
Disponible el libro de abstracts
Fotos: Ignacio López Goñi, Alicia M. Muro Pastor y Ramón Santamaría XI REUNIÓN DEL GRUPO DE MICROBIOLOGÍA MOLECULAR Sevilla, 6-8 de septiembre de 2016 XI REUNIÓN DEL GRUPO DE MICROBIOLOGÍA MOLECULAR Sevilla, 6-8 de septiembre de 2016 Comité Científico Bruno González Zorn (Universidad Complutense de Madrid) Junkal Garmendia García (CSIC/UPNA/G. de Navarra; Instituto de Agrobiotecnología) Susana Campoy Sánchez (Universidad Autónoma de Barcelona) José Berenguer Carlos (Universidad Autónoma de Madrid) Mª Ángeles de la Torre Ruiz (Universidad de Lérida) José Antonio Aínsa Claver (Universidad de Zaragoza) Félix Sangari García (Universidad de Cantabria) Josep Casadesús (Universidad de Sevilla) José Antonio Gil Santos (Universidad de León) Adela González de la Campa (Instituto de Salud Carlos III) Alejandro Mira Obrador (Centro Superior de Investigación en Salud Pública, Valencia) Víctor Jiménez Cid (Universidad Complutense de Madrid) Juan Alfonso Ayala Serrano (Universidad Autónoma de Madrid) José Antonio Bengoechea Alonso (Queen’s University Belfast, UK) Alicia M. Muro Pastor (CSIC-Universidad de Sevilla) Francisco Ramos Morales (Universidad de Sevilla) Joaquín J. Nieto Gutiérrez (Universidad de Sevilla) Eduardo Santero Santurino (Universidad Pablo de Olavide, Sevilla) Jesús Blázquez Gómez (CSIC, Instituto de Biomedicina de Sevilla) Comité Organizador Alicia M. Muro Pastor (CSIC-Universidad de Sevilla) Francisco Ramos Morales (Universidad de Sevilla) Joaquín J. Nieto Gutiérrez (Universidad de Sevilla) Josep Casadesús Pursals (Universidad de Sevilla) Página web Manuel Brenes Álvarez (Universidad de Sevilla-CSIC) Todas las sesiones y actividades de la Reunión se celebrarán en el Hotel Silken Al Andalus Palace, situado en el barrio de Heliópolis, zona sur de la ciudad, en Avenida de la Palmera, esq. c/ Paraná, junto al Estadio Benito Villamarín. El cóctel de clausura tendrá lugar en la terraza del Hotel Los Seises, junto a la Catedral. XI REUNIÓN DEL GRUPO DE MICROBIOLOGÍA MOLECULAR Sevilla, 6-8 de septiembre de 2016 Programa Martes, 6 de septiembre de 2016 15:00-17:30 Entrega de documentación y colocación de paneles 17:30-18:00 Ceremonia de inauguración 18:00-19:00 Conferencia inaugural La curva de crecimiento CRISPR: inoculación, latencia, fase exponencial, ¿fase estacionaria…? Francis J.M. Mojica Dpto. de Fisiología, Genética y Microbiología. Universidad de Alicante 19:00-21:00 Sesión I: Biotecnología microbiana Moderadora: Belén Floriano Panel 9 • Ingeniería metabólica de sistemas en la bacteria extremófila Chromohalobacter salexigens: estrategias para la mejora racional de cepas productoras de ectoínas Montserrat Argandoña Panel 12 • Engineering a hydrogen biosensor: selection of overproducing nitrogenase variants for biohydrogen production Emma Barahona Panel 19 • An enhanced folding reporter to select thermostable protein variants Sandra Bosch Panel 22 • Recombinant attenuated Mycobacterium tuberculosis for heterologous expression and secretion of DTP antigens: A rationale to construct a multivalent vaccine Esther Broset Panel 35 • Nuevas herramientas para reprogramar y estudiar el flujo de fosfoinosítidos en las membranas celulares de Saccharomyces cerevisiae Julia M. Coronas-Serna Panel 65 • New-tagged brucellosis vaccines and associated diagnostic tests Estrella Martínez-Gómez I Panel 86 Panel 105 21:00 • Novel c-di-GMP binding domain involved in production a mixed-linkage β-Glucan in Sinorhizobium meliloti Daniel Pérez-Mendoza • A regioselective synthesis of acylated carbohydrates and polyphenols with a novel acetylesterase (AE-6L) from Bacillus sp. HR21-6 Leyre Sánchez Barrionuevo Cóctel de bienvenida Miércoles, 7 de septiembre de 2016 9:00-11:00 Panel 21 Panel 26 Panel 40 Panel 48 Panel 50 Panel 57 Panel 60 11:00-11:30 Sesión II: Regulación génica Moderador: Fernando Govantes • Acercamientos globales a la identificación de RNAs no codificantes bacterianos Manuel Brenes-Álvarez • Caracterización funcional de las chaperonas de RNA en Staphylococcus aureus Carlos Caballero • Regulación de las proteínas LPXTG de superficie de Listeria monocytogenes en condiciones de crecimiento a 4 ºC Alberto Díaz-Romero • StdE y StdF, reguladores globales de la transcripción en Salmonella enterica Lucía García-Pastor • Implicación del pequeño ARN SuhB en la represión catabólica de los genes de degradación de tetralina en Sphingopyxis granuli estirpe TFA Inmaculada García-Romero • Protein-assisted riboregulation in the legume symbiont Sinorhizobium meliloti José I. Jiménez-Zurdo • Ni is able to induce the switch between cytochrome c6 and plastocyanin in Synechocystis sp. PCC 6803 Luis López-Maury Café 11:30-13:30 Sesión III: Diferenciación microbiana Moderador: Luis Ángel Fernández Panel 17 • Protein interactions in the Escherichia biosynthesis apparatus Joaquín Bernal II coli cellulose Panel 23 Panel 24 Panel 30 Panel 78 Panel 84 Panel 87 • Regulación ambiental de la síntesis del polisacárido PIA/PNAG en Staphylococcus aureus resistentes a meticilina Saioa Burgui • Compartmentalized cyanophycin and arginine catabolism in the filamentous heterocyst-forming cyanobacterium Anabaena Mireia Burnat • Análisis de la maquinaria molecular implicada en multicelularidad de Bacillus cereus Joaquín Caro Astorga • Nuevos elementos reguladores de la movilidad y la adhesión en Pseudomonas putida Blanca Navarrete • Biofilm oxygen gradients and ribonucleotide synthesis: Anaerobic ribonucleotide reductases are essential for proper biofilm formation Lucas Pedraz • Autophagy as an adaptive response to stress in Chlamydomonas reinhardtii M. Esther Pérez-Pérez 14:00-16:00 Almuerzo 16:00-17:30 Sesión IV: Antimicrobianos y resistencias Moderadora: Junkal Garmendia Panel 3 • Identifying molecules involved in aminoglycoside tolerance in Vibrio cholerae through genome- wide fitness/transcriptional profiling Sebastian Aguilar Pierlé Panel 18 • Vitamin K3 induces Stenotrophomonas maltophilia SmeVWX multidrug efflux pump Paula Blanco Torres Panel 59 • Targeted killing of bacteria using logic gates Rocío López-Igual Panel 91 • Killing effect of R-pyocins on susceptible and multiresistant Pseudomonas aeruginosa clinical isolates Anabel Prieto Panel 118 • Chimeric Csl2 enzybiotic: towards the tailor-made antiinfectives Roberto Vázquez 17:30-18:00 Café III 18:00-19:00 Sesión V: Genómica comparada y evolución Moderador: José Antonio Aínsa Panel 1 • A deletion impairs the proper typing of Mycobacterium africanum Estefanía Abascal Panel 34 • A comparative genomic strategy for target discovery and evolutionary inferences in pathogenic bacteria: Stenotrophomonas sp. as a case study Oscar Conchillo-Solé Panel 72 • Evolución pato-adaptativa de Haemophilus influenzae en el sistema respiratorio del paciente con Enfermedad Pulmonar Obstructiva Crónica Javier Moleres 19:00-21:00 Beer poster session Jueves, 8 de septiembre de 2016 9:00-11:00 Panel 11 Panel 16 Panel 20 Panel 41 Panel 62 Panel 123 Sesión VI: ADN móvil Moderadora: Montserrat Argandoña • CptA, an ICE-associated ATPase involved in DNA donation between Thermus thermophilus cells Ignacio Baquedano • Las secuencias de inserción facilitan la adaptación de un plásmido multicopia a un nuevo hospedador Cristina Bernabé-Balas • Incremento del número de copias de plásmido por aumento de la temperatura de crecimiento en Escherichia coli Emilia Botello • Rewinding the tape of evolutionary innovation of integron integrases José Antonio Escudero • La regulación dependiente de fase de crecimiento de la conjugación de R27 implica el circuito regulador plasmídico TrhR/TrhY-HtdA y el regulón cAMP Cristina Madrid • Mobilization mechanism of pathogenicity islands by endogenous phages of Staphylococcus aureus isolates from cystic fibrosis patients Miglė Žiemytė 11:00-11:30 Café IV 11:30-13:30 Sesión VII: Patogénesis Moderador: Alejandro Toledo-Arana Panel 25 • Identificación por PCR de genes asociados a la virulencia de Helicobacter pylori en biopsias gástricas Cochabamba – Bolivia Zulema Bustamante Panel 29 • Tn-seq identification of meningococcal factors required for in vitro and in vivo colonization of human cells Elena Capel Panel 32 • Effectors encoded outside of the locus of enterocyte effacement of enteropathogenic E. coli are essential for attaching and effacing (A/E) lesion formation in human intestinal biopsies Massiel Cepeda Panel 36 • Identification of a specific epitope on the surface of virulent Haemophilus parasuis strains Florencia Correa-Fiz Panel 61 • Biestabilidad en la expresión de genes importantes para la virulencia de Pseudomonas syringae Nieves López-Pagán Panel 75 • Identification of glycoproteins involved in virulence in Ustilago maydis Ismael Moreno-Sánchez Panel 121 • Rifampicin resistant rpoB alleles, or multicopy thioredoxin/thioredoxin reductase, suppress the lethality of the global stress regulator spx in Staphylococcus aureus Maite Villanueva 14:00-16:00 16:00-17:00 17:00-17:15 17:15-18:00 18:00-18:30 18:30-19:30 20:30-22:00 22:00 Almuerzo Sesión de paneles Entrega de premios a las mejores comunicaciones Entrega del premio Biomedal y Clausura Asamblea del Grupo de Microbiología Molecular Asamblea general de la SEM Paseo en autobús panorámico por la ciudad Cóctel de clausura. Terraza del Hotel Los Seises V RESÚMENES DE LAS COMUNICACIONES A deletion impairs the proper typing of Mycobacterium africanum Estefanía Abascal, Diana Maricela Herrera, Marta Herranz, Sheyla Santantón, Miguel Martínez Lirola, Griselda Tudó, Juliá Gonzalez, Juan Eyene, Emilio Bouza, Laura PérezLago, Darío García-de-Viedma Instituto de Investigación Sanitaria Gregorio Marañón, Madrid. [email protected] Genotyping of the Mycobacterium tuberculosis complex by 24-MIRU-VNTR is the reference method used for molecular epidemiology analysis. In certain isolates a complete pattern is not obtained by failures in the amplification of some loci, excluding those isolates from epidemiological analysis. Five isolates from Almería were suspected to be part of a recent transmission cluster, based on 23 loci results, but QUB-11b locus failed to be amplified in all of them. By applying external primers, an 88-bp deletion, which eliminated the annealing region of the forward primer used in the standard VNTR procedure for this locus, was observed. The analysis of the MIRU-VNTR pattern of these isolates in the MIRU-VNTRplus platform indicated that they corresponded to the Mycobacterium africanum Lineage 5, which was ratified by identifying the marker SNP for this lineage. A revision of the literature revealed that the deletion is not a lineage 5 marker, however lack of amplification in the QUB-11b locus was a feature rather frequent, as described in two studies including M. africanum isolates (100% and 51% of the isolates from Nigeria and USA respectively). We reanalyzed the isolates in our collection with no amplification in QUB11b and the deletion was identified in five of them, all immigrants from West Africa, where the Lineage 5 is overrepresented. To extend the magnitude of our observation, we genotyped a collection of 68 isolates obtained in Equatorial Guinea fifteen years ago. Twenty of them (29,41%), corresponding to all the Lineage 5 isolates in this sample, had the 88-bp deletion. QUB-11b is a MIRU-VNTR locus with a high discriminative value, what led us to design an alternative protocol to assure its analysis in the isolates harbouring the QUB-11b deletion. New allelic values were calculated, allowing the proper allelic assignation for all the strains harbouring the deletion in our study. The allelic assignation of the QUB-11b locus was essential to either confirm the Almería cluster, which had been overlooked, and to split some potential clusters in the sample from Equatorial Guinea. Funding: Ministry of Economy and Competitiveness ISCIII FIS (15/01554), cofinanced by ERDF Funds from EC: “A way of making Europe”. Panel 1 AS-48, una bacteriocina con actividad contra Mycobacterium tuberculosis. Clara Aguilar 1,2, *, Begoña Gracia 1,2,, Asunción Vitoria 1,2,3,5, Rubén Cebrián 4, Mercedes Maqueda 4, José Antonio Aínsa 1,2,5,6,* 1 Departamento de Microbiologia, Facultad de Medicina, Universidad de Zaragoza, c/Domingo Miral s/n, 50009-Zaragoza, Spain. 