convencional. El lecho fluido con CIP tiene mayor reproductividad. Hoy en día, prácticamente todos los equipos de producción a partir de cierto volumen tienen la opción en su fabricación de montar un skid de limpieza CIP. Igualmente, también existen en el mercado skids de limpieza, CIPs independientes, tantos fijos como móviles, que permiten acoplarse a un equipo o incluso varios para realizar limpiezas efectivas. Estos equipos pueden partir de una estructura e instrumentación básica hasta equipos más complejos incluso con intercambiadores de calor que pueden tratar los diferentes suministros necesarios para la limpieza, adecuándolos a sus condiciones de trabajo. Igualmente, existe incluso la posibilidad de tener equipos de limpieza modular, ampliable, o incluso equipos con un reservorio para facilitar la recirculación del caudal de limpieza si el propio equipo o cuerpo no puede ejercer como acumulador. A la hora de acoplar a equipos existentes nuevas skids es necesario tener dos consideraciones: a. Conocimiento de la receta de limpieza. Normalmente, todos los equipos a mejorar se limpian manualmente, por lo que ya existe un conocimiento previo del proceso a limpiar, de las zonas de difícil acceso o del detergente a utilizar. Esto no implica que no se deban realizar pruebas para obtener la receta de limpieza adecuada a ese producto. Para ello, es muy útil contar con una unidad móvil de limpieza que permita alternar las fases y detergentes. El objetivo es encontrar la receta óptima. b. Distribución del fluido de limpieza. En equipos que no están adaptados de fábrica para una limpieza automática es fundamental analizar cómo el fluido de limpieza debe alcanzar todo el interior del cuerpo. Existen numerosas alternativas y variantes, aunque basadas en nozzles o boquillas de espraya54 NOVIEMBRE/DICIEMBRE14 UN PROCESO DE LIMPIEZA AUTOMÁTICO Y VALIDABLE GARANTIZA UNA PRODUCTIVIDAD EFICIENTE REPRODUCIBLE Y TRAZABLE, PARÁMETROS AMBOS EXIGIBLES EN CUALQUIER PROCESO DE FABRICACIÓN FARMACÉUTICA do, ya sean rotativas, fijas, retráctiles, calefactadas, etc. En cuanto a la validación de la limpieza, esta depende mucho del cliente, pero hay varias alternativas: Tiempo. Se desarrolla la receta de limpieza y se ejecuta en varias ocasiones con resultado positivo, estableciendo ese tiempo de cada paso de la receta como óptimo y garantía de un correcto proceso de limpieza. En este caso, no se utiliza de manera habitual ningún instrumento en línea o análisis posterior que verifique el proceso de limpieza. Conductividad/pH. En esta opción, el proceso de limpieza finaliza cuando la calidad del agua del último aclarado es la adecuada para su vertido a la red. En este caso, hay que discernir si el vertido se puede realizar a la red sanitaria de desagüe convencional o si el producto previamente limpiado debe ser tratado con un tratamiento posterior, ya sea de filtración o térmico. Por último, un factor más a tener en cuenta, que no por prescindible deja de ser importante, es la interfaz del usuario. Las interfaces de usuario pueden ofrecer mucha y variada información, pero es deseable que, al menos, ofrezca la siguiente, a fin de facilitar la operatividad del operario y el seguimiento del proceso de limpieza: Indicación de los pasos a seguir por parte del operario para la limpieza, incluyendo comentarios y aceptación por parte del operario. Sinóptico real del estado del equipo en cada instante. Lectura del valor de la medición en línea de parámetros si existe. Conclusión El valor añadido que ofrece un proceso de limpieza automático queda rápidamente evidenciado independientemente del coste económico que los diferentes productos CIP del mercado puedan tener o incluso la inversión en tiempo necesario por parte del usuario para la determinación del mejor proceso y receta de limpieza que se ajuste a su equipo y producto. Un proceso de limpieza automático y validable garantiza una productividad eficiente reproducible y trazable, parámetros ambos exigibles en cualquier proceso de fabricación farmacéutica FARMESPAÑA INDUSTRIAL Tratamiento por rayos gamma Co-60 Dedicados al servicio de la irradiación Método efectivo Fácil de monitorizar No precisa cuarentena Rápido y fiable Esterilización final Productos sanitarios Ausencia de residuos Productos farmacéuticos La primera empresa española en tratamiento por rayos gamma. Desde 1970. Autorizaciones: 4824PS NCF: 4156-E RSIPAC B-001/05 Productos cosméticos Productos alimentarios Certificación: ISO 13485:2003 Investigación (polen, electrónica,...) La única planta industrial de tratamiento por rayos gamma de Co-60 en España Aragogamma, S.A. Crta. de Granollers-Cardedeu, Km. 3,5 08520 Les Franqueses del Vallés Barcelona +34 93 849 66 39 Oficinas: Salvador Mundí, 11 08017 Barcelona +34 93 204 97 03 www.aragogamma.com [email protected] VALIDACIONES Y CERTIFICACIONES María Ángeles Pérez de la Cruz Moreno. Kinestat/ Toxicología y Preclínica, Azierta. Alberto Carazo Fornieles. Ingelyt / Azierta En el siguiente artículo se analiza la necesidad del uso de datos toxicológicos para la realización de validaciones de los procesos de limpieza en instalaciones dedicadas a la industria farmacéutica y afines. El toxicólogo: una figura emergente. Validaciones de limpieza y compatibilización de productos Introducción Desde que, en 1988, la FDA confrontase el primer caso de contaminación cruzada analíticamente trazable entre colestiramina y un pesticida agrícola, las alarmas se dispararon en cuanto a la necesidad de validación de los procesos de limpieza1. Las primeras alusiones a la necesidad de establecer criterios de limpieza en conjunción con expertos toxicólogos y autoridades médicas datan de 1989, cuando Doug Mendenhall (Laboratorios Abbott) escribió un artículo expresando la necesidad de establecer límites aceptables en combinación con márgenes adicionales de seguridad. Eventos tales como el resultado del juicio en 1993 entre la FDA y los laboratorios Barr, la encuesta simultánea llevada a cabo en 1992 por la asociación de fabricantes farmacéuticos (phRMA) respecto a los límites comúnmente aceptados por la industria farmacéutica y/o el artículo de Foruman y Mullen (Laboratorios Elli Lilly); llevaron a establecer los primeros criterios estándares basados bien en criterios absolutos (ejemplo no más de 10 ppm de restos de un producto en otro, no más de 3 ppm, basado en el límite de la toxicidad de Arsenio, etc) o dependientes de la dosis (no más de 1/1000 de la dosis de un producto aparecería en la dosis de otro, etc.) A raíz de esta serie de eventos, la regulación de la GMP fueron reescritas con objeto de aumentar el detalle y la claridad y se empezó a hablar de la necesidad de establecer instalaciones dedicadas para compuestos como antibióticos betalactámicos o agentes patógenos vivos y la necesidad de establecer límites científicamente aceptables para el resto de productos. 56 NOVIEMBRE/DICIEMBRE14 Eudralex Volume 4. Good Manufacturing Practice (GMP) Guidelines Capítulos 3.6 y 5.182 EUROPA (EMA) EMA /INS /GMP /809387 /2009 Update on revision of Chapters 3 and 5 of the GMP Guide: “Dedicated facilities”3 21 CFR 211.42(d): Separation of facility and equipment 21 CFR 211.46(d): Separate air handling systems (HVAC) USA (FDA) 21 CFR 211.176: Test for traces of penicillin where possible exposure cGMP Guidance for industry non-penicillin beta-lactam drugs4 Guidance Manual Program 7356.002 for Drug Manufacturing Inspections5 Normativa ICH ICH Good manufacturing practice guide for active pharmaceutical ingredients Q76 Normativa WHO /OMS Annex 3 WHO Good Manufacturing Practices for Pharmaceutical Products Containing Hazardous Substances7 Annex 2 WHO Good Manufacturing Practices for Active Pharmaceutical Ingredients8 PICS PICS GMP GUIDE (Part I: Basic Requirements for Medicinal Products) PE 009-10 (PartI) Tabla 1. Incertidumbre A los que estamos en el ámbito de la fabricación farmacéutica siempre se nos ha planteado la duda de cuándo hay que tener una instalación dedicada o cuándo es posible compatibilizar en un área de fabricación distintos productos, especialmente cuando alguno de los mismos son esteroides, hormonas sexuales, corticoides, citostáticos, citotóxicos, fármacos con alta potencia, inmunodepresores, prostaglandinas… De hecho, en 2006, la PDA (Parenteral Drug Association) realizó una encuesta sobre el uso de límites que dejó patente que los criterios seguían siendo inconsistentes y arbitrarios. Pese a que las nuevas guías han clarificado algunos puntos, para ciertos productos no está claro si se pueden cumplir las especificaciones para asignar un compuesto a una instalación dedicada o compartida. Esta indefinición se presta a interpretaciones variadas por parte de las compañías, agencias regulatorias, consultores, etc., lo que hace que la incertidumbre sea aún mayor en la toma de decisión de si requiere una instalación dedicada o no para determinados productos. En paralelo, se ha evolucionado mucho en la tipología de productos, de los procesos de fabricación, de los equipos de fabricación, de los métodos de contención y de la precisión de los métodos analíticos, entre otros. Por ejemplo, actualmente hay una clara tendencia a desarrollar y fabricar productos de alta potencia. Se estima que los productos de alta potencia representan actualmente el 35% del mercado total y que el 50% de los productos en desarrollo se pueden considerar como productos de alta potencia. Si bien estos productos pueden ser muy beneficiosos en el tratamiento de ciertas enfermedades, presentan un reto sustancial para la industria farmacéutica. Los retos son variados: protección del personal y medio ambiente, demostrar la idoneidad de los controles establecidos para evitar la contaminación cruzada, satisfacer las exFARMESPAÑA INDUSTRIAL Compresores de aire centrífugos Subhead to come Centac®information serie C de 350 -1300 kW (500 -1750 cv) TECNOLOGÍA LÍDER EN AIRE COMPRIMIDO [email protected] www.ingersollrandproducts.com Rotary Compressors 3 Casas de Miravete, 