2 Instituto de Investigacion Sanitaria de Aragon (IIS-Aragon). 3 Servicio de Microbiología, Hospital Clínico Universitario Lozano Blesa, avda. San Juan Bosco, 15, 50009 Zaragoza, Spain. 4 Departamento de Microbiología, Facultad de Ciencias, Universidad de Granada, 18071 Granada, Spain. 5 CIBER de Enfermedades Respiratorias (CIBERES), Instituto de Salud Carlos III Departamento de Microbiología, Facultad de Ciencias, Universidad de Granada, 18071 Granada, Spain. 6 Instituto de Biocomputacion y Fisica de Sistemas Complejos, BIFI, Universidad de Zaragoza *Corresponding author: [email protected]; [email protected] El aumento de la incidencia de las cepas multiresistentes de Mycobacterium tuberculosis y los pocos fármacos antimicrobianos disponibles para su tratamiento, fomentan la necesidad de desarrollar nuevas líneas de antimicrobianos. Siguiendo esta idea, hemos investigado el péptido antibacteriano AS-48, producido por E. faecalis, para el que se propone que su diana es la membrana bacteriana y que presenta una gran actividad contra varias bacterias Gram positivas. Teniendo en cuenta la cantidad de rutas biosintéticas relacionadas con la membrana, es de esperar que la posibilidad de seleccionar una mutación que confiera resistencia a AS-48 será menor que en el caso de antibióticos frente a otras dianas, lo que supone una ventaja. Este estudio evalúa el efecto bactericida de AS-48 en cepas de M. tuberculosis, incluyendo H37Rv, cepas de referencia y de origen clínico. También se ha estimado este efecto frente a cepas de micobacterias no tuberculosas de origen clínico. Hemos demostrado que AS-48 tiene efecto bactericida frente a M. tuberculosis y otras especies de micobacterias. La combinación de AS-48 y lisozima o diferentes fármacos de primera línea, que habitualmente se usas para el tratamiento de tuberculosis, incrementa la acción bactericida de AS-48, mostrando una relación de sinergismo entra ambos compuestos. Teniendo en cuenta estas condiciones, AS-48 es capaz de matar M. tuberculosis a una concentración más baja cuando se encuentra en combinación con lisozima o etambutol. De esta manera la MIC (Minimal Inhibitory Concentration) de AS-48 llega a tener valores cercanos a los principales fármacos antituberculosos. Además, se ha ensayado la citotoxicidad de AS-48 frente a THP-1 y MHS, líneas celulares de macrófagos. Hemos determinado que en las concentraciones cercanas a la MIC AS-48 no presenta ningún efecto citotóxico. Resumiendo, consideramos que la bacteriocina AS-48 tiene un potencial interesante en el tratamiento de la tuberculosis ya que presenta actividad frente a distintas especies de micobacterias y una baja citotoxicidad en las líneas celulares ensayadas. PALABRAS CLAVE: AS-48, péptido antimicrobiano, actividad antituberculosis, sinergismo. Funded by: Secretaría de Estado de Investigación, Desarrollo e Innovación del Ministerio de Economía y Competitividad de España Ref: BES-2014-067962 Panel 2 Identifying molecules involved in aminoglycoside tolerance in Vibrio cholerae through genome- wide fitness/transcriptional profiling Sebastian Aguilar Pierlé, Zeynep Baharoglu, Evelyne Krin, Sean Kennedy and Didier Mazel Institut Pasteur [email protected] The extensive use of antibiotics worldwide has established different concentration gradients at an ecological scale. This exacts broad selective pressure on bacteria. Despite the initial focus on resistance as a result of exposure to lethal doses of antibiotics, an increasing amount of evidence supports a role for sublethal concentrations in its development. Vibrio cholerae is a gram-negative bacterium that represents a public health burden, particularly in developing countries. Sub-minimum inhibitory concentrations (subMICs) of antibiotics are now known to trigger the SOS response and mutagenesis in V. cholerae. Such responses can sometimes lead to the acquisition of traits through horizontal gene transfer, single nucleotide polymorphisms and recombination, which can play into the development of resistance. Despite recent advances, little is known about the genes involved in the response to sub-MICs of antibiotics in V. cholerae. We combined two highthroughput techniques to identify such genes. RNA-seq was used to identify transcriptional alterations during treatment with sub-MICs of tobramycin. Then, Tn-seq was applied to define the fitness of V. cholerae genes in the context of the aforementioned treatment. Analysis of RNA-seq data revealed downregulation of several carbohydrate transport genes and a sRNA that is predicted to regulate them. Consequently, Tn-seq data showed an advantage in fitness for those genes and their regulator through expansion of mutants in the context of tobramycin exposure. Downregulation of these transcripts, paired with a fitness advantage seen during antibiotic exposure led us to propose that this system has a role in the transport of aminoglycosides/carbohydrates. Deletion mutants of selected carbohydrate transport genes and their putative regulator allows V. cholerae to resist lethal concentrations of aminoglycosides. Secondary structure conservation analyses revealed that, despite sequence diversity, this sRNA is present throughout the Vibrionales. This suggests coevolution of the sRNA and its targets, and a common route of entry for aminoglycosides in this Order. Validation of this species specific mechanism opens the door for carbohydrate enhanced antibiotic treatments and targeted killing of Vibrio species. Panel 3 Regulación del gen srfJ de Salmonella enterica Julia Aguilera Herce y Francisco Ramos Morales Departamento de Genética, Facultad de Biología, Universidad de Sevilla [email protected], [email protected] Las bacterias del género Salmonella enterica son bacilos móviles Gram negativos, pertenecientes a la familia Enterobacteriacea. Su patogenicidad se debe a la presencia de genes de virulencia, agrupados en regiones cromosómicas llamadas islas de patogenicidad. Las más importantes son la isla de patogenicidad 1 (SPI1) y la isla de patogenicidad 2 (SPI2), que codifican a los sistemas de secreción de tipo III (T3SS). El T3SS1 transloca efectores a través de la membrana citoplasmática de la célula hospedadora durante la primera fase de la infección. El T3SS2 transloca efectores a través de la membrana vacuolar, donde la bacteria se encuentra alojada, lo que permite su proliferación y supervivencia. SrfJ es uno de los efectores secretados por el T3SS2 [1]. Aunque el gen srfJ no forma parte de la SPI2, su expresión depende del sistema de regulación de dos componentes SsrA-SsrB, el principal regulador de esta isla de patogenicidad, por lo tanto coordinado con la expresión de T3SS2. La expresión de srfJ está asociada con otra condición fisiológica completamente distinta: la presencia de mio-inositol en el medio [1]. Esto ocurre porque este gen se localiza en la isla de utilización del mio-inositol, controlada por el represor IolR [2]. Para entender este sistema de doble regulación, hemos analizado la estructura de la unidad transcripcional que codifica srfJ, y las posibles regiones promotoras que pueden afectar a su expresión. Para alcanzar este objetivo, usamos un plásmido [3] con el que se crearon fusiones transcripcionales con el operón luxCDABE de Photorhabdus luminescens. La expresión de srfJ está bajo el control de dos promotores que están activados bajo dos condiciones distintas. El primero es la región promotora del gen iolE cuya expresión está regulada por IolR. Además, srfJ tiene su propia región promotor, cuyo regulador principal es SsrB, así que es por eso que responde a medios que simulan condiciones intravacuolares. La regulación de estas dos regiones promotoras se ha estudiado en detalle usando líneas mutantes para reguladores de Salmonella que pudieran afectar a la expresión de SPI1 y SPI2. 1. Cordero-Alba M, Bernal-Bayard J, Ramos-Morales F. SrfJ, a Salmonella type III secretion system effector regulated by PhoP, RcsB, and IolR. J Bacteriol. 2012;194: 4226–36. doi:10.1128/JB.00173-12 2. Kröger C, Fuchs TM, Kro C, Fuchs TM, Kröger C, Fuchs TM. Characterization of the myo-inositol utilization island of Salmonella enterica serovar Typhimurium. J Bacteriol. 2009;191: 545–54. doi:10.1128/JB.01253-08 3. Winson MK, Swift S, Hill PJ, Sims CM, Griesmayr G, Bycroft BW, et al. Engineering the luxCDABE genes from Photorhabdus luminescens to provide a bioluminescent reporter for constitutive and promoter probe plasmids and mini-Tn5 constructs. FEMS Microbiol Lett. 1998;163: 193–202. doi:10.1016/S0378-1097(98)00173-6 Financiación: MINECO SAF2013-46229-R Panel 4 NsrR1, un pequeño RNA implicado en la respuesta al estrés de carencia de nitrógeno en cianobacterias Isidro Álvarez-Escribano, Agustín Vioque, Alicia M. Muro-Pastor Instituto de Bioquímica Vegetal y Fotosíntesis (IBVF) [email protected] Los RNAs pequeños no codificantes (sRNAs) están implicados en la regulación de la mayoría de los aspectos del metabolismo bacteriano. Generalmente, estas moléculas actúan a nivel post-transcripcional, complementando la regulación ejercida por los reguladores transcripcionales. Los sRNAs ejercen su acción mediante diversos mecanismos, que en la mayoría de los casos implican la interacción con otros mRNAs, denominados dianas. Es bastante habitual que la interacción de los sRNAs con sus dianas tenga lugar en la región en la que se inicia la traducción del mRNA, provocando que su estabilidad y niveles de traducción se vean afectados. Nuestro objetivo es identificar los mecanismos reguladores orquestados por sRNAs en el marco de la regulación global de la asimilación de nitrógeno en Nostoc (Anabaena) sp. PCC 7120, una cianobacteria capaz de diferenciar heterocistos, células especializadas en la fijación de nitrógeno atmosférico. A partir de un análisis transcriptómico mediante dRNASeq pudimos identificar todos los transcritos cuya expresión responde a la disponibilidad de nitrógeno, incluyendo sRNAs y transcritos antisentido, que podrían tener un papel regulador (1). NsrR1 (Nitrogen stress repressed RNA 1), es un pequeño RNA (112 nt) cuya expresión se reprime en ausencia de nitrógeno combinado. El promotor de NsrR1 está regulado por NtcA, una proteína de la familia FNR/CRP que es el regulador global de la asimilación de nitrógeno en cianobacterias. Las secuencias que codifican NsiR1 aparecen conservadas en los genomas de la mayoría de las cianobacterias formadoras de heterocistos, lo que sugiere que se trata de un sRNA funcional (2). La aplicación del algoritmo CopraRNA (3) combinado con un análisis global del transcriptoma de una estirpe mutante en NsrR1, comparada con la estirpe silvestre, nos ha permitido identificar alguna de sus posibles dianas, que actualmente estamos analizando en el marco de los mecanismos de adaptación controlados por NtcA. En concreto, NsrR1 podría modular la traducción de All1871 (una proteína conservada de función desconocida cuya transcripción está regulada por la intensidad luminosa) y de NblA, una proteína implicada en la degradación de ficobilisomas (un proceso encaminado a “reciclar” el nitrógeno disponible en forma de aminoácidos en estos complejos proteicos que funcionan como antenas para los fotosistemas). 1. Mitschke J, Vioque A, Haas F, Hess WR, Muro-Pastor AM. 2011. Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc Natl Acad Sci U S A 108:20130-20135. 2. Brenes-Álvarez M, Olmedo-Verd E, Vioque A, Muro-Pastor AM. 2016. Identification of conserved and potentially regulatory small RNAs in heterocystous cyanobacteria. Front Microbiol 7:48. 3. Wright PR, Richter AS, Papenfort K, Mann M, Vogel J, Hess WR, Backofen R, Georg J. 2013. Comparative genomics boosts target prediction for bacterial small RNAs. Proc Natl Acad Sci U S A 110:E3487-3496 Financiación: MICINN (BFU2010-14821), MINECO (BFU2013-48282-C2-1; Contrato predoctoral BES-2014-068488_Isidro Álvarez Escribano) Panel 5 Brucella microti hydroxylated ornithine lipids. A case of genomic reduction in the adaptation of brucellae to intracellular life? Beatriz Aragón-Aranda*, Marina Bárcena*, Miriam Salvador, Leyre Palacios-Chaves, Miguel Ángel Vences, Estrella Martínez, Ignacio Moriyón, Maite Iriarte*, Raquel CondeÁlvarez* *These authors contributed equally to the work Departamento de Microbiología y Parasitología de la Universidad de Navarra [email protected] The brucellae are facultative intracellular pathogens of mammals that cause brucellosis, a worldwide-extended zoonosis. As described for Brucella abortus and other classical Brucella species, their pathogenicity relates in part to a cell envelope composition that facilitates resistance to bactericidal peptides and other effectors/receptors of innate immunity. However, B. microti, a recently described species infecting rodent-like mammals, is more readily detected by innate immunity and, consistent with a more ancestral phylogenetic position close to plant symbionts rhizobia and free-living soil αProteobacteria, grows under a wider range of conditions. Among other amino lipids, both rhizobia and brucellae display ornithine lipids (OL). Complete OL carry one α-amide linked acyl-oxyacyl residue that results from the sequential action of olsB and olsA. In rhizobia, the hydroxyl-linked acyl residue is C2 hydroxylated by an olsC-encoded hydroxylase, and hydroxylation has been found important in pH adaptation in these bacteria. Since, olsC is mutated in B. abortus and other classical species but not in B. microti, the relevance of this difference was studied by deleting olsB and olsC in B. microti. The mutants did not show changes in growth rates, pH and bactericidal peptide sensitivity, although differences in these properties with B. abortus were consistently observed. Moreover prelimilary data suggest that OL or hydroxylated-OL (OH-OL) are not involved in B. microti virulence. Therefore the presence of OH-OL in B. microti is unlikely to account for the differences with B. abortus and the lost of olsC in classical Brucella species might represent a case of genomic reduction in the adaptation of brucellae to intracellular life. Research at the laboratories of the authors is supported by grants from the Ministerio de Economía y Competitividad of Spain (AGL2014-58795-C4-1-R). Fellowship support for B.A-A from the Ministerio de Economía y Competitividad of Spain (FPI) is gratefully acknowledged. Panel 6 Aumento transitorio de la resistencia a antibióticos por un SNP natural en el plásmido pB1000 Manuel Ares-Arroyo, Alfonso Santos-Lopez, Cristina Bernabe-Balas, Alvaro San Millan, Rafael Ortega-Huedo, Natalia Montero y Bruno Gonzalez-Zorn. Departamento de Sanidad Animal y Centro de Vigilancia Sanitaria Veterinaria (VISAVET), Facultad de Veterinaria, Universidad Complutense de Madrid, Spain. [email protected] pB1000 es un plásmido pequeño de la superfamilia ColE1 que codifica el gen blaROB-1. Ha sido descrito en varios patógenos de la familia Pasteurellaceae, donde es estable y confiere alta resistencia a β-lactámicos. En este trabajo estudiamos el hallazgo de un aislado clínico de Pasteurella multocida donde se describió el plásmido pB1000 original cohabitando con otro plásmido pB1000 cuya única diferencia era un SNP en el origen de replicación. Para averiguar cómo afectaba la presencia de esta mutación a la biología del plásmido utilizamos Haemophilus influenzae Rd KW20 como modelo. Las técnicas que empleamos para caracterizar el comportamiento de ambos alelos, tanto de forma independiente como en cohabitación, fueron PCRs cuantitativas, ensayos de competición, tests de susceptibilidad a antibióticos y evoluciones experimentales. Nuestros resultados mostraron que el nuevo alelo del plásmido con el SNP presenta un mayor número de copias que el plásmido original. Esta nueva variante también aporta a la bacteria una mayor concentración mínima inhibitoria frente a ampicilina, así como un mayor coste metabólico. Sorprendentemente, cuando ambos plásmidos coexisten en la misma bacteria, el número de copias, la concentración mínima inhibitoria y el coste metabólico representan la suma de ambos alelos por separado. Este resultado indica que pB1000 original y su nueva variante actúan como plásmidos independientes. Posteriormente, realizamos evoluciones experimentales en presencia y en ausencia de antibiótico. Cuando se mantenía la presencia de ampicilina ambos plásmidos permanecían en las bacterias más de 300 generaciones, gracias a la ventaja que supone el incremento del número de copias. Sin embargo, en ausencia de ampicilina, el nuevo alelo con el SNP en el origen de replicación se perdía, evitándose así el coste que supone mantener un mayor número de copias plasmídicas para la bacteria. En conclusión, hemos descrito un mecanismo por el cual una mutación natural en el origen de replicación de pB1000 puede aportar a las bacterias un mayor nivel de resistencia a antibióticos, siendo un mecanismo reversible para evitar así la desventaja competitiva que supone en ausencia de antibióticos. Financiación: Beca de colaboración UCM Panel 7 Pentapeptide repeat proteins involved in filamentation and heterocyst differentiation in the cyanobacterium Anabaena Sergio Arévalo, Mireia Burnat, Félix Ramos-León and Enrique Flores Instituto de Bioquímica Vegetal y Fotosíntesis, CSIC and Universidad de Sevilla, Sevilla [email protected] The pentapeptide repeat protein (PRP) family consists of proteins containing at least eight repetitions in tandem of a consensus sequence of amino acids ([STAV] [D/N] [L/F] [S/T/R] [X]) and is widely distributed in nature. Some cyanobacteria (oxygenic phototrophs) can grow forming filaments of cells that, under nitrogen deprivation, contain two cell types: the CO2 fixing vegetative cells and the N2 fixing heterocysts. Heterocysts differentiate from vegetative cells with a pattern of about two heterocysts separated by 10 to 20 vegetative cells. The model heterocyst-forming cyanobacterium Anabaena sp. PCC 7120 presents a large number of PRPs, some of which are involved in filamentation (1), heterocyst differentiation (2) or heterocyst patterning (3, 4). The FraF protein is needed to restrict filament size under nitrogen deprivation, constituting the only known negative filamentation factor (1). FraF is a 222-residue protein and essentially consists of a pentapeptide repeat domain (aa31 to aa195). The fraF gene is located immediately downstream of—in the opposite DNA strand—the fraC operon, which consists of three genes needed for Anabaena to make long filaments under nitrogen deprivation. FraF might influence filamentation by counteracting the action of positive filamentation proteins. To study protein-protein interactions, we have purified a His-tagged FraF protein and are preparing antibodies against it. HglK is involved in heterocyst maturation, since an hglK mutant is impaired in deposition of the glycolipid layer of the heterocyst envelope (2). HglK is a 727residue protein that consists of two sections: an integral membrane section bearing four transmembrane segments (aa1 to aa303) and a periplasmic section (aa304 to aa 727) that contains a long peptapeptide repeat domain (aa430 to aa664). The predicted structure of this domain is strongly similar to that of FraF. Observed by transmission electron microscopy, the hglK mutant exhibits large spaces between adjacent cells in the filament (2), suggesting that HglK plays a role in the architecture of the intercellular septa. To study further the phenotypes associated with inactivation of hglK we have isolated mutants of hglK in different genetic backgrounds, and to study its subcellular localization we have generated strains producing His-tagged HglK and HglK-GFP fusions. Work supported by grant no. BFU2014-56757-P from Plan Nacional de Investigación, Spain, co-financed by the European Regional Development Fund. (1) (2) (3) (4) Merino-Puerto et al. (2013) J. Bacteriol. 195:3957-3966. Black et al. (1995) J. Bacteriol. 177:6440-6448. Liu & Golden (2002) J. Bacteriol. 184:6873-6881. Liu & Wolk (2011) J. Bacteriol. 193:6070-6074. Panel 8 Ingeniería metabólica de sistemas en la bacteria extremófila Chromohalobacter salexigens: estrategias para la mejora racional de cepas productoras de ectoinas Montserrat Argandoña1, Francine Piubeli1, Ali Tahrioui1, Rosa García-Valero1, Emilia Naranjo1, Lourdes Martínez-Martínez1, Manuel Salvador2, Joaquín J. Nieto1, Carmen Vargas1. 1 Departamento de Microbiología y Parasitología. Facultad de Farmacia. Universidad de Sevilla 2 Faculty of Health and Medical Sciences, University of Surrey, Guildford, UK [email protected] El desarrollo en paralelo de la Biología de Sistemas y la Biología Sintética en los últimos años ha permitido combinar ambas en una nueva especialidad conocida como Ingeniería Metabólica de Sistemas, permitiendo así un notable avance en el desarrollo racional de cepas mejoradas para su aplicación industrial. En relación a la Biología de Sistemas, los modelos metabólicos a escala genómica utilizados y aplicados a la Ingeniería metabólica han ayudado a predecir diversas modificaciones en determinados genes/rutas permitiendo re-direccionar los flujos metabólicos hacia el producto de interés mediante numerosas herramientas computacionales. Actualmente el esfuerzo está centrado en integrar o añadir a los modelos metabólicos, los modelos transcripcionales, ampliando así el conocimiento del sistema y permitiendo encontrar nuevas dianas que no pueden ser detectadas mediante modelos metabólicos. Sin embargo, en la mayoría de los casos se tiene poco conocimiento experimental de los microorganismos de interés, por lo que la obtención de los modelos de regulación se hace una tarea muy complicada. Sin embargo, estrategias que integren datos experiementales globales con análisis in silico pueden ayudar a solventar en parte esta limitación. C. salexigens es una bacteria halófila que produce compuestos de interés biotecnológico (ectoínas) de la que se tenían pocos conocimientos a nivel global. Por ello nuestro Grupo inició el desarrollo de la Biología de Sistemas mediante el análisis integrado de datos experimentales multiómicos y genómicos, utilizando diversas herramientas computacionales para poder llevar acabo un diseño racional de mejora de cepas. Así, hemos logrado desarrollar un modelo metabólico a escala genómica de alta precisión, capaz de simular en diferentes condiciones de salinidad, y una red de regulación transcripcional preliminar, previa al modelo de regulación, que integra factores transcripcionales y genes diana que intervienen en la adaptación metabólica celular en función de la demanda de ectoinas. Actualmente estos modelos y redes están siendo ampliados y mejorados, incluyendo nuevas rutas metabólicas o elementos reguladores clave como sistemas de dos componentes, reguladores metabólicos específicos o reguladores globales, combinando estudios in silico y experimentales, de RNA-seq, metabólicos o de interactómica dirigida. Financiación: BIO2015-63949-R (MINECO/FEDER, UE) y Junta de Andalucía (P11-CV17293 MO). Panel 9 Hydroxylamine derivatives as a new paradigm in the search of antibacterial agents Laia Miret1, Esther Julián2, Aida Baelo3, Josep Astola3, Ariadna Lobo-Ruiz1, Fernando Albericio1,4, Eduard Torrents3 1 Department of Organic Chemistry, University of Barcelona, Barcelona, Spain. 