22B 28031 Madrid 91 627.74.00 VALIDACIONES Y CERTIFICACIONES pectativas de las autoridades regulatorias, etc. Estos retos e incertidumbre tienen un impacto tremendo en las inversiones a realizar por una compañía así como en las oportunidades comerciales que pueda tener o no tener una empresa para el desarrollo y fabricación de determinados productos. Normativa Los puntos específicos de las normativas más relevantes (EMA, FDA, ICH, WHO y PICs) con respecto a la compatibilización de productos en las líneas de fabricación y a la necesidad de tener o no instalaciones dedicadas se recogen en la tabla 1. Posibles malas interpretaciones de las normativas En 2005 se propuso una revisión de la guía GMP debido a falta de claridad y hoy en día el debate sigue patente. De hecho, estas normativas generan toda una serie de “malas interpretaciones” o indefiniciones en la necesidad de zonas dedicadas o no para una serie de productos y de la posibilidad de compatibilizar productos o no en una misma área. Mezclan los conceptos GMP encaminados a proteger a los pacientes de potenciales contaminaciones cruzadas con los conceptos de seguridad e higiene encaminados a proteger de la exposición a los trabajadores y al medioambiente. La clasificación basada en los OELs (occupational exposure limits), PELs (permisible exposure limits), TLVs (threshold limit values), STEL (short term exposure limits), etc. va encaminada a proteger al operario no al paciente final, que es el objetivo de las GMP. Existen diferentes clasificaciones y criterios (Safebridge, Merck, ISPE, International Society of Pharmaceutical Ingeneering, INSHT, Instituto Nacional de Seguridad e Higiene del Trabajo…) utilizadas para definir la potencia de un medicamento. Hay que tener en cuenta que las GMPs no definen qué es un producto potente ni refieren a ninguna guía. No indican qué es una instalación dedicada. Muchos lo entienden como instalación segregada y llevándolo al extremo edificios dedicados, cuando todo indica que una instalación dedicada está encaminada a la fabricación de una tipología determinada de producto, pudiendo estar en un mismo edificio, siempre que no haya riesgo de contaminación cruzada, y que haya una correcta segregación de 58 NOVIEMBRE/DICIEMBRE14 Para el resto de productos, los fabricantes, cuando introducen un nuevo producto en una zona de producción, deben evaluar el producto y el proceso para valorar si se puede fabricar en una instalación multiproducto. Esta evaluación debe basarse en datos toxicológicos del producto. Cuando se sabe que el producto tiene un potencial sensibilizante o es altamente potente o tóxico, las autoridades supervisoras deben ser consultadas para discutir las medidas de gestión del riesgo. Este documento por un lado acerca posturas con los criterios de la FDA en cuanto a requerimientos de instalaciones dedicadas y por otro introduce la necesidad de juzgar criterios científicos basados en datos toxicológicos del producto. flujos de personas, materiales y HVAC, el grado de segregación y medidas de protección deberán venir definidos por el análisis de riesgos. Las definiciones simplistas, como hormonas sexuales o citostáticos o fármacos de alta potencia o esteroides o incluso términos como algunas hormonas o algunos esteroides, son definiciones imprecisas y hacen dar respuestas reactivas y conclusiones subjetivas y simplistas. Por ejemplo, no parece lógico que se etiquete de la misma manera (hormonas sexuales) al Etinilestradiol con concentración en la forma final de 30 μg y un NOAEL (No Observed Adverse Effect Level) de 3,5 μg/ día con la Progesterona, con una concentración final en la forma farmacéutica de 200 mg y un NOAEL de 19,8 mg/día ¿Se han de tratar igual?, ¿existe el mismo riesgo?. Las decisiones se han de tomar de una manera científica producto a producto, teniendo en cuenta las herramientas de análisis de riesgos descritas en la ICH Q9. No definen los casos excepcionales y al aceptarse el principio de fabricación por campaña han dejado la puerta abierta para que cualquier compañía con unos criterios adecuados de validación de limpieza pueda fabricar estos productos en una instalación multiproducto, con el riego de que cualquier inspector, cliente, consultor lo cuestione. Criterios científicos/toxicológicos Para intentar aclarar el tema, la EMA publicó en 2009 el documento EMA /INS / GMP /809387 /2009 Update on revision of Chapters 3 and 5 of the GMP Guide: “Dedicated facilities”, respecto a la necesidad o no de instalaciones dedicadas. En este documento el grupo de trabajo de inspectores GMP/GDP de la EMA acordó: Que se requieren instalaciones dedicadas para antibióticos betalactámicos y cuando se manipulan organismos patógenos vivos. Datos toxicológicos Los datos toxicológicos son utilizados con una triple finalidad: Para definir unos límites de limpieza que deben cumplir las validaciones de limpieza. Para evaluar la posibilidad de compatibilizar o no un producto en una línea multiproducto. Como criterio objetivo para definir el riesgo de contaminación cruzada a través del ARL (Nivel aceptable de residuo), que se define como la máxima concentración permitida de un residuo en el producto siguiente. La normativa y guías que se emplean para definir estos datos toxicológicos y los criterios para calcular estos límites de limpieza son los siguientes: ICH Quality Management Q9. ISPE Volumne-7 Risk-based Manufactured of Pharmaceutical Product. PDA Technical report 29. EMA/CHMP/SWP/598303/2011 Concept Paper on the development of toxicological guidance for use in risk identification in the manufacture of different medicinal products in shared facilities. EMA/CHMP/ CVMP/ SWP/169430/2012 Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities. Note for Guidance on Impurities: Residual Solvents (CPMP/ICH/283/95 junto con CPMP/ICH/1507/02, CPMP/ ICH/1940/00, CPMP/QWP/450/03, EMEA/CVMP/511/03 y CPMP/ QWP/8567/99). FARMESPAÑA INDUSTRIAL VALIDACIONES Y CERTIFICACIONES VICH GL18(R): Impurities: Residual solvents in new veterinary medicinal products, active substances and excipients (EMA/CVMP/VICH/502/99-Rev.1). EMA/CHMP/QWP/251344/2006 Limit on genotoxic impurities. Appendix 3 of ICH Q3C (R4) “Impurities: Guideline for Residual Solvents” Appendix 3 of VICH GL 18 on “Residual solvents in new veterinary medicinal products, active substances and excipients (Revision)”. Cálculo de los límites de limpieza. Importancia de los datos toxicológicos Los datos toxicológicos y el experto toxicólogo son claves para la definición de los valores de partida para el cálculo del límite de limpieza. La función del experto es realizar una evaluación del riesgo de los potenciales efectos adversos en la salud, tras la exposición del fármaco en estudio. Se trata de una caracterización científica sistemática para identificar, describir y medir el riesgo tras la revisión de los datos toxicológicos relevantes disponibles relacionados con el producto en estudio9. Para ello es necesario validar los datos toxicológicos existentes y hacer un balance de los efectos tóxicos encontrados. Esta revisión se hará a través del acceso y análisis de la información obtenida en bases de datos médicas (Pubmed, scifinder…), así como bases de datos específicas de toxicología (TOXNET, páginas específicas de cada país…). En el informe final y en la definición de los valores toxicológicos se tendrán en cuenta, además de la revisión bibliográfica, todos los factores que marcan las guías internacionales de cara a definir los valores toxicológicos como el potencial genotóxico y carcinogénico, factores de seguridad (que aseguran por ejemplo la extrapolación entre toxicología animal y humana), vía de administración (que entre otros puede modular la exposición ligada a la administración por vía oral y tópica, por ejemplo), biodisponibilidad, tipología de pacientes, etc. El experto toxicológico tiene que avalar los datos relativos a la caracterización de dosis-respuesta (NOAEL, LD 50, LC50, ED50, LOAEL…), que servirán para el cálculo de otros parámetros claves tales como ADI (ingesta diaria segura), MRLs (Nivel de riesgo mínimo), RfD (dosis de referencias), RfC (concentraciones de referencias). Así, la NOAEL (nivel sin efectos adversos observables) y ADE (exposición diaria aceptable) de partida se calculan para los API y la LD50 (Dosis letal 60 NOVIEMBRE/DICIEMBRE14 50) o ADI para agentes de limpieza o intermedios. Estos valores son claves para saber el ARL (nivel aceptable de residuo). Valores de proceso. Distintos escenarios A partir del ARL se calculan otras variables como el MAC (Máximo remanente admisible) o el SAL (Limite de superficie), que permite calcular los límites de limpieza (swabs o enjuagues). Para el cálculo de estas variables se utilizan valores de proceso (tamaños de lote y de superficie compartida de equipos entre productos). Estos valores de proceso permiten tener distintos escenarios, de tal manera que cuanto mayores son los lotes y menores son las superficies compartidas de contacto (por ejemplo mediante equipos dedicados o desechables), mayores son los límites de limpieza. Racional final Una vez obtenidos los límites de limpieza hay que cruzarlos con los límites de cuantificación de los métodos analíticos de limpieza y los valores reales de limpieza obtenidos. Estos tres factores, representados en la siguiente tabla, definirán la compatibilidad de los productos. Factores que definen la compatibilidad o no de los productos (A) Límites teóricos de limpieza a obtener. (B) Límites de cuantificación del método analítico de limpieza. (C) Límites reales obtenidos por el procedimiento de limpieza. Tabla 2. De las casuísticas posibles presentadas (ver Tabla 3. Casuísticas posibles –tras la obtención de los límites–), el mejor escenario es que los límites de cuantificación y los valores reales de limpieza obtenidos sean menores que el límite preestablecido obtenido. Si los límites de cuantificación o de limpieza son mayores que el límite preestablecido se podría hacer un recálculo con lotes mayores o con menor superficie de contacto para incrementar el límite teórico; si aun así alguno de los criterios no cumple se requerirían instalaciones dedicadas. Conclusiones Tanto la FDA como la EU requieren instalaciones dedicadas para antibiótico betalactámicos y cuando se manipulan organismos patógenos vivos. Para el resto de productos, la segregación e instalaciones dedicadas deben ser requeridos cuando los controles físicos y procedimentales no pueden demostrar la capacidad de controlar a niveles aceptables el peligro potencial de contaminación cruzada. El ARL (Nivel aceptable de residuo) es el criterio objetivo para definir el riesgo de contaminación cruzada y se define como la máxima concentración permitida de un residuo en el producto siguiente. El cálculo del ARL (nivel aceptable de residuos) y los límites de limpieza están basados en los datos de partida toxicológicos de los productos involucrados; estos datos toxicológicos deben estar avalados científicamente por un experto en toxicología e idealmente también por un médico experto en evaluación de medicamentos. Los datos de limpieza, junto con el análisis de riesgo correspondiente, son los factores que determinan la introducción de nuevos productos a las líneas de fabricación Referencias 1. Walsh A. Cleaning_Validation_for_the_21st_Century:_ Acceptance_Limits_for Active Pharmaceutical Ingredients (APIs): Part_I. Pharmaceutical Engineering July/August 2011. Pp 74-83. 