2 Group of Mycobacteriology. Department of Genetics and Microbiology. Facultat de Biociències. Universitat Autònoma de Barcelona, Bellaterra, Barcelona, Spain 3 Bacterial Infections and antimicrobial therapies. Institute for Bioengineering of Catalonia (IBEC), Barcelona, Spain 4 CIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine, Barcelona, Spain [email protected] / [email protected] Ever since the discovery and introduction of antibiotics, bacterial pathogens have been developing resistance, reducing drastically or eliminating the effectiveness of such antibacterial drugs for the treatment of several infectious diseases. Multi-drug-resistant bacteria causing severe infections mainly grow in complex bacterial communities known as biofilms, in which bacterial resistance to antibacterial agents and to the host immune system is strengthened. As drug resistance is becoming a threatening problem, it seems necessary to develop new antimicrobial agents that target pathogens essential processes. Ribonucleotide Reductase (RNR) enzyme plays a key role in the DNA replication process, providing the building blocks for DNA synthesis and repair, thus being essential for bacterial proliferation during infection. Here we show the synthesis and evaluation of the antibacterial activity of a small library of N-Hydroxylamines, a family of radical scavenger compounds known to inhibit RNR. We demonstrate the broad antimicrobial effect of several of the compounds together with low toxicity to eukaryotic cells, by determining both the antibacterial activity against different gram-negative and gram-positive pathogenic species and the eukaryotic cytotoxic effect. Furthermore, the most promising compounds were evaluated on both Pseudomonas aeruginosa and Staphylococcus aureus static biofilms, showing that they are able to block biofilm formation from planktonic culture as well as reduce the biomass of an established biofilm. So as to test some of the compounds in conditions that better resemble the in vivo infections, a continuous flow P. aeruginosa biofilm was tested against two of the most effective compounds, demonstrating the capacity of the compounds to decrease biofilm biomass. This study settles the starting point for further research in N-hydroxylamines compounds as potential effective antibacterial agents to fight drug-resistant pathogenic bacteria. This work was supported in part through grants to ET from the Ministerio de Economia y Competitividad (BFU2011-24066 and BIO2015-63557-R), Generalitat de Catalunya (2014 SGR01260), Spanish Cystic and from fundació La Caixa y CAIXAIMPULSE. AB is thankful to the Ministerio de Educación Cultura y Deporte for its financial support through the FPU programme (FPU13/06083). Panel 10 CptA, an ICE-associated ATPase involved in DNA donation between Thermus thermophilus cells Ignacio Baquedano1, Alba Blesa1, Nieves García-Quintans1, Mario Mencía1, Carlos P. Mata2, José R. Castón2, and José Berenguer1 1 Centro de Biología Molecular Severo Ochoa (UAM-CSIC). 2Centro Nacional de Biotecnología (CSIC). Campus Universidad Autónoma de Madrid. 28049-Madrid. [email protected] Integrative conjugative elements (ICEs) are self-transmissible genetic mobile elements that integrate into the chromosome of its bacterial hosts. They encode the capability to excise from the chromosome under specific metabolic conditions, an origin of transference (oriT) and, in many cases, the mobilization genes required for their conjugative transfer to a recipient cell though a type IV secretion system (T4SS) (1). In Thermus thermophilus HB27 a highly efficient conjugation-like mechanism allows the transfer of large DNA fragments. The mechanism is bidirectional, and involves both active DNA pushing from the donor cell and active DNA pulling from the recipient cell (2). As the pulling step depends on the natural competence machinery, this push-pull mechanism has been baptized as “transjugation”. A bioinformatic search for putative ATPases of the FtsK family that could be involved in DNA pushing led to the identification of the cptA gene. As expected, recombinant CptA protein shows ATPase activity and forms hexameric rings which central pore seems enough as to allow dsDNA passage. In T. thermophilus, CptA binds to the membrane at the cell poles, and its presence is required for DNA pushing during transjugation. The cptA gene is encoded within a four genes operon which includes a putative nuclease of the NurA family, a type II restriction enzyme, and a putative DNA-methylase. Genes around this operon include transposases, pseudogenes, and a phage-like recombination specific nuclease of the XerC family. The whole 14,857 bp region has a G+C DNA content below (59%) that of the chromosome (68,7%) and is limited by 47-bp-long direct repeats that corresponds to the 3’ part of isoleucine tRNA. These data,along the absence of the whole region in the closely related strain T. thermophilus HB8, suggested its nature as a putative ICE-like element. To demonstrate this, different assays have been carried showing that the region is able to excise from the chromosome leading to an apparently non replicative circular form. Transfer of the whole element and specific integration are being checked. 1) Bellanger X, Payot S, Leblond-Bourget N, Guédon G. 2004. Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol Reviews 38: 720-760 2) Blesa Estaban A. 2016. Horizontal gene transfer in Thermus thermophilus: mechanisms and barriers. phD. Universidad Autónoma de Madrid Acknowledgements: This work has been supported by grant BIO2013-44963-R from the Spanish Ministry of Economy and Competence. Panel 11 Engineering a hydrogen biosensor: selection of overproducing nitrogenase variants for biohydrogen production Emma Barahona, Emilio Jiménez-Vicente and Luis M. Rubio Centro de Biotecnología y Genómica de Plantas. Universidad Politécnica de Madrid (UPM) - Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA) . Campus Montegancedo UPM. 28223-Pozuelo de Alarcón (Madrid), Spain [email protected]. A number of microorganisms are being studied as potential producers of H2 through biophotolysis, indirect biophotolysis, photo-fermentations or dark-fermentations. Microorganisms produce H2 by the activity of either hydrogenases or nitrogenases. Hydrogenases catalyze the reaction: 2H+ + 2e- ↔ H2, whereas nitrogenases catalyze the reduction of N2 with the following limiting stoichiometry: N2 + 8H+ + 8e- ↔ H2 + 2NH3. In this work, we have coordinated aspects of both pathways to develop optimized biocatalysts for H2 production using two genetic modules: 1. Engineering an H2 responsive genetic circuit in the purple non-sulphur N2-fixing bacterium Rhodobacter capsulatus SB1003. R. capsulatus carries a system to detect H2 that is composed of three proteins: a H2-sensor hydrogenase (HupUV), a histidine kinase (HupT) and a response regulator HupR (Vignais et al., 2005). In the presence of H2, this sensor triggers expression of hydrogenase structural and biosynthetic genes. Taking advantage of this system, we have introduced a reporter gene under the control of hupS promoter and removed the uptake hydrogenase, generating a new biological-sensor strain capable of producing a measurable and proportional signal when H2 is present in the cell. 2. Generating in vitro evolution of the molybdenum nitrogenase structural genes nifH, nifD and nifK by random mutagenesis. The resulting variants were cloned under nifH promoter control into a broad-host-range vector (Kovach et al., 1995) optimized for diazotrophic conditions. Libraries obtained (around 4 x 106 clones) were introduced and expressed in the strain carrying the modified biological hydrogen sensor. The combination of both tools results in the development of a genetic circuit for the highthroughput screening of H2 overproducing nitrogenase variants thus allowing detection and isolation of clones that present a significant signal increased, through the use of cellsorting cytometry. Thus far, around 1500 clones have been successfully selected by this method; and several of them were able to produce until 18 times more H2 than the control strain. This methodology has potential to be implemented in other biological systems to increase H2 production yields. References Kovach, M.E., Elzer, P.H., Steven Hill, D., Robertson, G.T., Farris, M.A., Martin Roop, R., Peterson, K.M., 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antiobiotic-resistance cassettes. Gene. 166, 175-171. Vignais, P.M., Elsen, s., Colbeau, A., 2005. Transcriptional regulation of the uptake [NiFe] hydrogenase genes in Rhodobacter capsulatus. Biochem. Soc. Trans. 33, 28-32. Panel 12 Estudio de la regulación global del sistema CbrAB en Pseudomonas putida y búsqueda de secuencias consenso de unión al ADN Rocío Barroso, Sofía M. García-Mauriño, Eduardo Santero e Inés Canosa Centro Andaluz de Biología del Desarrollo, Universidad Pablo de Olavide [email protected] El sistema CbrAB es un sistema de regulación global exclusivo del género Pseudomonas que parece responder a cambios de disponibilidad de nutrientes en el medio ambiente. Los análisis de microarrays en Pseudomonas putida realizados en el laboratorio permitieron determinar que CbrAB participa en procesos tan diversos como en el transporte y utilización de aminoácidos como fuente de carbono, tolerancia a metales pesados, movilidad bacteriana o represión catabólica. El elemento regulador del sistema, CbrB, consta de tres dominios: un dominio receptor, un dominio central con actividad ATPasa y dominio de unión a DNA. Para identificar las dianas a las que CbrB reconoce de forma directa, se realizó un ensayo de ChIP-Seq, que ha permitido identificar regiones promotoras de distintos genes a las que se une CbrB para controlar su expresión. Además se pretende encontrar patrones comunes en estas regiones para definir una secuencia consenso de unión de CbrB. Las regiones promotoras de algunos de los genes diana identificados presentaron tres secuencias repetidas de 6 pb (F1 en orientación directa, y R1 y R2 en orientación inversa), que podrían constituir un elemento común a todos ellos. Para comprobar la relevancia de estas secuencias en la activación transcripcional por CbrB de estos genes, se realizaron fusiones transcripcionales de las regiones promotoras al gen lacZ, y se analizó la expresión como medida de la actividad βgalactosidasa en distintas condiciones de activación del sistema. También se estudió la activación cuando la región promotora contenía sustituciones en los posibles subsitios de unión F1, R1 y R2. Además se estudió la unión de CbrB a la región promotora del operón diana PP2810-13 mediante ensayos de retardo en gel (EMSA), y a la que contenía los sitios de unión de CbrB mutados. El operón PP2810-13 codifica una posible bomba de extrusión tipo RND. Con el objeto de dilucidar su función y la relación con los procesos en los que interviene el sistema CbrAB, se ha construido un mutante de inserción en el gen PP2812 y se ha realizado una caracterización fenotípica del mismo. Panel 13 Un tránscrito antisentido conecta el estrés celular con la respuesta SOS Laurène Basteta, Carlos Caballeroa, Maite Villanuevab, Iñigo Lasab y Alejandro ToledoAranaa a Laboratorio de Regulación Génica Bacteriana. Instituto de Agrobiotecnología, CSICUPNA. Avda. Pamplona 123, 31192-Mutilva, Navarra. b Laboratorio de Biofilms Microbianos. Instituto de Agrobiotecnología, CSIC-UPNA. Avda. Pamplona 123, 31192-Mutilva, Navarra. [email protected] La determinación precisa de los mapas transcriptómicos bacterianos ha revelado que el número de regiones cromosómicas con transcripción solapante es mucho mayor de lo que habíamos imaginado1. Estos RNAs antisentidos (asRNAs) se pueden generar de dos maneras diferentes: i) por la presencia de un promotor situado en la cadena contraria a una región codificante (CDS) ó ii) por la deficiencia en la parada de transcripción entre genes convergentes. El segundo caso ocurre cuando no existe un terminador de transcripción (TT) entre los genes o la estructura del TT es inestable, generando así regiones 3’ solapantes1,2,3. También, existe la posibilidad de que la formación de la estructura terminadora pueda ser modificada por factores externos tal y como ocurre en algunos riboswitches. Todo ello hace que el nivel de antisentido dependa del promotor de los genes convergentes y, si existe, del TT presente entre ellos. En cualquier caso, la transcripción solapante en las regiones 3’, incrementa la complejidad de la organización de los genes en el cromosoma al establecer interconexiones entre los mismos además de la establecida por los operones. En un trabajo previo, descubrimos que esta transcripción solapante afecta a la expresión de varios reguladores de Staphylococcus aureus. Uno de ellos correspondía al gen lexA, que codifica para el principal regulador de la respuesta SOS1. Mediante Northern blot determinamos que el transcrito antisentido se genera a partir del promotor SigB del small RNA sbrB. Dicho transcrito incluye la CDS de SbrB, una región que solapa completamente con el mRNA de lexA y la CDS de SosA. Para que este RNA de 1,4 Kb se genere, la RNA polimerasa debe evitar el TT de sbrB, finalizando su transcripción en el TT de sosA. El reemplazo del promotor SigB de sbrB por uno constitutivo, provoca cambios en la expresión de LexA cuando la respuesta SOS es inducida por Mitomicina, lo que demuestra la interconexión entre ambas vías de regulación. Aunque son necesarios más experimentos para comprender este mecanismo de regulación, este estudio ejemplifica la complejidad transcritómica presente en las bacterias, donde un mismo transcrito puede contener múltiples elementos funcionales. Referencias: 1) Lasa I, et al. 2011. PNAS 108:20172–20177. 2). Lasa I, et al. 2012. RNA Biology 9:1039–1044. 3) Ruiz de los Mozos, et al, PLOS Genetics 9(12): e1004001. Agradecimientos: al Ministerio de Economía y Competitividad y al Consejo Europeo de Investigación por la financiación de los proyectos de investigación BFU2014-56698-P y ERC-2014-CoG-646869, respectivamente. A la Universidad Pública de Navarra por la financiación del contrato pre-doctoral de C.C. Panel 14 Development of random transposon mutagenesis system in Thermus thermophilus 1 Alba Blesa1, Didier Mazel2 & José Berenguer1 Centro de Biología Molecular Severo Ochoa. CSIC-UAM. Madrid. 2 Institute Pasteur. Paris. [email protected] Thermus thermophilus is acknowledged as the best thermophile bacterial model due to its fast growth under laboratory conditions and its ease for genetic manipulation. Indeed, its extraordinary natural competence has allowed the development of an increasingly sophisticated genetic toolbox, including cloning and expression plasmids, thermostable resistance markers and promoter proof vectors working at high temperature. Unfortunately, generation and sorting of genomic libraries in a cost-effective way remains a bottleneck. Transposons have became versatile tools for genetic manipulation and analysis of bacteria, reaching new fields such as genomics and transcriptomics by its application in the generation of mutant libraries with random insertions. This work aimed the construction of a thermostable transposon-based system which would enable performing random mutagenesis in Thermus strains. In contrast to previously reported akin systems1, no previous in vitro transposition was arranged, whereas a natural jump of the transposon was expected. Two systems were developed following the pUT model based on antibiotic resistant suicide constructs2, each one grounded on a DDE transposase (ISBstI and ISNth2) from two thermophilic bacteria. To ensure a genome-wide analysis, we estimated an accrual of the mutant collection of about 10,000 clones, figure which statistically suggests that the transposon has been inserted in every gene throughout the genome. Detection of the insertion sites in 10 clones revealed a random distribution, but massive sequencing by TN-seq3 will be required to assess this in a whole genome scale. TN-seq allows simultaneous sequencing of the transposon insertion sites in different mutants, and to compare the patterns of such insertion among library subjected to different selection stresses, for instance. This way, a whole collection of the genes involved in a given process can be detected at the same time. The availability of these new in vivo transposition tools not only widens Thermus genetic toolbox but also results from this study will provide priceless insights of undiscovered gene functions required for stress-survival, lateral gene transfer or any process assayed in T. thermophilus. References 1. Carr, J. F., Gregory, S. T., & Dahlberg, A. E. (2015). Transposon mutagenesis of the extremely thermophilic bacterium Thermus thermophilus HB27. Extremophiles, 19(1), 221-228. 2. De Lorenzo, V., Herrero, M., Jakubzik, U. & Timmis, K. N. (1990). Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. Journal of Bacteriology 172, 6568-6572. 3. Van Opijnen, T; Bodi, KL; Camilli, A. (2009). Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nature Methods. 6. 767-772 Acknowledgements This work has been funded by Grants BIO2013-44963-R, and FP7-PEOPLE-2012-IAPP nº 324439. A. Blesa held a FPI contract. Panel 15 Las secuencias de inserción facilitan la adaptación de un plásmido multicopia a un nuevo hospedador Cristina Bernabe-Balas, Alfonso Santos-Lopez, Manuel Ares-Arroyo, Rafael OrtegaHuedo, Natalia Montero y Bruno Gonzalez-Zorn. Departamento de Sanidad Animal y Centro de Vigilancia Sanitaria Veterinaria (VISAVET), Facultad de Veterinaria, Universidad Complutense de Madrid, España. [email protected] La transferencia horizontal de plásmidos entre distintas especies bacterianas supone un agravante del rápido aumento de la resistencia a antibióticos que existe actualmente. Sin embargo, no se conocen con claridad los mecanismos implicados en la adaptación y estabilización de un plásmido cuando es adquirido por un nuevo hospedador. Para explorar la capacidad adaptativa de los plásmidos transformamos el plásmido tipo ColE1 pB1000, adaptado a la familia Pasteurellaceae, en Escherichia coli DH5α, donde es altamente inestable, y lo evolucionamos con presión selectiva. Tras extraerlo y transformarlo de nuevo en E. coli, analizamos la estabilidad, el número de copias (NCP) y el nivel de resistencia (CMI) en el nuevo hospedador. La secuenciación de los plásmidos evolucionados reveló la adquisición de dos secuencias de inserción, IS1 e IS10, desde el cromosoma de E. coli a diferentes replicones. Ambas ISs se insertaron entre las relaxasas y el gen de resistencia, con tan solo dos nucleótidos de diferencia entre ellas. Así mismo, algunos plásmidos presentaron mutaciones puntuales en el origen de replicación (oriV). Sorprendentemente, la estabilidad de los plásmidos que portaban una IS o un SNP en el oriV se vio incrementada significativamente, siendo 100% estables cuando ambas modificaciones estaban en el mismo replicón. Mediante PCR cuantitativa comprobamos que un SNP producía un incremento del NCP en E. coli, sin embargo, el NCP se mantenía inalterado cuando pB1000 codificaba una IS. La CMI de las bacterias portadoras de plásmidos con un SNP también se vio significativamente aumentada, mostrando una relación con el aumento del número de plásmidos. Para investigar el mecanismo por el cual las ISs estabilizaban pB1000 clonamos dos fragmentos de ADN del mismo tamaño que las ISs en la misma posición de pB1000 donde éstas se insertaban. La estabilidad de estas construcciones fue muy similar a la de los plásmidos con una secuencia de inserción. Este resultado parece indicar que la interrupción de esa región plasmídica es clave para la estabilización de pB1000 en E. coli. Ésta es la primera descripción de la estabilización de un plásmido en un nuevo hospedador gracias a la adquisición de una secuencia de inserción, demostrando el importante papel que pueden tener los elementos móviles en la adaptación y diseminación de plásmidos en nuevos ambientes. Trabajo financiado por las Ayudas de Ministerio de Educación para la Formación de Profesorado Universitario (FPU). Referencia: FPU13/06215. Panel 16 Protein interactions in the Escherichia coli cellulose biosynthesis apparatus Joaquín Bernal, Petya Krasteva, Laetitia Travier; Fernando Ariel Martin, Rémi Fronzes and Jean-Marc Ghigo Unité de Génétique des Biofilms- Institut Pasteur [email protected] Biofilms are microbial communities characterized by three-dimensional growth resulting from the ability of individual cells to adhere to each other as well as to produce an extracellular matrix that ensures biofilm physical cohesion. Numerous Gram-negative bacteria produce cellulose as a biofilm matrix polymer, a property relying on the expression of bacterial cellulose synthesis (Bcs) proteins, forming the cellulose synthesis apparatus. In E. coli and others Enterobacteracea, BcsQ, gene of unknown function is located upstream of the bcs genes. Previous studies in our laboratory showed that BcsQ is essential for cellulose biosynthesis in E. coli and it localizes at the cell pole (1). We also showed cellulose is secreted by this pole (1). However, the specific role of BcsQ in the cellulose production remains unclear. We hypothesize that BcsQ may be required for polar localization or partitioning of the cellulose biosynthesis apparatus in E. coli. To address this question we carried out a functional and structural analysis of the cellulose biosynthesis apparatus. First, combining pull-down and bacterial two-hybrid (2) approaches we analyzed the interactome of BcsQ. We succeeded to purify stable and active cellulose synthesis apparatus and analysis of protein interactions suggests the existence of a multi-protein megacomplex, in which BcsQ interacts with almost all bcs proteins. We then confirmed these results using NS-EM technology (Negative StainedElectronic Microscopy) and we solved for the first time the 3D-structure of the full innermembrane cellulose synthesis apparatus. Finally, we tested the activity of this purified cellulose complex and we showed that it is functional and able to synthetize de novo cellulose in vitro. I will discuss the particularity of the structure of the cellulose secretion complex and its relationships with other systems involved in exopolysaccharide secretion. 1. Le Quéré, B. and Ghigo, J.M. (2009) BcsQ is an essential component of the Escherichia coli cellulose biosynthesis apparatus that localizes at the bacterial cell pole. Mol. Microbiol. 