2. http://ec.europa.eu/health/files/eudralex/vol-4/pdfsen/cap3_en.pdf 3. http://www.ema.europa.eu/docs/en_GB/document_ library/Report/2010/01/WC500039911.pdf 4. http://www.fda.gov/downloads/Drugs/Guidances/ UCM246958.pdf 5. http://www.fda.gov/AboutFDA/CentersOffices/ OfficeofMedicalProductsandTobacco/CDER/ ucm095598.htm 6. http://www.ich.org/products/guidelines/quality/ quality-single/article/good-manufacturing-practiceguide-for-active-pharmaceutical-ingredients.html 7. http://www.who.int/medicines/areas/quality_safety/ quality_assurance/ 8. http://apps.who.int/prequal/ info_general/ documents/TRS957/TRS957_ Annex2_GMP-API.pdf 9. Human Health Risk Assessment. Ronald E. Baynes. A Textbook of Modern Toxicology, Third Edition, edited by Ernest Hodgson ISBN 0-471-26508-X 2004 John Wiley & Sons, Inc Casuísticas posibles (tras la obtención de los límites) A<B No compatibles Productos no compatibles con el método analítico de verificación de limpieza debido al límite de cuantificación de los métodos analíticos. A<C No compatibles Productos no compatibles con el proceso de fabricación y de limpieza debido a que el procedimiento de limpieza no alcanza los límites preestablecidos de limpieza. A>B A >C Compatibles Productos compatibles con el proceso de fabricación y de limpieza así como con el método analítico de verificación de limpieza. Tabla 3. FARMESPAÑA INDUSTRIAL NORMATIVA Jose Racionero, Managing Director en AIS Vision Systems Existen numerosas razones por las cuales ser capaces de trazar un artículo puede suponer grandes ventajas. Si el artículo es un fármaco, a las ventajas debemos añadir razones de seguridad y prevención de falsificaciones que podrían afectar a la salud de ciudadanos de todo el mundo. Sobre las legislaciones en la trazabilidad de fármacos E ntre los años 2007 y 2012 se incautaron más de 30 millones de fármacos falsificados en las fronteras de la Unión Europea y las estimaciones de la European Alliance for Safe Medicines en referencia a los fármacos distribuidos por internet son de un índice de falsificación del 62%. Es evidente que las falsificaciones han encontrado una vía de entrada a la cadena de distribución de fármacos legítimos. Las ya mencionadas y similares cifras obtenidas en otras regiones justifican la creación de regulaciones que permitan garantizar la autenticidad de un fármaco con el fin de preservar la salud de los pacientes. Durante la última década, organismos relacionados con la salud pública de diversos países han decidido poner en marcha sistemas de trazabilidad de obligado cumplimiento que permitan reducir el número de fármacos falsificados que circulan en el mercado, así como una retirada efectiva de todas las unidades que pudieran eventualmente contener alguna anomalía perjudicial para la salud. Estas normas traen consigo otros beneficios tales como una mejora en la distribución y un mejor control de los productores de principios activos empleados en la fabricación de fármacos. Con independencia del país o región del globo a la que hagan referencia cada una de los sistemas, todos ellos se sirven de uno u otro modo de codificación para garantizar la trazabilidad, cobrando por tanto una importancia crucial el data carrier empleado (generalmente un código de barras) y la calidad del mismo, que deberá garantizarse para hacer posible una posterior recuperación de los datos que contiene. 62 NOVIEMBRE/DICIEMBRE14 A lo largo de este artículo realizaremos un recorrido sobre las principales normas mencionando sus principales rasgos y beneficios, así como la tecnología que será necesario introducir en las líneas de producción para garantizar su cumplimiento. Regulaciones Mundiales French CIP 13 El 16 de marzo del año 2007 la AFSSAPS (Agence française de sécurité sanitaire des produits de santé) publicó una nota en el Journal officiel de la République française, B.O.E. Francés, en la que se anunciaba la entrada en vigor a partir del 31 de Diciembre de 2010 de la normativa comúnmente conocida como French CIP 13. La AFSSAPS había escogido el Data Matrix ECC200 como data carrier para el código de artículo de 13 dígitos (CIP 13) el lote y la caducidad de todos los fármacos de prescripción médica. Todos los estuches individuales deberían contener este código además del contenido en formato humanamente legible por tal de que su distribución en el país francés fuera permitida. La norma no imponía ni a la serialización de los estuches ni tampoco a un marcaje concreto en las agrupaciones empleadas en la distribución (cajas, kits o palés). Los datos codificados deberían obedecer a la sintaxis GS1-128, permitiendo así un alto nivel de estandarización y homogeneización de las codificaciones, con la presencia obligada de los identificadores de aplicación 01 (GTIN/CIP 13), 17 (caducidad) y 10 (lote), además de permitir el uso de otros IAs para la adición de información complementaria. La elección del Data Matrix ECC200 como data carrier se demostró un movimiento muy acertado gracias a la capacidad de esta simbología para albergar una gran cantidad de datos en un espacio pequeño, punto crucial cuando se trata de marcar estuches de fármacos, así como su gran robustez que permite la lectura del código pese a que el 30% del mismo esté dañado. La tecnología necesaria en la línea se limita a equipos de visión que garanticen que el marcaje, dada una cierta fabricación con lote fijo, es constante en el tiempo y al uso de verificadores off-line que aseguren la calidad del código en base a los estándares ISO/IEC. Aunque con algunas carencias, la norma francesa aunaba puntos clave y suponía un punto de partida hacia los sistemas de trazabilidad modernos que acabarán imponiéndose en el continente europeo. China Drug Electronic Supervision (DES) El CFDA (China Food and Drug Administration) comenzó a trabajar en la creación de una red de monitorización de la cadena de suministro de fármacos y otros productos relacionados con el ámbito de la salud que tendrá implicaciones de obligado cumplimiento para todos los productores que exporten al país asiático a partir del 31 de Diciembre de 2015. La CFDA ha escogido la simbología de códigos de barras Code 128C como data carrier para la codificación de los 20 dígitos que componen el PIATS (Product Identification, Authentication and Tracking System). Este código estará presente en todos los estuches, cajas y palés distribuidos en el país y sus 20 dígitos servirán para identificar FARMESPAÑA INDUSTRIAL Los sistemas de trazabilidad integral tendrán una importancia crucial en el cumplimiento de la normativa. LA NORMA ES MUY LAXA Y NO IMPONE EL USO DE UN DATA CARRIER ESPECÍFICO O LA PRESENCIA DE DETERMINADOS IDENTIFICADORES DE APLICACIÓN, DE MODO QUE ESTAS DECISIONES QUEDAN EN MANOS DEL ETIQUETADOR al producto de forma inequívoca, además de contener dígitos de control que dificultan la creación no autorizada de códigos y suponen una barrera a las falsificaciones. La estructura del código ha sido ideada por la misma CFDA y será fija. Su calidad deberá, también en este caso, garantizarse mediante el uso de sistemas de verificación ISO. El organismo de salud chino ha creado una base de datos que servirá de interacción a todos los participantes en la cadena de suministro, desde el productor hasta el paciente, y que permitirá descargar los códigos autorizados por la CFDA previa inserción en el estuche durante su producción así como comprobar la autenticidad del producto tras adquirirlo en farmacia mediante el envío de un SMS o el ingreso de los datos en una web creada con esta finalidad. En este caso la tecnología de control necesaria consta de sistemas de visión inteligente que puedan garantizar la exactitud de los números PIATS empleados así como la calidad de marcaje de cada uno de los códigos de barras que atraviesan la línea y FARMESPAÑA INDUSTRIAL un software de trazabilidad que interaccione con la base de datos de números disponibles. Trazabilidad completa al servicio de los más de 1.300 ciudadanos de la República Popular China mediante un sistema que supone un esfuerzo significativo a los laboratorios farmacéuticos de todo el mundo y que permitirá que con un simple teléfono móvil podamos verificar la autenticidad de un fármaco. FDA - UDI (Unique Device Identifier) La agencia norteamericana FDA (U.S. Food and Drug Administration) convirtió en ley la norma UDI el 7 de Septiembre de 2007 mediante la firma de los Food and Drug Administration Amendments Act of 2007. La nueva norma no gira en torno a los fármacos, a diferencia de las comentadas anteriormente, sino