72, 724–740 2. Karimova, G., J. Pidoux, A. Ullmann and D. Ladant. (1998). A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U.S.A. 95 :5752-5756. Panel 17 Vitamin K3 induces Stenotrophomonas maltophilia SmeVWX multidrug efflux pump Paula Blanco Torres, Fernando Corona Pajares, M. Blanca Sánchez, José Luis Martínez Menéndez Centro Nacional de Biotecnología (CSIC) [email protected] Stenotrophomonas maltophilia is a gram-negative bacillus associated with plants and aquatic environments. It is also an opportunistic pathogen with increasing prevalence able to cause infections in immunocompromised patients or in those with a previous pathology. The treatment of the infections caused by this bacterium is often complicated due to the several intrinsic resistance mechanisms that this species presents to currently used antibiotics. One of better studied resistance mechanisms in S. maltophilia are multidrug efflux pumps (MDR) belonging to RND (resistance nodulation division) family, which cause resistance to a wide range of antibiotics and toxic compounds. Some of them have a basal expression level but, in general, MDR expression is often repressed and only reach high expression levels when the local regulator is mutated or when bacteria are in the presence of the right effectors. In this study we have developed a YFP-based sensor with the aim to identify effectors able to induce SmeVWX, a S. maltophilia efflux pump involved in quinolones, chloramphenicol and tetracyclines resistance. With this purpose, we tested a variety of naturally different compounds and we analyzed the fluorescence signal given by the expression of YFP under the control of the smeVWX promoter. Among the tested compounds, vitamin K3, an agent with increasing importance in cancer research and present in some haemostatic drugs, was able to induce SmeVWX efflux pump with the consequence of a decrease in the susceptibility of S. maltophilia to ofloxacin. This indicates that transient situations of phenotypic resistance to antibiotics currently in use may happen when patients are treated with formulations containing vitamin K3. Panel 18 An enhanced folding reporter to select thermostable protein variants Sandra Bosch, José Berenguer, Aurelio Hidalgo Centro de Biología Molecular Severo Ochoa, Universidad Autónoma de Madrid (Madrid, Spain) [email protected], [email protected] The folding interference principle [1] enables an activity-independent method for the selection of thermostable mutants of any protein [2]. This method consists in the expression of a fusion of two proteins, the protein of interest at N terminus and a thermostable antibiotic selection marker (the reporter) at C terminus. If the protein of interest is not correctly folded, the reporter will not fold properly, thus the clone will not survive. Candidate thermostable reporters used to correlate protein stability with host survival are the engineered kanamycin nucleotidyltransferase from Staphylococcus aureus (Kat), bleomycin binding protein from Streptoalloitechus hindustanus (ShBle) and the engineered hygromycin B phosphotransferase from Eschericha coli (Hph5) [3]. Kat exhibited positive but weak correlation due to the dimeric nature of Kat. ShBle did not provide selection due to the overstabilization caused by the tetrametic nature of ShBle. Hph5 has a monomeric structure, but is not stable enough. Therefore, we tried to obtain a more thermostable variant of Hph5 by error-prone PCR (epPCR). After selection at 70 °C using Thermus thermophilus as host, the thermostability of 20 selected clones was verified using a serial dilution assay on plate. The clone that showed the highest relative thermostability on plate, named Hph17, was characterized kinetically and structurally. Hph17 has 5 amino acid substitutions with respect to the parental Hph5, but only 1 of them seems to stabilize the structure of Hph17 according to bioinformatics simulations. Hph5 and Hph17 did not show a significantly different melting temperature. However, Hph17 has a higher activity than Hph5, at all temperatures tested. Finally, Hph17 was used as a folding reporter to screen for thermostable variants of industrially relevant enzymes. Bibliography 1. Waldo, G. S., Standish, B. M., Berendzen, J., & Terwilliger, T. C. (1999). Rapid protein-folding assay using green fluorescent protein. Nature biotechnology, 17(7), 691695. 2. Chautard, H., Blas-Galindo, E., Menguy, T., Grand'Moursel, L., Cava, F., Berenguer, J., & Delcourt, M. (2007). An activity-independent selection system of thermostable protein variants. Nature methods, 4(11), 919-921. 3. Nakamura, A., Takakura, Y., Kobayashi, H., & Hoshino, T. (2005). In vivo directed evolution for thermostabilization of Escherichia coli hygromycin B phosphotransferase and the use of the gene as a selection marker in the host-vector system of Thermus thermophilus. Journal of bioscience and bioengineering, 100(2), 158-163. Acknowledgments: This work has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement 635595 "CarbaZymes" and the Spanish Ministry of Economy and Competitiveness under grant BIO2013-44963-R. Panel 19 Incremento del número de copias de plásmido por aumento de la temperatura de crecimiento en Escherichia coli B. Mendoza-Chamizo, R. González-Soltero y E. Botello Área de Genética. Depto. de Bioquímica y Biología Molecular y Genética. Facultad de Ciencias. Universidad de Extremadura. 06006 Badajoz. [email protected] Un aumento de la temperatura de incubación a un cultivo de Escherichia coli induce replicaciones cromosómicas extras. Esta replicación inducida por calor, HIR (HeatInduced Replication), inicia en oriC y presenta requerimientos funcionales diferentes a los de la replicación cíclica (1, 2, 3). HIR no es exclusiva de E. coli, sino que también tiene lugar en otras especies bacterianas (4). Para determinar si HIR es una respuesta específica de oriC o un mecanismo de estrés extensible a otros replicones, se ha estudiado si esta replicación tiene lugar en minicromosomas y plásmidos. La emisión de fluorescencia por la proteína GFP puede utilizarse para cuantificar la dosis génica (5, 6, 7). Se ha aprovechado dicha emisión para estudiar HIR en distintos plásmidos portadores del gen GFPmut2 clonado bajo el control del promotor del operón arabinosa, pBAD, y de su proteína reguladora, AraC (6, 8). Esta construcción permite la determinación del número de copias de plásmido (pcn) mediante técnicas como la espectrofluorimetría (pcn/masa), la citometría de flujo (pcn/célula) y la qPCR (pcn/reacción). Se han estudiado diferentes replicones con distintos mecanismos de control de la replicación: un minicromosoma oriC sopABC, plásmidos derivados de F, P1 ΔincA y pSC101, con control de la replicación por iterones, y de R1, p15A y pBR322, con control de la replicación por asRNA (6). A diferencia de los cromosomas bacterianos, los replicones estudiados no presentan HIR. El aumento de temperatura induce la replicación de minicromosoma y plásmidos como una adaptación a la nueva temperatura de crecimiento, en función de la velocidad de crecimiento. Se ha observado una correlación directa entre los parámetros pcn/masa y pcn/célula con la velocidad de crecimiento determinada por la temperatura: a mayor temperatura, mayor número de copias. La replicación del minicromosoma no depende de la actividad de RecA; en plásmidos, la inducción de la replicación por aumento de temperatura es superior en ausencia de RecA y en crecimiento exponencial, RecA funcional favorece la replicación. La comparación de las técnicas utilizadas lleva a proponer la espectrofluorimetría como el método más sencillo, rápido y preciso para la determinación del pcn en plásmidos con el gen GFPmut2 (4, 7). 1. Botello E, Jiménez-Sánchez A (1997). A temperature shift-up induces initiation of replication at oriC on the Escherichia coli chromosome. Mol Microbiol 26:133-144. 2. González-Soltero R, Botello E, Jiménez-Sánchez A (2006). Initiation of heat-induced replication requires DnaA and the L-13mer of oriC. J Bacteriol 188:8294-8298. 3. González-Soltero R, Jiménez-Sánchez A, Botello E (2008). Functional requirements for heat induced genome amplification in Escherichia coli. Process Biochem 43:1162-1170. 4. Mendoza-Chamizo B (2016). Replicación inducida por estrés térmico en bacterias y plásmidos. Tesis doctoral. Universidad de Extremadura. 5. González-Soltero R (2007). Replicación inducida por estrés térmico en cromosoma y plásmidos de Escherichia coli. Tesis doctoral. Universidad de Extremadura. ISBN 978-84-7723-764-8. 6. Lobner-Olesen A (1999). Distribution of minichromosomes in individual Escherichia coli cells: implications for replication control. EMBO J 18:1712-1721. 7. Mendoza-Chamizo B, González-Soltero R, Botello E (2012). Plasmid copy number quantification by spectrofluorometry. En: Mendez-Vilas A, ed. Microbes in applied research: current advances and challenges. World Scientific Publishing Co. Pte. Ltd., Singapore. pp 432-436. 8. Cormack BP, Valdivia RH, Falkow S (1996). FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173:33-38. Panel 20 Acercamientos globales a la identificación de RNAs no codificantes bacterianos Manuel Brenes-Álvarez, Elvira Olmedo-Verd, Agustín Vioque, Alicia M. Muro-Pastor Instituto de Bioquímica Vegetal y Fotosíntesis (CSIC-Universidad de Sevilla) [email protected] La aplicación de nuevas tecnologías, fundamentalmente RNASeq, al análisis del transcriptoma ha traído consigo una nueva percepción de los mecanismos de regulación de la expresión génica en bacterias. Actualmente está bien establecido que la transcripción de pequeños RNAs reguladores (sRNAs) y transcritos antisentido (asRNAs) constituye una parte esencial de los transcriptomas bacterianos. En nuestro laboratorio estamos interesados en los mecanismos reguladores ejercidos por RNAs no codificantes en el contexto de la adaptación a la carencia de nitrógeno, utilizando como modelo la cianobacteria Nostoc (Anabaena) sp. PCC 7120, un organismo capaz de diferenciar heterocistos, un tipo celular especializado en la fijación de nitrógeno atmosférico. A partir de un análisis transcriptómico mediante dRNASeq (1), disponemos de datos relativos a todos los inicios de transcripción (TSS) en el genoma de PCC7120, así como de su dinámica de uso en respuesta a la carencia de nitrógeno combinado y de su dependencia de los reguladores transcripcionales NtcA y HetR. Al objeto de identificar posibles sRNAs hemos diseñado una estrategia computacional que combina la información transcriptómica previa con una predicción de terminadores independientes de Rho y un análisis filogenético de 89 genomas cianobacterianos. El análisis filogenético muestra que alguno de los sRNAs predichos por el algoritmo tienen distribución universal, mientras que otros están conservados únicamente en los genomas de cianobacterias con capacidades concretas, como la diferenciación de heterocistos, por lo que estos sRNAs podrían estar asociados a dichas funciones. Hemos validado experimentalmente la transcripción de varios sRNAs que aparecen conservados en genomas de cianobacterias formadoras de heterocistos, incluyendo NsiR8 (nitrogen stress inducible RNA 8), cuya transcripción tiene lugar exclusivamente en este tipo celular (2). El algoritmo que hemos diseñado es en principio aplicable a cualquier grupo de bacterias para el que se disponga de varios genomas relacionados y de información relativa a los inicios de transcripción en uno de ellos, utilizado como referencia. Por otro lado, estamos iniciando acercamientos para la identificación de asRNAs que pudieran estar relacionados con la diferenciación de heterocistos. Disponemos de datos correspondientes a la hibridación de microarrays diseñados en base a los datos de RNASeq y que incluyen un total de 55.299 sondas para todos los transcritos, incluyendo transcritos antisentido. La construcción de redes de co-expresión a partir de los datos anteriores nos permitirá identificar grupos de transcritos (incluyendo asRNAs) que compartan patrones de expresión asociados a distintos aspectos de la adaptación al estrés de nitrógeno y la diferenciación celular. 1. Mitschke J, Vioque A, Haas F, Hess WR, and Muro-Pastor AM (2011). Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc. Natl. Acad. Sci. USA 108, 20130-20135. 2. Brenes-Álvarez M, Olmedo-Verd E, Vioque A, Muro-Pastor AM (2016). Identification of conserved and potentially regulatory small RNAs in heterocystous cyanobacteria. Front Microbiol 7:48. Financiación: MICINN (BFU2010-14821), MINECO (BFU2013-48282-C2-1), MECD (Contrato predoctoral FPU014/05123_Manuel Brenes Álvarez). Panel 21 Recombinant attenuated Mycobacterium tuberculosis for heterologous expression and secretion of DTP antigens: a rationale to construct a multivalent vaccine Esther Broset1, 2, Alex I. Kanno3, Luciana C.C.Leite3, Carlos Martín1,2, Jesús GonzaloAsensio1,2 1 Grupo de Genética de Micobacterias, Universidad de Zaragoza, Zaragoza, Spain 2 CIBER enfermedades respiratorias. Instituto de Salud Carlos III, Madrid, Spain 3 Centro de Biotecnologia, Instituto Butantan, São Paulo, SP, Brazil [email protected] Live attenuated BCG vaccine is effective in reducing severe forms of tuberculosis (TB) in children but confers variable protection against pulmonary TB in adults (1). To overcome this handicap of BCG, we have developed the live vaccine MTBVAC, based on the attenuation of Mycobacterium tuberculosis by deletion of the phoP and fadD26 virulence genes (2). MTBVAC is currently in phase Ib clinical trials in South Africa.(NTC02729571) Due to its adjuvant capacity, BCG has been traditionally proposed as vector to deliver viral, bacterial or parasite antigens (3). In this context, we propose the study of MTBVAC as a multivalent recombinant vaccine against different infectious diseases including diphtheria, tetanus and whooping cough. Prior to construct recombinant MTBVAC (rMTBVAC) strains; we studied the possible immune interference between MTBVAC and the current vaccines against diphtheria, tetanus and whooping cough (DTP). Actually two formulations of DTP are administered depending on the region: either the pertussis inactivated toxins (DTaP) or the pertussis whole-cell vaccine (DTwP). In this context, we tested both DTP formulations in mice previously vaccinated with MTBVAC. Results were compared with mice independently vaccinated with MTBVAC or DTP alone. Mice conserved similar immune response against MTBVAC or DTP in all cases, which proves absence of interference between both vaccines. To construct rMTBVAC, genetically inactivated and codon optimized genes for diphtheria (CRM197), pertussis toxoids (S1), or Fragment C of tetanus toxin (FC) were synthesized. Each DTP antigen was cloned under the control of three phage derived promoters with different strength (4). To promote secretion of these antigens, the signal sequence of Ag85A (a known mycobacterial secreted protein) was placed in frame before each DTP construct. Further, a FLAG peptide was placed in the C-terminus of these proteins to facilitate downstream detection by Western Blot. Protein expression and secretion of CRM197, S1 and FC was confirmed by Western blot using antigen-specific and anti-FLAG antibodies. Preliminary experiments in mice showed that vaccination with rMTBVAC expressing the S1 toxoid protects against Bordetella pertussis colonization. On-going experiments aim to detect anti-DTP antibodies in mice vaccinated with these rMTBVAC. Taking together, these results provide proof-of concept that MTBVAC is an efficient antigenic vector for heterologous antigen expression and put forward rMTBVAC to protect against different infectious diseases. References: (1) Fine, P.E. (1995). Lancet 346(8986):1339-45. (2) Arbués et al. (2013). Vaccine 31(42):4867-73 (3) Bastos et al. (2009). Vaccine 27(47): 6495-503. (4) Kanno et al. (2016). Appl. Environ. Microbiol 82(8): 2240-2246 Funding: Esther Broset was recipient of BES-2012-052937 grant by Ministerio de Economía y Competitividad de España. This work was funded by BIO2014-5258P and TBVAC2020 projects Panel 22 Regulación ambiental de la síntesis del polisacárido PIA/PNAG en Staphylococcus aureus resistentes a meticilina Saioa Burgui1, Cristina Solano1, Jaione Valle1 e Iñigo Lasa1 1 Navarrabiomed-Universidad Pública de Navarra, IdiSNA, Pamplona-31008, Navarra, Spain [email protected] Staphylococcus aureus es una de las principales bacterias causantes de infecciones nosocomiales asociadas a la implantación de dispositivos médicos. La dificultad de tratar estas infecciones se asocia principalmente a la aparición de cepas con resistencia a la meticilina (MRSA) y a otros antibióticos inducida por su capacidad de formar biofilm. Uno de los principales componentes de la matriz del biofilm de S. aureus es el exopolisacárido de adherencia intercelular PIA/PNAG, cuya síntesis depende de las enzimas codificadas en el operón icaADBC y regulada por el producto del gen icaR que se transcribe de manera opuesta al operón. La mayoría de aislados clínicos de S. aureus resistentes a meticilina (MRSA) no son capaces de sintetizar el polisacárido PIA/PNAG en las mismas condiciones de laboratorio en las que las cepas meticilina sensibles lo sintetizan, sugiriendo la existencia de un mecanismo de regulación especifico en estas cepas. Analizando distintas condiciones ambientales que podrían afectar la síntesis de PIA/PNAG, hemos encontrado que la temperatura es un elemento clave en la represión de la síntesis de PIA/PNAG. La cepa S. aureus MW2 (MRSA) no produce niveles detectables de PIA/PNAG a 37ºC, sin embargo, a 28ºC produce elevados niveles de dicho exopolisacarido. Utilizando una colección de mutantes en cada uno de los sistemas de dos componentes de la cepa MW2 hemos determinado que el sistema de dos componentes ArlRS es responsable de activar la síntesis de PIA/PNAG a 28ºC, ya que la cepa deficiente en ArlRS deja de producir PIA/PNAG a esta temperatura. Un análisis detallado de la cascada de regulación utilizando fusiones transcripcionales del promotor del operón icaADBC e icaR con la proteína reportera Gfp no han mostrado diferencias significativas en los niveles de transcripción de ambos a las distintas temperaturas. Sin embargo, el análisis de los niveles de expresión de las proteínas del operon Ica, muestran una mayor acumulación de los niveles de la proteína IcaC a 28ºC en la cepa salvaje, acumulación que desaparece cuando se muta el sistema ArlRS Los resultados indican que el sistema de dos componentes ArlRS regula a nivel posttranscripcional la expresión del operón icaADBC mediada por la temperatura. Panel 23 Compartmentalized cyanophycin and arginine catabolism in the filamentous heterocyst-forming cyanobacterium Anabaena Mireia Burnat and Enrique Flores Instituto de Bioquímica Vegetal y Fotosíntesis, Consejo Superior de Investigaciones Científicas (CSIC) and Universidad de Sevilla (US), Sevilla [email protected] Some cyanobacteria grow as filaments that, under nitrogen deprivation, contain two cell types: the vegetative cells that perform CO2 fixation and the heterocysts specialized for N2 fixation (1). During diazotrophic growth heterocysts and vegetative cells exchange nutrients. Many cyanobacteria synthesize cyanophycin, a nitrogen-rich reserve polymer that accumulates conspicuously at the heterocyst poles. Cyanophycin is degraded by cyanophycinase producing a dipeptide, β-aspartyl-arginine, that is hydrolyzed by isoaspartyl-dipeptidase. In the model heterocyst-forming cyanobacterium Anabaena sp. PCC 7120, whereas cyanophycinase is expressed in heterocysts, isoaspartyl-dipeptidase concentrates in vegetative cells. Thus, under diazotrophic conditions, cyanophycin degradation is compartmentalized: cyanophycin is hydrolyzed in heterocysts producing a dipeptide, ß-aspartyl-arginine, that is transferred to vegetative cells, where it is further hydrolyzed to aspartate and arginine that are used in metabolism (2). Although arginine catabolism has been scarcely investigated in cyanobacteria, an arginase-like pathway appears to be important in these organisms (3). An arginase-encoding gene remained, however, to be identified. We have recently found that inactivation of a gene encoding a member of the guanidine-modifying protein superfamily, here denoted ArgE, impairs the production of proline and glutamate not only from radiolabeled arginine but also from radiolabelled ornithine. This observation and complementary results show that ArgE is a novel enzyme that generates proline from arginine in a two-step reaction in which ornithine is an intermediate. During diazotrophic growth, an ArgE-GFP fusion protein concentrates in vegetative cells rather than in heterocysts. In the arginine decarboxylase pathway, arginine decarboxylase (SpeA) produces agmatine, and agmatinase (SpeB) produces urea and putrescine, which is used for polyamine synthesis. We have previously identified the agmatinase-encoding speB gene of Anabaena, whose inactivation leads to accumulation of agmatine resulting in deleterious effects (4). We have now constructed an Anabaena speA mutant and found that it is impaired specifically in diazotrophic growth, indicating an important role of the arginine decarboxylase pathway under these conditions. During diazotrophic growth, SpeA-GFP and SpeB-GFP fusions also concentrate in vegetative cells. Our results have identified a novel arginase-like pathway in nature and show that, during diazotrophic growth, arginine catabolism in Anabaena is compartmentalized taking place mainly, if not exclusively, in vegetative cells. Work supported by grant no. BFU2014-56757-P from Plan Nacional de Investigación, Spain, co-financed by the European Regional Development Fund. (1) (2) (3) (4) Flores & Herrero (2010) Nat Rev Microbiol 8:39-50. Burnat et al (2014) Proc Natl Acad Sci USA 111:3823-3828. Quintero et al (2000) J Bacteriol 182:1008-1015. Burnat el al (2014) MicrobiologyOpen 3:777-792. Panel 24 Identificación por PCR de genes asociados a la virulencia de Helicobacter pylori en biopsias gástricas Cochabamba – Bolivia Bustamante, Z1.; Yañez R1.; Saravia J2. Quiroga D1., Funes1 F. Zabalaga S1.; Pinto J1. Rodriguez S1., Fernandez R1.; Sejas E1., Koller J2.; Cartagena K2.; Laserna J.L.2. ; Pacheco C2 1 Facultad de Ciencias Farmacéuticas y Bioquímicas Universidad Mayor de San Simon Cochabamba - Bolivia 2 Clínica Los Olivos-Cochabamba [email protected] Helicobacter pylori es un bacilo gramnegativo microaerófilo que coloniza de manera persistente la mucosa gástrica de humanos, implicada en numerosas enfermedades gastroduodenales. La detección de genes específicos, permite conocer el grado de patogenecidad de las bacterias que infectan la mucosa gástrica. En Bolivia no se cuentan con datos sobre la prevalencia de los tipos virulentos de Helicobacter pylori que se encuentran infectando la mucosa gástrica de pacientes con diferente grado de gastropatías. El objetivo principal de este trabajo fue identificar genes de virulencia en biopsias que dieron positiva la prueba de la ureasa y pruebas moleculares de pacientes que acudieron a la Clínica los Olivos de septiembre a diciembre del año 2015 y determinar la asociación con diferentes patologías gástricas. Se trabajó con el ADN de 78 biopsias positivas para H. pylori., se identificaron por reacción en cadena de la polimerasa, amplificando genes de virulenci; se realizó una PCR multiple, para amplificar los genes, cag A, bab A2, otra para amplificar los genes vacA y su alelos s1, s2, m1,m2 ; la determinación de ice A1 e ice A2 se realizó en reacciones separadas. Se encontró mayor prevalencia de los genes vacAs1m1 con 36/78 casos, seguido del gen iceA1 en 29/78 muestras y el gen cagA en 28/78; vacAs2m2 en 23/78; iceA2 en 20/78, con menos frecuencia los genes babA2 en cuatro muestras. Se observó asociación significativa entre los genes vacAs1m1 con gastropatías agudas y crónicas, también se asoció el gen babA2 con metaplasia y gastropatías crónicas. Los resultados además mostraron que en varias muestras gástricas positivas a Helicobacter, coexisten dos o más genes simultáneamente, siendo más frecuentes la asociación entre cagA,iceA1, vacAs1m1 en 8 muestras; cagA; vacAs1m1 en 8; iceA1, vacAs2m2 en 8 , iceA1 ; vacAs1m1 en 5; iceA2; vacAm2s2 en 6. La asociación más importante fue entre los genes cagA y vacs1m1, que se encontraron en 22 casos relacionados con gastropatias agudas y crónicas. La determinación de estos genes brinda una información muy importante sobre los tipos circulantes de este microorganismo y el grado de virulencia, que debería alertar para tomar medidas de prevención y tratamientos más acertados. Panel 25 Caracterización funcional de las chaperonas de RNA en Staphylococcus aureus Carlos Caballero1, Arancha Catalán1, Marta Vergara2, Begoña García2, Cristina Solano2, Iñigo Lasa2 y Alejandro Toledo-Arana1 1 Laboratorio de Regulación Génica Bacteriana. Instituto de Agrobiotecnología, CSICUPNA. Avda. Pamplona 123, 31192-Mutilva, Navarra. 