a los dispositivos médicos empleados en el tratamiento o diagnóstico de pacientes (desde gasas a bisturís o marcapasos) y tiene como propósito incrementar la seguridad de estos mediante la identificación de todos ellos con un único identificador de dispositivo Verificador de etiquetas en línea para impresoras y aplicadores automáticos. (el UDI, Unique Device Identification), que comenzó a aparecer en los dispositivos a partir del 24 de Septiembre de 2014 en forma humanamente y automáticamente legibles. El nombre de la norma puede llevar a engaño, por lo que es importante aclarar que no se trata de un sistema de trazabilidad completa y por tanto no todos los dispositivos tendrán un número de serie único. Por contra, el UDI estará formado por un identificador de producto, el Device Identifier (GTIN si se emplea sintaxis GS1), y un identificador de producción, el Production Identifier, que contendrá información relacionada con la fabricación y que cambiará de una partida a otra; el lote o la caducidad serían buenos ejemplos. La norma es muy laxa y no impone el uso de un data carrier específico o la presencia de determinados Identificadores de Aplicación, de modo que estas decisiones quedan en manos del etiquetador. La figura del etiquetador es en la mayoría de los casos equivalente a la del fabricante, aunque se emplea este término para aclarar que la responsabilidad del cumplimiento recae sobre el último manipulador del producto antes de su llegada a los puntos de uso o venta. Dada la cantidad de información que deberá codificarse en el data carrier escogido, parece inevitable el uso de Data Matrix o Code 128. En ambos casos, lo razonable sería emplear la sintaxis GS1 para identificar claramente el identificador del producto y los identificadores de producción mediante los Identificadores de Aplicación 01, 10, 17, entre otros. Los UDI aparecerán en todos los niveles de agregación: dispositivo, estuche, caja y palet y el código de barras deberá tener una calidad mínima de C para que se autorice su comercialización. La FDA ha creado una base de datos para concentrar la información de los dispositivos que llegan al mercado estadounidense. Ésta recibe el nombre de GUDID (Global Unique Device Identification Database). Los etiquetadores deberán rellenar formularios estándar con la información general de los productos y no será necesario actualizar los datos para cada partida que llegue al mercado. Esta base de datos servirá para que los diversos agentes que intervienen o tienen responsabilidades en la distribución de los dispositivos puedan realizar consultas o actualizar información en caso de necesidad. Será necesario establecer sistemas de control de calidad en línea tales como veriNOVIEMBRE/DICIEMBRE14 63 NORMATIVA LAS INICIATIVAS EN POS DE UN MAYOR CONTROL Y TRAZABILIDAD EN LA DISTRIBUCIÓN DE FÁRMACOS HAN TOMADO FORMA BAJO LA CONOCIDA COMO FALSIFIED MEDICINES DIRECTIVE (FMD), QUE APLICARÁ A LOS FÁRMACOS DISTRIBUIDOS EN EUROPA A PARTIR DE 2018 ficadores ISO de códigos de barras y sistemas de visión artificial en línea que garanticen el marcaje y la legibilidad de los DI y PI marcados en formas humanamente legible e interpretables por una máquina. La norma UDI obliga a los fabricantes a ponerse manos a la obra con la estandarización en los dispositivos médicos y lo hace de un modo razonable y flexible que minimiza el impacto que su cumplimiento pueda suponer en los fabricantes de dispositivos. FMD Europea La Comisión Europea para la Salud Pública ha querido también lanzar iniciativas en pos de un mayor control y trazabilidad en la distribución de fármacos. Estas iniciativas han tomado forma bajo la conocida como Falsified Medicines Directive (FMD), que aplicará a los fármacos distribuidos en Europa a partir de 2018. El objetivo es, una vez más, luchar contra las falsificaciones de fármacos e incrementar la seguridad del paciente implicando a todos los participantes en la cadena de suministro; desde el laboratorio que produce el fármaco hasta la farmacia que lo dispensa. Quedan aún por decidir algunos aspectos pero todo apunta a que el data carrier escogido será el Data Matrix ECC200 con sintaxis GS1, continuando así con la tendencia marcada por el French CIP 13 de 2011. Se conocen ya con seguridad algunos hechos sobre las medidas de seguridad: el estuche del fármaco estará serializado y contará con una prueba de manipulación que revelará si ha sido abierto desde su salida del laboratorio productor. Todos los niveles de agregación contarán con un número de serie que los distinga inequívocamente del resto y la nota mínima de los códigos de barras según los estándares ISO/IEC será de C. 64 NOVIEMBRE/DICIEMBRE14 El FMD contará con una base de datos central, la Union Database, y bases de datos regionales que comunicarán con la central para garantizar la inmediatez de los datos ante una consulta sobre la autenticidad de un fármaco. Esta consulta podría llevarse a cabo por las autoridades, hospitales o incluso el propio paciente mediante plataformas de consulta basadas en la web (aunque el plazo de creación de estas plataformas sigue por concretar). La norma va más allá y se propone auditar también a los productores de principios activos (cumplimiento de GMP) y garantizar las buenas prácticas de los distribuidores de fármacos (GDP) así como dotar a las farmacias online de un distintivo vinculado a la web de la UE que permita al comprador verificar la reputación de la tienda en cuestión. Para garantizar el cumplimiento en línea de producción serán necesarios sistemas de visión inteligente capaces de comunicar con un software que gestione una base de datos de números de serie y garantice la ausencia de duplicados o códigos ilegibles (verificación ISO) además de la correcta colocación de los sistemas de prueba de manipulación. Será necesario también verificar las etiquetas de caja y palet para asegurar que la trazabilidad es posible a todos los niveles de agregación. El de la FMD es un propósito muy razonable para alcanzar cotas de seguridad únicas en el mercado mundial, pero su definición mediante los Delegated Acts de la comisión europea está demorándose en exceso y el coste de implementación para los laboratorios será considerable al caer sobre ellos un elevado número de responsabilidades en el cumplimiento de la norma. Qué nos depara el futuro Sistemas de trazabilidad unitaria con números de serie no predecibles se acabarán imponiendo en todos los mercados mundiales. Iniciativas como el DES de China o el FMD europeo abogan de forma acertada por la implicación de todos los agentes en la cadena de fabricación y distribución del fármaco pero nos alejan de un sistema unificado a nivel planetario y de las ventajas que esto supondría. En un mercado cada vez más globalizado tendría sentido unificar los data carriers y la sintaxis así como interconectar las bases de datos de los diversos continentes para permitir consultas óptimas y facilitar la adopción de normativas por parte de los productores de fármacos. EL PROPÓSITO DE LA FMD ES MUY RAZONABLE PARA ALCANZAR COTAS DE SEGURIDAD ÚNICAS EN EL MERCADO MUNDIAL, PERO SU DEFINICIÓN MEDIANTE LOS DELEGATED ACTS DE LA COMISIÓN EUROPEA ESTÁ DEMORÁNDOSE EN EXCESO Y EL COSTE PARA LOS LABORATORIOS SERÁ CONSIDERABLE Pese a la ausencia de una solución global, el futuro es muy prometedor en cuanto a las capacidades implícitas en el hecho de tener a disposición bases de datos de tal envergadura y su interconexión con, por ejemplo, bases de datos de interacciones o los historiales de los pacientes. Tales sistemas podrían reducir drásticamente el número de administraciones con interacción negativa en el paciente, lo que ahorraría importantes sumas a los sistemas públicos de sanidad y contribuiría a salvar anualmente miles de vidas Glosario – – – – – Sistema de visión con cámara inteligente para la supervisión integral de la productividad. – – Data Carrier: Dispositivo capaz de contener información legible para una máquina. Simbología: Conjunto de instrucciones sobre cómo configurar las barras oscuras y claras que conforman un código de barras (ejemplos: EAN 13, Code 128 o Data Matrix). Data Matrix: Código de barras bi-dimensional con gran capacidad de compresión. Serialización: Proceso por el cual cada uno de los productos fabricados en una línea de producción contienen una matrícula única, el número de serie, que los diferencia del resto. Identificadores de Aplicación: Prefijos empleados en la sintaxis GS1-128 para dar sentido a la información que los sigue dentro de un código de barras. GMP: Good Manufacturing Practices GDP: Good Distribution Practices FARMESPAÑA INDUSTRIAL AVANCE PROGRAMA DE CONFERENCIAS 2015 www.farmaforum.es 4 de marzo 10:00 - 10:45 Apertura 10:45 - 11:15 FORO CPPTIC Nuevos retos en la contratación pública Impulso a la innovación. Aspectos destacados de la transposición de las nuevas Directivas de Contratación, así como del nuevo periodo de Fondos Estructurales 2014-2020, con un apoyo claro a la innovación. Beatriz Juliá Directora General Kamalan Consulting y del Foro de colaboración público - privada en tecnología 11:15- 11:30 COFFE BREAK 11:30 - 12:15 AEFI La cadena de distribución de medicamentos. Las Buenas Prácticas de Distribución Beatriz Artalejo Presidenta AEFI 12:15 - 13:00 ANTARES CONSULTING Bridging entre Academia e Industria: qué hacer para convertir un proyecto en atractivo y vendible. Análisis de un caso de éxito Josep Lluís Falcó Brenes Senior Manager Antares Consulting 13:00 - 13:45 CINFA Biosimilares, un reto para la industria y para el regulador Dr. Julio Maset #HQDBSNQBHDMS¨ƥBN 13:45 - 14:30 IMS La evolución del mercado farmacéutico después de la crisis Concha Almarza #HQDBSNQ.ƤDQHMF#DUDKNOLDMS@MC#@S@2NTQBDR 14:30 - 15:30 LUNCH 15:30 - 16:15 AEPIMIFA 1HZWHFKQRORJ\WUHQGVLQLQMHFWDEOHVPDQXIDFWXULQJƩOOLQJ Mr. Jean Nomes $W#HQDBSNQCD(MFDMHDQ¨@CDK@OK@MS@CD/ƥYDQDM/TTQR!DKFHB@ 16:15 - 17:00 INGELYT, AZIERTA Criterios de compatilidad de productos en áreas de producción Alberto Carazo Responsable de asesoría farmacéutica de Ingelyty y consultor independiente de otras compañías nacionales e internacionales María Ángeles Pérez de la Cruz Responsable dela área preclínica y toxicológica de AZIERTA 17:00 - 17:45 GRIFOLS Soluciones estériles: La elección del CMO, una decisión estratégica. Marga Viñes International Product Manager IV Therapy & Contract manufacturing de Grifols 17:45 - 18:30 5 de marzo 09:15 - 10:00 IMCO High containment en proceso de producción de sólidos. Peter Merimeche OLLEROS Y ABOGADOS Las estrategias de patentes defensivas a la luz de las últimas decisiones de las autoridades de competencia Isabel Martínez 10:00 - 10:45 TELSTAR 4XDOLW\E\'HVLJQSDUDODRSWLPL]DFL²QGHUHFHWDVGHOLRƩOL]DFL²Q Alicia Tebar ,@M@FDQ0 0A#^ 10:45 - 11:15 BEC Risk Analysis Manuel Peña Consultor senior de formación y mejora de procesos 11:15- 11:45 COFFE BREAK 11:45 - 12:30 FARMAVENIX Modelo de innovación y desarrollo logístico Rafael García Casal Director General de Farmavenix 12:30 - 13:15 ISPE "ISPE"s Drug Shortage Prevention Plan Thomas Zimmmer Vicepresidente de Operaciones Europa 13:15 - 14:00 AEMPS Producción de instalaciones compartidas Cristina Gomez Chacón Jefe de área, área de Inspección en Agencia Española de medicamentos y Productos Sanitarios 14:00 - 14:45 ROVI Cambio cultural: del mundo ideal a la realidad Pilar García-Morato Directora de planta Farmacéutica 14:45 - 15:45 LUNCH 15:45 - 16:30 SKAN La importancia de los aisladores en el mundo Farmacéutico Mario Bielsa Sales Manager 16:30 - 17:15 STERIS El enfoque del ciclo de vida de la validación de limpieza Elizabeth Rivera Technical Service Specialist 17:15 - 18:00 CMP Nuevas tendencias en las buenas prácticas de fabricación de cosméticos Eduardo Rodríguez Pons- Esparver Director técnico + Revista profesional de Proveedores de la Industria Farmacéutica y Tecnología de Laboratorio PROGRAMAEDITORIAL2015 CIERRE 17/07/2015 JULIOAGOSTO ESPECIAL BIOTECNOLOGÍA ESPECIAL CPHI MADRID, OCTUBRE FILTRACIÓN II DRUG DELIVERY SYSTEMS ENEROFEBRERO ESPECIAL FARMAFORUM CIERRE 20/02/2015 MADRID, 4-5 MARZO SOFTWARE PARA LABORATORIOS I APIS REGULATORY AFFAIRS FABRICACIÓN A TERCEROS AUTOMATIZACIÓN DE PROCESOS INGENIERÍA FARMACÉUTICA Y MANTENIMIENTO II ROBÓTICA EFICIENCIA ENERGÉTICA EN LOS LABORATORIOS TECNOLOGÍA DE LABORATORIOS I GUÍA DE PROVEEDORES DE LA INDUSTRIA FARMACÉUTICA SOFTWARE PARA LABORATORIOS I SEPTIEMBREOCTUBRE INGENIERÍA FARMACÉUTICA I ESPECIAL PHARMAPROCESS ETIQUETADO Y CODIFICACIÓN I ESPECIAL BIOLATAM FILTRACIÓN I TECNOLOGÍA DE LABORATORIOS II MARZOABRIL CIERRE 10/04/2015 ESPECIAL CONGRESO ANUAL AEFI ESPECIAL FERIA HISPACK ESPECIAL SIL CIERRE 16/10/2015 BARCELONA, 27-28 OCTUBRE SANTIAGO DE CHILE, 16-17 NOVIEMBRE EQUIPAMIENTO PARA SALAS BLANCAS MOBILIARIO PARA LABORATORIO BARCELONA, 21-24 ABRIL BARCELONA, 9-11 JUNIO ESPECIAL FERIA EMPACK ENVASE Y EMBALAJE II TECNOLOGÍA COSMÉTICA ETIQUETADO Y CODIFICACIÓN II LOGÍSTICA Y DISTRIBUCIÓN LOGÍSTICA Y DISTRIBUCIÓN II ENVASE Y EMBALAJE I NOVIEMBREDICIEMBRE AUTOMATIZACIÓN DE ALMACENES SALAS BLANCAS II SALAS BLANCAS I ESTERILIZACIÓN II TRATAMIENTO DE AGUAS I VALIDACIONES Y CERTIFICACIONES II HIGIENE, SEGURIDAD Y MEDIO AMBIENTE CONSULTORÍA Y FORMACIÓN MAYOJUNIO CIERRE 05/06/2015 TRATAMIENTO DE AGUAS II ESPECIAL ACHEMA FRANKFURT, 15-19 JUNIO ENSAYOS CLÍNICOS VALIDACIONES, CERTIFICACIONES I FABRICACIÓN A TERCEROS II NORMATIVA, REGISTROS Y LEGISLACIÓN TRATAMIENTO DE SÓLIDOS MICROBIOLOGÍA ESPECIAL LOGÍSTICA FARMACÉUTICA TRATAMIENTO DE GASES ENVASE Y EMBALAJE ESTERILIZACIÓN I ETIQUETADO Y CODIFICACIÓN CLIMATIZACIÓN LOGÍSTICA Y DISTRIBUCIÓN COSMÉTICA: FABRICACIÓN A TERCEROS CENTROS DE DISTRIBUCIÓN FARMACÉUTICA ¿QUIÉN ES QUIÉN EN LA INGENIERÍA FARMACÉUTICA? ¿QUIÉN ES QUIÉN EN LA LOGÍSTICA FARMACÉUTICA? CIERRE 11/12/2015 CIERRE DICIEMBRE lifescienceslab Suplemento de Investigación, Analítica, Diagnóstico y Control en el Laboratorio NOV-D)C37 supplying water ción: Para más informa edia.com info@grupo-omnim Tel. 617 708 514 Soluciones modulares para el suministro de agua AGUA POTABLE AGUA RESIDUAL REGENERACIÓN sss./4)#)!3 Dara Pharmaceutical Packaging presenta su equipo SX-50 SpeedFill. Dosificadora peristáltica de sobremesa El SERGAS y Boehringer Ingelheim firman un acuerdo para la promoción de la investigación científica y la formación El Servicio Gallego de Salud y el laboratorio Boehringer Ingelheim han firmado un convenio de colaboración para la promoción de actuaciones en los ámbitos de la investigación y la formación científica orientadas a la mejora de la atención y los resultados en salud de pacientes. El convenio contempla especialmente patologías crónicas, como diabetes y EPOC (Enfermedad Pulmonar Obstructiva Crónica), y otras áreas terapéuticas como oncología. El acuerdo ha sido firmado por la Consejera de sanidad gallega, Rocío Mosquera, y por las representantes de Boehringer Ingelheim España, Alicia Gil y Kàtia Piñol. Este acuerdo se enmarca dentro de la estrategia del Sergas 2014 en la que se establecen una serie de líneas de acción prioritarias en relación a la cronicidad y los objetivos de la Consejería de Sanidad de Galicia para impulsar, planificar, coordinar y evaluar las acciones llevadas a cabo para mejorar el proceso de atención y los resultados en salud con la participación de todos los agentes del sistema de salud y el sistema social, con el fin de establecer un marco de actuación global. 68 NOVIEMBRE/DICIEMBRE14 El dosificado peristáltico se ha convertido en poco tiempo en el sistema preferido por la industria farmacéutica, ya que aporta esterilidad, flexibilidad y fácil limpieza, gracias a que el fluido únicamente está en contacto con el interior del tubo y la aguja de dosificado. La máquina que les presentamos es una llenadora peristáltica de sobremesa, para un dosificado exacto y de alta calidad, en frascos, tubos y viales. El diseño del equipo se ha realizado para su uso en área estéril, conforme a las normas cGMP y de la US-FDA en especial correspondencia con las exigencias de la industria farmacéutica. La SX-50 está propulsada mediante un potente servomotor, capaz de aceptar hasta 3 cabezales en serie. El cuerpo del cabezal es monobloque, está construido en una sola pieza para una mejor asepsia. El cabezal peristáltico bombea los fluidos de manera suave y progresiva, sin romper la cadena molecular de los mismos, es excelente para dosificar sólidos en suspensión, vacunas, tejidos celulares y otros fluidos sen- sibles. Se trata de un equipo construido con los mejores componentes estándar de mercado, así elementos como el servomotor, reductor, pantalla táctil o PLC de control son elementos de fácil reposición en su área geográfica, facilitando un mantenimiento sencillo y rápido. Los tubos “Platinium Cured Silicone” se fabrican en sala aséptica y son de fácil esterilización mediante autoclave o mediante radiación por rayos gamma. El equipo está diseñado para funcionar de forma independiente o ser parte de una máquina llenadora industrial, totalmente automática. Todos los parámetros de producción son almacenados en la memoria del sistema, parámetros tales como: tVolumen de dosificado. tCinemática del proceso de llenado, control individual de las rampas de aceleración y desaceleración. tIntervalo de tiempo entre dosificación. tParámetros de calibración. tFunción anti-goteo “dripfree”. tVelocidad de producción. Se regulan automáticamente y son asociados a cada programa de dosificado. El equipo puede almacenar hasta 200 programas de lotes de producción diferentes. Equipamientos opcionales tAgujas, soporte y tubos de dosificado. tCabezal peristáltico adicional, hasta 3 cabezales en serie. tConexión a micro balanza y re-calibrado automático del volumen a dosificar. tCabezal peristáltico construido íntegramente en acero inoxidable AISI316L. tSoftware para la documentación o impresión de todos los datos relevantes de la producción. tEjecución eléctrica antideflagrante Xproof. tValidación IQ / OQ. Para más información www.dara-pharma.com Barcelona tendrá una sede de la nueva comunidad de conocimiento e innovación de la UE en salud La Unión Europea (UE) a través del Instituto Europeo de Innovación y Tecnología (EIT) anunció el martes el proyecto ganador de la convocatoria de la nueva Comunidad de Conocimiento e Innovación (KIC) centrada en vida saludable y envejecimiento activo. El proyecto ganador ha sido Innolife, una propuesta liderada en el estado español por la Universidad de Barcelona (UB), nuclea- da a partir de su campus de excelencia internacional en salud, HUBc, y con el impulso de Biocat. Innolife contará con una sede en España que estará ubicada en el Parc Científic de Barcelona (PCB). FARMESPAÑA INDUSTRIAL SYGNIS firma un acuerdo no exclusivo con la compañía alemana BioCat para la distribución en Europa de su primer producto para la amplificación de ADN SYGNIS AG ha anunciado la firma de un acuerdo de distribución para TruePrime™ con la compañía alemana BioCat GmbH, especializada en la comercialización de productos innovadores para investigación en ciencias biológicas. Con este acuerdo, SYGNIS otorga a BioCat derechos (no exclusivos), para promocionar y comercializar el kit TruePrime™ Single Cell WGA para la amplificación de ADN a partir de una única célula. Se trata del primero de una serie de productos de la familia de productos TruePrime™ de SYGNIS diseñados para la amplificación de ADN o ARN y que cuentan con múltiples aplicaciones, especialmente en el ámbito de la secuenciación de nueva generación. Se espera que este primer producto sea lanzado al mercado en enero de 2015. Como proveedor de soluciones altamente especializadas, BioCat ofrece en la actualidad una amplia gama de reactivos de alta calidad para su uso en investigación, así como tecnologías de vanguardia en el campo de la genómica, la proteómica y la biología celular, que comercializa entre clientes de toda Europa. “Estamos muy satisfechos con este acuerdo de distribución para nuestro primer producto, TruePrime™, con un socio tan relevante como BioCat. Con una amplia red de clientes en el ámbito de la biología molecular y una capacidad logística contrastada, BioCat facilitará a los investigadores la posibilidad de aprovechar los beneficios de nuestra nueva tecnología para que éstos puedan utilizarla en el creciente campo de la secuenciación de nueva generación (NGS)”, señala Pilar de la Huerta, CEO de SYGNIS. En esta línea añade que poner a partir de ahora a disposición del mercado en Europa el kit TruePrime™ Single Cell WGA “es, sin duda, un logro relevante en la nueva estrategia de producto y comercialización que hemos iniciado en SYGNIS. Además, la firma de un contrato de estas características antes del lanzamiento del producto, demuestra el atractivo de este kit, así como la confianza que en su potencial tienen los expertos en el mercado, como BioCat”. Por su parte, el Dr. Michael Ehret, CEO de BioCat GmbH, señala que “estamos muy satisfechos con la inclusión de productos de SYGNIS en nuestro portfolio de soluciones innovadoras para investigadores que trabajan en el campo de la biología molecular. FARMESPAÑA INDUSTRIAL NOVIEMBRE/DICIEMBRE14 69 sss./4)#)!3 Roche adquiere Ariosa Diagnostics y entra así en los mercados del cribado prenatal no invasivo (CPNI) y los servicios de análisis de ADN acelular Roche ha dado a conocer la compra de Ariosa Diagnostics, Inc. (Ariosa), una empresa no cotizada en bolsa con sede en San José (California, Estados Unidos). Ariosa es un proveedor de servicios analíticos de diagnóstico molecular que ofrece un sistema de cribado prenatal no invasivo (CPNI) de gran selectividad y precisión, basado en tecnología de ADN acelular y realizado en su laboratorio, que cumple los requisitos CLIA. La prueba HarmonyTM Prenatal de Ariosa es un análisis de sangre que se realiza ya a las 10 semanas de iniciado el embarazo. En esta prueba se analiza ADN acelular del feto en la sangre materna para evaluar el riesgo de síndrome de Down y otras anomalías genéticas. Concretamente, la prueba valora el riesgo de trisomía fetal (presencia de un cromosoma adicional que puede provocar trastornos genéticos graves) para los cromosomas 13, 18 y 21. La prueba HarmonyTM Prenatal está validada según los criterios CLIA por un sólido conjunto de datos clínicos y respaldada por los resultados de estudios clínicos obtenidos en más de 22.000 mujeres de todas las edades y categorías de riesgo*. Anualmente, en todo el mundo se producen más de 200 millones de embarazos1, y el cribado prenatal del síndrome de Down se ha convertido en una práctica habitual en muchos países. Las pruebas de cribado habituales dan lugar a falsos positivos en hasta un 5%2 de los casos, mientras que HarmonyTM presenta una tasa de falsos positivos inferior al 0,1%3. «La adquisición de Ariosa es un ejemplo más del compromiso de Roche con el diagnóstico molecular avanzado», ha declarado Roland Diggelmann, director operativo (COO) de la División Diagnostics de Roche. «El ADN acelular circulante alberga la promesa de proporcionar información diagnóstica precoz con un simple análisis de sangre en muchos segmentos importantes, como el embarazo, el cáncer o los trasplantes, algo que encaja muy bien con nuestra estrategia de medicina personalizada y nuestro com- promiso de marcas nuevas pautas en la atención sanitaria». «Estamos encantados de unir nuestras fuerzas a las de Roche para seguir adelante en nuestro compromiso de impulsar un análisis genético asequible de alta calidad que mejore la atención sanitaria de los pacientes de todo el mundo», ha manifestado por su parte Ken Song, director general (CEO) de Ariosa. Se prevé que la operación se cierre a lo largo del mes de diciembre de 2014. *Los datos no han sido presentados a los organismos reguladores ni evaluados por ellos, y la prueba no se comercializa como diagnóstico in vitro en Estados Unidos ni en la Unión Europea. El 82% de los asistentes a Biospain identificaron oportunidades reales de negocio El 7º Encuentro Internacional de Biotecnología, BioSpain 2014 (www.biospain2014. org), el principal evento de biotecnología que se celebra en España y uno de los más importantes a nivel internacional, se celebró el pasado mes de septiembre en Santiago de Compostela. Este evento bienal, organizado por la Asociación Española de Bioempresas (ASEBIO) -y, en esta edición, también por la Xunta de Galicia, la Universidad de Santiago de Compostela, la Universidad de Vigo y el Concello de Santiago- ha reforzado un año más su carácter internacional: de las 855 entidades privadas y públicas asistentes a la feria, con un crecimiento del 12,2% respecto a la edición anterior, 289 son de fuera de España, procedentes de 37 países, lo que supone cerca del 34% del total, frente 70 NOVIEMBRE/DICIEMBRE14 al 28% de internacionalización que se alcanzó en la pasada edición, lo que supone un incremento de la internacionalización del 37,6% sobre la edición de 2012. El 66% de las empresas procedieron de España, seguidas de las de Reino Unido (6,21%), Francia (3,51%), Portugal (3,40%), EE.UU. (2,93%), Colombia (2,34%), México (2,34%), y Suiza, (1,87%), entre otras. Además, esta 7ª edición de Biospain, que coincide este año con la celebración del Año de la Biotecnología en España y con el XV aniversario de ASEBIO, contó con 200 expositores y con la presencia de 50 inversores -80% internacionales-, entre fondos de capital riesgo, fondos de inversión de grandes farmas, business angels, etc., que participaron en el Foro de Inversión, donde se analizaron los planes de negocio de 40 empresas de varios países. En el partnering, se alcanzaron las 3.327 reuniones de negocio, lo que supone un crecimiento del 20% respecto a la edición anterior, y se registraron 2.000 asistentes en total al evento (un 8% más), lo que consolida a Biospain 2014 en el quinto evento mundial por reuniones de desarrollo de negocio de este sector. En Biospain 2014 se celebraron 50 conferencias paralelas y científicas, 37 presentaciones de compañías, y se presentaron en el partnering 720 productos disponibles para ser licenciados. El Comité de Organización de Biospain ha realizado una encuesta de satisfacción entre las personas asistentes al evento, en el que han contestado un 25% del total. En la misma, contestada mayoritariamente por representantes de empresas biotecnológicas e instituciones de investigación, destaca que el 55% de los encuestados afirma que Biospain cumplió sus expectativas, y un 37% contestó que superó sus expectativas. El 82% identificó oportunidades reales de negocio, y en cuanto a la valoración general sobre el evento, el 54% afirmó es buena, el 19% afirmó que es muy buena y en la media el 24% lo que supone un 97% de satisfacción de los asistentes. FARMESPAÑA INDUSTRIAL ASEBIO considera que los precios de referencia no deberían ser aplicables a los biológicos La Asociación Española de Bioempresas (ASEBIO), junto con la European Association for Bioindustries (Europabio) ha organizado el I Encuentro EuropabioASEBIO sobre innovación y terapias biológicas en el Ministerio de Sanidad, Servicios Sociales e Igualdad, acto en el que se ha presentado el documento de posición de productos biológicos de ASEBIO. Las terapias biológicas han mejorado las expectativas y la calidad de vida de pacientes con enfermedades graves neurológicas, reumatológicas, endocrinas o tumorales, entre otras. El proceso de fabricación de estos fármacos es más complejo que el de los sintetizados químicamente, y normalmente, se administran mediante inyección o infusión en medio hospitalario. Sus características diferenciales le otorgan un tratamiento especial, tanto desde el punto de vista legislativo, como médico y económico. Cuando la patente de los medicamentos biológicos vence, el arsenal terapéutico se va ampliando con el desarrollo de los fármacos biosimilares, que son moléculas similares, pero no idénticas, no sustituibles, ya que provienen de líneas celulares distintas y de procesos de fabricación diferentes. En países como España, Francia, Alemania y Reino Unido no se permite la sustitución automática de un medicamento biológico original o biosimilar por otro, si no que dicha sustitución debe basarse en una decisión del médico prescriptor que trata al paciente. FARMESPAÑA INDUSTRIAL Estos medicamentos necesitan un tipo de regulación específica que permita diferenciarles de los químicos tradicionales, que son considerados intercambiables. Por ello, el Grupo de trabajo de medicamentos innovadores de ASEBIO ha trabajado en un documento de posición acerca de estas terapias para que sirva como guía de conocimiento de su acceso al mercado en España. En el mismo, ASEBIO considera que el sistema de precios de referencia no debería ser aplicable a los biológicos por sus especiales características, aunque actualmente así se recoge en la regulación. Como explica Regina Revilla, presidenta de ASEBIO, “cada terapia biológica es única e insustituible y puesto que la relación entre los productos biológicos y los biosimilares no es equiparable a la de los productos de marca y sus genéricos, ni su desarrollo, costes de fabricación y manera de competir en el mercado es igual, su régimen de precios tampoco debe ser igual que el régimen de precios de los genéricos. Creemos, por tanto, que la regulación específica debe recoger la importancia de la prescripción por marca sin que se produzca sustitución y respetando la toma de decisión clínica personalizada, basada en la evidencia científica”. Como ejemplo de las diferencias que existen entre los productos genéricos y los biosimilares, se puede tener en cuenta que para el desarrollo de un medicamento genérico se requieren entre dos y tres años y una inversión de uno a tres millones de euros, mientras que para el desarrollo de un biosimilar se necesitan de 100 a 300 millones de euros y entre cinco y ocho años, según datos del Libro Blanco de los Medicamentos Biosimilares en España: Calidad Sostenible La garantía del acceso universal a medicamentos clave. Los medicamentos biológicos y biosimilares de nueva generación (anticuerpos monoclonales) son todos de uso hospitalarios y compiten económicamente mediante descuentos, compras centralizadas u otros procedimientos de contratación pública competitiva, por lo que tiene mucho más sentido aplicar un régimen competitivo a este tipo de medicamentos no sustituibles y no un régimen de precios de referencia. El ISCIII y AstraZeneca suman fuerzas con el fin de promover la formación científica y la investigación en salud AstraZeneca y el Instituto de Salud Carlos III (ISCIII) han firmado un convenio marco con el fin de promover la formación científica y la investigación en el ámbito de la salud. Gracias a este acuerdo ambas entidades estrecharán sus relaciones para aunar esfuerzos y establecer normas amplias de actuación que encaucen e incrementen su colaboración dentro de este marco. “La finalidad de este convenio es establecer un marco de actuación entre AstraZeneca y el ISCIII en actividades docentes, soporte científico y tecnológico así como en biomedicina. La formación continuada y el impulso de la ciencia son dos objetivos prioritarios para nuestra compañía y apostar por ellos supone trabajar con referentes en este ámbito, como lo es ISCIII”, señala Ludovic Helfgott, presidente de AstraZeneca España. Dentro de este acuerdo se incluyen diferentes modalidades de colaboración entre estas dos entidades entre las que se encuentra el desarrollo de proyectos de investigación tanto en el ámbito nacional como internacional o la cooperación en programas de formación. El convenio firmado entre el ISCIII y AstraZeneca tendrá una duración mínima de dos años y refleja la creación de una comisión mixta integrada por miembros de ambas entidades. Su objetivo será establecer propuestas de colaboración en temas científico-tecnológicos y velar por el cumplimiento de los fines que han dado lugar a la firma del convenio así como realizar un seguimiento de los convenios específicos que se suscriban. NOVIEMBRE/DICIEMBRE14 71 sss")/).&/2-4)#! Una medicina nueva, más inteligente y menos invasiva sss,ASREVOLUCIONESCIENTÓlCOTECNOLØGICASSUFRIDASENESTOSÞLTIMOS A×OSHANPOSIBILITADOELDESARROLLOEXPONENCIALDEHERRAMIENTASYTÏCNICAS CON LAS QUE SE ESTÉ INICIANDO UN CAMBIO DE PARADIGMA EN LA MEDICINA ACTUAL%LANÉLISISMASIVODEDATOSPROVENIENTESDELASØMICASPROPICIADO PORLASUPERCOMPUTACIØNYLAREDUCCIØNDECOSTESDELOSANÉLISISCATALIZA RÉNUNCAMBIORADICALENLAFORMADEABORDARLASALUDENESTESIGLO88) .ATALIA*IMÏNEZ,OZANO PhD Healthcare & Life Sciences Consultant “Los avances en genómica nos van a permitir saber cómo funcionan las enfermedades, quiénes son más susceptibles a padecerlas y cómo podemos tratarlas con éxito, reduciendo la incertidumbre y el estrés de los pacientes y sus familias”. Esta afirmación de Tom Fowler, Director de Salud Pública en Genomics England, realizada recientemente en la Jornada sobre “Medicina de Precisión” celebrada en el Centro de Investigación Príncipe Felipe de Valencia, sintetiza alguna de las consecuencias de los avances en la Medicina de Precisión, también conocida como Medicina P4. Según Fowler, una de cada diecisiete personas desarrolla una enfermedad rara y al menos el 80% de las enfermedades raras tienen un componente genético identificado, que actualmente puede detectarse rápidamente y a un 72 NOVIEMBRE/DICIEMBRE14 coste razonable. Las ciencias ómicas, la robótica, la simulación 3D y la realidad aumentada, el Big Data, la supercomputación… son los grandes impulsores de un cambio que nos lleva a este nuevo modelo médico orientado a mantener el bienestar de los ciudadanos, que se apoya en cuatro pilares: predicción, prevención, personalización y participación. Un modelo que no sólo es bueno para los ciudadanos, sino que también lo será para garantizar la sostenibilidad de un sistema sanitario, que es un componente clave de lo que conocemos como estado del bienestar. Un cambio que tiene una sólida base tecnológica, que facilita la aplicación en el entorno clínico de los avances en investigación, y cuyo éxito dependerá de que seamos capaces de alinear los esfuerzos que están llevando a cabo los investigadores, los profesionales sanitarios y la industria, para lograr un impacto positivo en la salud de los ciudadanos. Genómica y mucho más La genómica está en los cimientos de la Medina P4. Proporciona un mejor conocimiento de las causas de las enfermedades y de cómo las enfermedades se desarrollan en cada individuo. Hace realidad una medicina más personalizada que minimiza los efectos secundarios causados por los fármacos. En el caso de las enfermedades raras, la secuenciación del genoma aumenta las posibilidades de diagnóstico. También tiene un papel importante en el seguimiento de las enfermedades infecciosas, ya que puede explicar cómo se propagan las infecciones y, en muchos casos, permitir la localización exacta de la fuente. Pero hay más cosas. Es el caso de avances de la cirugía guiada por imagen personalizada. Así, según Jean-Luc Dimarcq, director Administrativo del Strasbourg Institute of Image-Guided Surgery, “La medicina moderna se basa cada vez más en los modelos de gestión y prevención de errores desarrollados por la aeronáutica”. Para él, los cirujanos, radiólogos y endoscopistas intervencionistas deben “entrenarse” como lo hacen los pilotos de aeronaves, utilizando simuladores donde podrán planificar el procedimiento en modelos virtuales específicos para el paciente, de la misma manera que los pilotos que abordan un plan de vuelo. Una simulación que puede realizarse antes de la intervención real, en la que estarán asistidos por sistemas de navegación basados en la imagen y equipos robotizados. Una nueva visión que exigirá optimizar las terapias personalizadas utilizando la realidad virtual y aumentada, mejorar el acceso mínimamente invasivo, incorporar la robótica y promover la formación en terapias híbridas. La cirugía del futuro será una fina mezcla de información, imagen real o virtual y robótica, con el objetivo principal de aumentar la seguridad del paciente y mejorar la calidad de vida. Unas técnicas que exigen un entrenamiento específico orientado a capacitar a una nueva generación de médicos que integre las habilidades de un cirujano mínimamente invasivo, de un radiólogo intervencionista y de un endoscopista. Big Data En esta nueva medicina, el conocimiento que facilita la información, extraída de billones de datos, será clave para la transformación de la salud. Según Julio Mayol, director de Innovación del Hospital Clínico San Carlos y co-director del consorcio M+Visión, “Si queremos crear sistemas sanitarios que respondan a necesidades de ciudadanos, tenemos que pasar de sanidad basada en el Conocimiento a sanidad basada en Inteligencia. Para eso tenemos &!2-%30!»!).$5342)!, que tener no Big Data sino All Big Data, todos los datos para responder a nuevos retos de la sanidad”. Este “All Big Data” incluirá el sensoroma (todas las variables de los individuos captadas mediante sensores en cualquier lugar) y el socioloma (todos los datos correspondientes al componente social de los individuos y los grupos en los que viven, trabajan y desarrollan su actividad). Un cambio de modelo que exige, sobre todo, liderazgo. “El presupuesto y la tecnología están ahí”. Según Mayol, la medicina debe estar apoyada en la inteligencia (hacer las preguntas correctas con todos los datos disponibles) y no solo en la prestación de servicios basados en un conocimiento limitado de nuestra práctica. El nuevo sistema sanitario debe saber hacer las preguntas que permitan dar las respuestas a nuevos retos de la sanidad. “Una Medicina 5P”, que añade el elemento poblacional a la predicción, la prevención, la personalización y la participación, porque no enferma igual una persona del norte que una del sur, o una con mejor formación, o más recursos. Nuevas infraestructuras Un Big Data debe apoyarse en unas nuevas infraestructuras bioinformáticas que aseguren a largo plazo la existencia de las base de datos y recursos que hoy posibilitan la investi&!2-%30!»!).$5342)!, gación en ciencias de la vida que se realiza en los diferentes países europeos, un objetivo que hoy asume el proyecto Elixir, que además de mantener esta información, dotará de interoperabilidad a los recursos que existen en los distintos países y promoverá determinados estándares de calidad. Un ejemplo de estos recursos es la base de datos EGA, cuyo desarrollo y mantenimiento está en manos del European Bioinformatics Institute (EBI) y del Centro de Regulación Genómica (CRG), uno de los diez nodos del Instituto Nacional de Bioinformática (INB). Esta base de datos almacena de forma segura datos genómicos e información sobre las características fenotípicas de los pacientes. Grandes proyectos como el Estudio de la Leucemia Linfática Crónica liderado por España dentro del Proyecto Internacional del Genoma del Cáncer (ICGC) almacena sus datos en este repositorio. Según Víctor de la Torre, Coordinador General del INB (Instituto Nacional de Bioinformática), ELIXIR tendrá un gran impacto. “El tratamiento genómico se resuelve de manera independiente en muchos hospitales. Una de las principales tareas de ELIXIR es enlazar estos esfuerzos y promover la adopción de estándares de calidad en los procesos. Hay mucha gente haciendo lo mismo, como si reinventáramos la rueda todos los días, es necesario apro- vechar al máximo los recursos y ofrecer biomedicina de calidad a escala europea”. Un nuevo entorno posible gracias a los avances tecnológicos, especialmente en supercomputación, que permiten abordar con éxito una medicina que maneja trillones de datos. Un ejemplo: según el doctor Francisco J. del Castillo, del Servicio de Genética de Hospital Ramón y Cajal, en un exoma completo encontramos de media unas 40.000 variantes genéticas y para que esta información sea relevante en la práctica clínica hacen falta equipos de bioinformáticos en los hospitales y una interacción muy estrecha entre el clínico, el genetista y el bioinformático. La Medicina P4 ofrece un diagnóstico y un tratamiento a medida de cada paciente y permitirá reducir costes, reacciones a fármacos y los tiempos de hospitalización. Explosión de datos La historia clínica electrónica, las pruebas de imagen multimodal y las nuevas técnicas de secuenciación del genoma están generando una explosión de datos en los entornos sanitarios que requiere de nuevas aproximaciones sobre cómo se gestiona esta información, con qué seguridad y cómo se garantiza la supervivencia en el tiempo para que pueda estar disponible a lo largo de la vida de los pacientes. Todo ello en un marco científico muy productivo y que obliga a una evolución constante de la tecnología para adaptarse a los nuevos estándares y formatos. En este escenario de futuro, la visión de Bull es convertirse en facilitador de la implementación de un modelo sanitario basado en la Medicina P4, construyendo un puente tecnológico que permita la traslación de las ciencias ómicas al ámbito clínico. Mantener el estado del bienestar Más allá de los avances científicos y técnicos, la Medicina P4 tiene un objetivo principal, mantener uno de los activos fundamentales del estado del bienestar: la salud de los ciudadanos, y aparece como una pieza clave en la sostenibilidad de un sistema mermado por los recortes económicos que amenazan su continuidad. Un nuevo estado del bienestar de ciudadanos sanos, que enferman menos y que cuando lo hacen reciben un tratamiento personalizado, más eficaz, menos invasivo y sin efectos secundarios. Una precisión que contribuirá a una mejora sustancial de la calidad de vida de los ciudadanos y a una reducción considerable de los gastos. Este último elemento deberá bastar para que el nuevo modelo reciba el impulso necesario de organismos, instituciones, empresas e industrias y la comprensión y colaboración de los profesionales de la salud NOVIEMBRE/DICIEMBRE14 73 GUÍA DE SERVICIOS FARMESPAÑA INDUSTRIAL pone a su disposición la forma más práctica de localizar a sus proveedores organizados por sectores de actividad. Las páginas de servicio más completas con empresas fabricantes de equipos, productos y de servicios para la industria farmacéutica y sectores afines. INFÓRMESE EN EL TELÉFONO 986 19 04 88 Y EN NUESTRA WEB www.farmaindustrial.com Tarifas 2013 inserción anual Módulo sencillo (55 x 65 mm) Módulo doble (55 x 150 mm) Módulo doble (117,5 x 65 mm) Módulo triple (180 x 65 mm) Datos a cumplimentar en los módulos Razón social Teléfono y fax Domicilio www Distrito/Población mail CONSULTORÍA Y VALIDACIONES 550 800 800 1200 euros euros euros euros DESINFECCIÓN AQUÍ SU PUBLICIDAD POR 800 € / AÑO 74 NOVIEMBRE/DICIEMBRE14 FARMESPAÑA INDUSTRIAL ASESORÍA Y VALIDACIONES EQUIPAMIENTO PARA LABORATORIO FARMESPAÑA INDUSTRIAL ENVASE Y EMBALAJE EQUIPAMIENTO PARA LABORATORIO ESTERILIZACIÓN NOVIEMBRE/DICIEMBRE14 75 GUÍA DE SERVICIOS EQUIPAMIENTO DE LABORATORIOS ETIQUETA FILTRACIÓN HIGIENE INDUSTRIAL AQUÍ SU PUBLICIDAD POR 800 € / AÑO 76 NOVIEMBRE/DICIEMBRE14 FARMESPAÑA INDUSTRIAL EXCIPIENTES INGENIERÍA, SALAS Y EQUIPOS FARMACÉUTICOS INGENIERÍA Y CONSULTORÍA MAQUINARIA MAQUINARIA DE ENVASADO Y PROCESO Pharmaceutical Filling Lines 7!/3(,!0(! $% .0-#%1- 6 %,4!1!$- !32-+92(#- .!0! *! (,$3120(! &!0+!#:32(#! "(-2%#,-*<'(#!6#-1+:2(#! 79/3(,!1 (,$(4($3!*%1 - *;,%!1 #-+.*%2!1 .!0! %* **%,!$- 6 #%00!$- $% .0-$3#2-1 *;/3($-1 1%+( *;/3($-1 6 %, .-*4- %, #-,$(#(-,%1%12:0(*%1 79/3(,!1 **%,!$-0!1#%00! $-0!1 .!0! &0!1#-1 4(!*%1 #90.3*!1)%0(,'!1 !'18 7*3)-1 *!+(,!0%1 1 7(1*!$-0%1 $% !*2! #-,2%, #(<, 79/3(,!1 !32-+92(#!1 $% (,1.%##(<, $% !+.-**!1 4(!*%16)%0(,'!1 Dara Pharmaceutical Packaging -*,$-**$%*!!,6! !*(*%-!*(*%( 0!,-**%01 %* !5 www.dara-pharma.com FARMESPAÑA INDUSTRIAL NOVIEMBRE/DICIEMBRE14 77 GUÍA DE SERVICIOS INGENIERÍA Y VALIDACIONES LIMPIEZA Y DESINFECCIÓN LOGÍSTICA LA ALBERCA S.A. www.ingenieriayqualificacion.com Sistemas de Mantenimiento y Limpieza Expertos en validaciones de equipos y sistemas. Proyectos de calidad de ingeniería en la industria farmaceútica. Limpieza y desinfección de todo tipo de zonas y áreas 21 CFR Part 11 Compliance. 10 años de experiencia en el sector. [email protected] MATERIAL DE LABORATORIO 91 402 32 99 www.limpiezaslaalberca.com MATERIAL DE LABORATORIO ROBÓTICA AQUÍ SU PUBLICIDAD POR 550 € / AÑO 78 NOVIEMBRE/DICIEMBRE14 FARMESPAÑA INDUSTRIAL SALAS LIMPIAS SALAS LIMPIAS FARMESPAÑA INDUSTRIAL SALAS LIMPIAS SEGURIDAD INDUSTRIAL NOVIEMBRE/DICIEMBRE14 79 GUÍA DE SERVICIOS OUTSOURCING TRANSPORTE INTERNACIONAL Y ADUANAS TRATAMIENTO DE AGUAS transportes internacionales y aduanas Carga aérea Tráfico Marítimo Servicio Internacional terrestre Agente Aduanas OEA Consultoría OEA Consultoría aduanera Comercio exterior www.ibercondor.com SERVICIOS SUMINISTROS INDUSTRIALES SUMINISTROS INDUSTRIALES El reflejo de una Gran Experiencia. Departamento propio de Ingeniería. Fabricamos Equipos de Proceso e Ingenier’a para Laboratorios. Camino de Valdecabañas, 30 28500 Arganda del Rey - Madrid Tel: +34 91 872 83 36 - Fax: +34 91 870 64 22 [email protected] - www.prenitor.com T I N S M A C A L® 80 NOVIEMBRE/DICIEMBRE14 FARMESPAÑA INDUSTRIAL TRADUCCIONES VALIDACIONES De especialistas para especialistas Proyectos multilingües 25 años de experiencia Servicios lingüisticos y editoriales para la industria farmacéutica Tlf: +34 www.amr-traducciones.com [email protected] SUMINISTROS INDUSTRIALES FARMESPAÑA INDUSTRIAL VALIDACIÓN Y CONSULTORÍA VALIDACIONES NOVIEMBRE/DICIEMBRE14 81 ANUNCIANTES AD-TECH . . . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios AMR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios ARAGOGAMMA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55 AZIERTA . . . . . . . . . . . . . . . . . . . . . . . . 9, Guía de servicios BMPIBERICA FASTDOOR . . . . . . . . . . . . . . . . . . . . . . . . 13 BURDINOLA . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios CAMPAK. . . . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios CILIT-BWT. . . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios COMPLIANCE . . . . . . . . . . . . . . . . . . . . . Guía de servicios CONSTANTIA TOBEPAL . . . . . . . . . . . . . . . . . . . . . . . . . 15 CONTEC . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 DARA PHARMACEUTICAL . . . . . . . . . . . . Guía de servicios ELIS. . . . . . . . . . . . . . . . . . . . . . . . . . . 45, Guía de servicios ETIQUETAS VINALOPO . . . . . . . . . . . . . . Guía de servicios EXPOSÓLIDOS. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61 FARMAFORUM. . . . . . . . . . . . . . . . . . . . . . . Contraportada FARMESPAÑA EVENTOS . . . . . Interior de contraportada FETTE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 FIKE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios FILTROS CARTÉS . . . . . . . . . . . . . . . . 11, Guía de servicios FREEZEDRY . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios GAS NATURAL FENOSA . . . . . . . . . . . . . . . . . . . . . . . . . 39 GRIFOLS ENGINEERING . . . . . . . . . . . . . . . . . . . . . . . . . 23 HALL TECH. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 IBERCONDOR . . . . . . . . . . . . . . . . . . . . . Guía de servicios IBERFLUID INSTRUMENTS. . . . . . . . . . . . . . . . . . . . . . . . 17 IGLOO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67 INGECLIMA . . . . . . . . . . . . . . . . . . . . 59, Guía de servicios INGENIERÍA Y QUALIFICACIÓN . . . . . . . Guía de servicios INGERSOLL RAND IBERICA. . . . . . . . . . . . . . . . . . . . . . . 57 IONISOS IBÉRICA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7 LA ALBERCA . . . . . . . . . . . . . . . . . . . . . . Guía de servicios MATACHANA . . . . . . . . . . . . . . . . . . . . . . Guía de servicios MERCK – MILLIPORE . . . . . . . . . . . . . . . Guía de servicios MOBELMOL . . . . . . . . . . . . . . . . . . . . 69, Guía de servicios MONTAJES IGUÑA . . . . . . . . . . . . . . . . . Guía de servicios MP CONTROL . . . . . . . . . . . . . . . . . . . . . Guía de servicios PANREAC QUIMICA. . . . . . . . . . . . . . . . . Guía de servicios PHARMAQUALITY EUROPE. . . . . . . . . . . Guía de servicios x PIC - PREVENCIÓN INDIVIDUAL Y COLECTIVA, . . . . . . 33 PRENITOR INOXIDABLES . . . . . . . . . . . . Guía de servicios QTI . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios QUALIPHARMA . . . . . . . . . . . . . . . . . . . . Guía de servicios QUILINOX . . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios SSI SCHAEFER . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41 STÄUBLI ESPAÑOLA . . . . . . . . . . . . . 37, Guía de servicios STX . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Interior de portada TCI . . . . . . . . . . . . . . . . . . . . . . . Portada, Guía de servicios TECNICA DE FLUIDOS . . . . . . . . . . . . . . Guía de servicios F FI TECNICOS EN CALIBRACIÓN Y MONTAJE . . . . . . . . . . . . . . . . . . . . . . Guía de servicios Revista Profesional para Proveedores de la Industria Farmacéutica y Tecnología de Laboratorio ULMA PACKAGING . . . . . . . . . . . . . . . . . Guía de servicios VALIDATEC . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios VESTILAB . . . . . . . . . . . . . . . . . . . . . . 43, Guía de servicios VIASTORE. . . . . . . . . . . . . . . . . . . . . . . . . Guía de servicios Porque sabemos lo que cuesta organizar un evento. Farmespaña Eventos JORNADAS DESAYUNOS DE TRABAJO CONVENCIONES [email protected] CONGRESOS 902 36 46 99 RUEDAS DE PRENSA MESAS REDONDAS Madrid, 4-5 de marzo de 2015 II Foro de la Industria Farmacéutica, Biofarmacéutica, Cosmética y Tecnología de Laboratorios Centro de Congresos Príncipe Felipe (Hotel Auditórium) Madrid +1.000 visitantes 200 congresistas RESUMEN EDICIÓN ANTERIOR MARZO 2014 200 reuniones bilaterales +60 expositores Patrocinadores Entidades colaboradoras *=NAKL9KGÍ;A9D=K Revista Profesional para proveedores de la Industria Farmacéutica [email protected] 986 19 12 93 [email protected] www.farmaforum.es 916 30 85 91
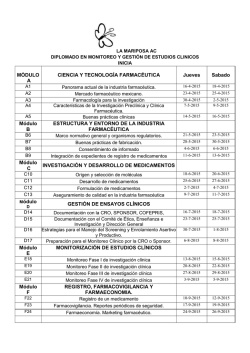
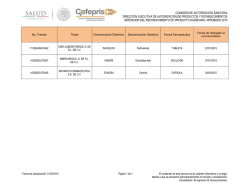
© Copyright 2026