2 Laboratorio de Biofilms Microbianos. Instituto de Agrobiotecnología, CSIC-UPNA. Avda. Pamplona 123, 31192-Mutilva, Navarra. [email protected] Las proteínas de unión a RNA (RBPs) son elementos imprescindibles para modular la expresión génica en cualquier ser vivo. Todos los organismos contienen numerosas RBPs repartidas en diferentes grupos en función de los dominios proteicos que contienen, por ejemplo, proteínas ribosomales, ribonucleasas y chaperonas de RNA. En nuestro grupo nos hemos centrado en el estudio de las chaperonas de RNA que incluyen Hfq, CvfB y la familia cold-shock (CSPs) utilizando como modelo a Staphylococcus aureus, uno de los patógenos más relevantes. El dominio cold-shock se encuentra altamente conservado y presente en todos los organismos. El número de CSPs es variable en las especies bacterianas, por ejemplo, el genoma de Escherichia coli contiene nueve CSPs mientras que el de S. aureus sólo tres. Se ha propuesto que las CSPs modulan la expresión génica mediante su unión a los RNA mensajeros (mRNAs), previniendo la formación de estructuras secundarias que impedirían la correcta traducción o modificando la estabilidad del mRNA, pero los mRNAs dianas son aún desconocidos. Hfq actúa como catalizador entre los small RNAs (sRNAs) y sus mRNAs diana para modular su traducción en Gramnegativos. Sin embargo, esta función no ha podido ser extrapolada a Gram-positivos. De igual modo, la función de CvfB es completamente desconocida. Para abordar el estudio de estas proteínas, hemos marcado cada una de ellas mediante un tag y analizado su expresión mediante Western-blots. De todas ellas, seleccionamos CspA e identificamos más de 300 mRNAs dianas mediante RIP-on-chip. Los análisis transcriptómicos y proteómicos revelaron que CspA estimula mayoritariamente la traducción de sus dianas sin afectar a los niveles de mRNAs. Uno de los procesos que modula CspA es la producción de estafiloxantina, el pigmento amarillo que protege a la bacteria del estrés oxidativo. Experimentos de complementación del mutante ΔcspA con plásmidos que expresan CspA, CspB o CspC, revelaron que la producción del pigmento sólo se recupera con CspA a pesar de que la identidad entre las CSPs es mayor al 80%. Esto indica que a pesar de la alta identidad proteica, las CSPs cumplirían funciones diferenciadas en las bacterias. Agradecimientos: al Ministerio de Economía y Competitividad y al Consejo Europeo de Investigación por la financiación de los proyectos de investigación BFU2011-23222 y ERC-2014-CoG-646869, respectivamente. A la Universidad Pública de Navarra por la financiación del contrato pre-doctoral de C.C. Panel 26 Caracterización funcional de las proteínas TasA y TapA en la formación de fibras de tipo amiloide en Bacillus subtilis Cámara-Almirón, J1. , Morel, B2., Conejero-Lara, F2, Romero, D1. 1 Instituto de Hortofruticultura Subtropical y Mediterránea “La Mayora”, Universidad de Málaga y Consejo Superior de Investigaciones Científicas (IHSM-UMA-CSIC). Departamento de Microbiología, Centro de Supercomputación y Bioinnovación, Universidad de Málaga, Calle Severo Ochoa 34, Parque Tecnológico de Andalucía, 29590 Málaga, España. 2 Departamento de Química Física, Facultad de Ciencias, Universidad de Granada, 18071, Granada, España. [email protected] Las proteínas amiloides son un grupo heterogéneo de proteínas diferentes en secuencia aminoacídica, pero similares en su estructura cuaternaria: fibras enriquecidas en láminas beta, con gran estabilidad, resistencia y capacidad de unión de colorantes específicos, como el rojo congo o la thioflavina T. Estas proteínas han estado tradicionalmente asociadas a patologías neurodegenerativas en humanos como el Alzheimer o el Parkinson. Sin embargo, los miembros dentro de esta familia están ampliamente distribuidas en la naturaleza, desde bacterias hasta humanos, e intervienen en un amplio rango de funciones biológicas, motivo por el que se han denominado “amiloides funcionales”. En bacterias, los amiloides funcionales son responsables de participar en funciones muy diversas como la interacción célula-célula, con superficies abióticas, y formación de biofilms. En Bacillus subtilis, la proteína amiloide TasA es el componente proteico mayoritario de la matriz extracelular del biofilm de este microorganismo y el principal elemento que constituye las fibras amiloides, mientras que la proteína auxiliar TapA, presente en mucha menor proporción, actúa favoreciendo el ensamblaje de las mismas. Estas actúan como un andamiaje proteico donde se disponen el resto de componentes de la matriz extracelular, lo que confiere a esta estructura una mayor estabilidad y, por consiguiente, proporcionan una mayor robustez al biofilm. En este este trabajo se llevó a cabo el análisis de regiones o dominios tanto de TasA como de TapA importantes para la amiloidogénesis, así como para la funcionalidad de ambas proteínas. Para ello, el estudio se enfocó desde un punto de vista multidisciplinar, combinando pruebas clásicas de caracterización de amiloides con técnicas de biología molecular y diversas pruebas biofísicas. Los resultados obtenidos hasta la fecha han demostrado la existencia de pequeñas secuencias, dentro de las proteínas TasA o TapA con capacidad para polimerizar en la forma de fibras, lo que muestra su importancia en el proceso de fibrilación y en la funcionalidad de ambas proteínas. En el caso de la proteína TapA, las regiones analizadas ponen de manifiesto la importancia de su extremo amino-terminal tanto en la funcionalidad de la proteína como en su interacción con TasA. Proyecto financiado por la Unión Europea (European Research Council – Starting Grant 8.06 UE/60.8003). Panel 27 Cell division in the filamentous cyanobacterium Anabaena, a true pluricellular bacterium Sergio Camargo, Laura Corrales-Guerrero, Ana Valladares, Silvia Picossi, Enrique Flores and Antonia Herrero Instituto de Bioquímica Vegetal y Fotosíntesis, Consejo Superior de Investigaciones Científicas and Universidad de Sevilla [email protected] Filamentous cyanobacteria are among the oldest pluricellular organisms in our planet, and some of them, such as the model strain Anabaena sp. PCC 7120, are capable of cell differentiation. Under conditions of nitrogen deficiency, the Anabaena filament is composed of two types of cooperative cells: the vegetative cells that fix CO2 performing oxygenic photosynthesis, and the heterocysts that perform N2 fixation. Heterocysts are separated by intervals of 10 to 20 vegetative cells. An intense exchange of metabolites and signaling molecules takes place between vegetative cells and heterocysts, thus setting the filament as the organismic and selective unit. Structurally, Anabaena is a Gramnegative bacterium. Its filaments are composed of cells with individual cytoplasmic membranes that are surrounded by a peptidoglycan layer, which is thickened in the septal regions between contiguous cells. In contrast, the cells share the periplasm, which is limited by an outer membrane that is continuous along the filament. In the septal regions proteinaceous complexes, called septal junctions, are found that are essential for maintaining long filaments and for intercellular molecular exchange, which can be probed with fluorescent tracers. The objective of this work is to understand the distinctive characteristics of cell division in Anabaena that allows propagation of a filament of communicated cells. Polymerization of FtsZ is the first recognizable event during cell division in most bacteria. FtsZ is highly conserved among bacteria. However, FtsZ of Anabaena possesses an N-terminal extension that is only found in other cyanobacteria and in chloroplasts, which are of cyanobacterial origin. To understand the role of this protein fragment, we have generated a mutant strain derived from Anabaena that conditionally expresses the native FtsZ and a version of this protein lacking the 50 Nterminal amino acids. Cells with different ratios of the two versions of the protein can be obtained under different growth conditions. Under conditions in which the short form of FtsZ is predominant, cell growth is maintained but the cells show a considerable increase in size and an altered morphology. This observation suggests a role of the deleted part of FtsZ in the coordination of cell growth and division. Panel 28 Tn-seq identification of meningococcal factors required for in vitro and in vivo colonization of human cells. Capel, Elenaa,b; Zomer, Aldert Lc; Barnier, Jean-Philippea,b,d; Nussbaumer, Thomase; Bole, Christinef; Izac, Brigitteb,g; Frapy, Erica,b; Meyer, Juliea,b; Euphrasie, Daniela,b ; BouzinbaSégard, Haniaab,g; Bille, Emmanuellea,b,d; Jamet, Annea,b,d,h; Cavau, Annea,b; Letourneur, Franckb,g; Bourdoulous, Sandrineb,g; Rattei, Thomase; Join-Lambert, Oliviera,b,d; Nassif, Xaviera,b,d and Coureuil, Mathieua,b. Institut Necker Enfants-Malades, INSERM U1151, Equipe 11, Paris, Francea; Université Paris Descartes; Sorbonne Paris Cité, Faculté de Médecine, Paris, Franceb; Department of Infectious Diseases and Immunology, Faculty of Veterinary Medicine, Utrecht University, The Netherlandsc; Assistance Publique – Hôpitaux de Paris, Hôpital Necker Enfants Malades, Paris, Franced; CUBE - Division of Computational Systems Biology, Dept. of Microbiology and Ecosystem Science, University of Vienna, Austriae; Plateforme génomique de l'Institut Imagine, Hôpital Necker, Paris, Francef; INSERM U1016, Institut Cochin, CNRS UMR8104, Paris, Franceg; Unidade de Microbiologia Molecular e Infecção, Instituto de Medicina Molecular, Lisbon, Portugalh. [email protected] Neisseria meningitidis is a leading cause of bacterial meningitis and septicemia affecting infants and adults worldwide. N. meningitidis is also a common inhabitant of the human nasopharynx and, as such, is highly adapted to its niche. During bacteremia, N. meningitidis gains access to the blood compartment where it adheres to endothelial cells of blood vessels causing dramatic vascular damages. Colonization of the nasopharyngeal niche and communication with the different human cell types is a major aspect of the N. meningitidis life cycle that is poorly understood. Here, highly saturated random transposon insertion libraries of N. meningitidis were engineered and the fitness of mutations during (i) routine growth, (ii) in vitro colonization of endothelial or epithelial cells in a flow device, and (iii) in vivo colonization of human vessels in a human skin grafted mice model was assessed, using Tn-seq analysis. This allowed the identification of genes essential for bacterial growth and genes specifically required for host cell colonization. In addition, after having identified the small non-coding RNAs (sRNA) located in intergenic regions, the phenotypes associated with mutation in those sRNAs were defined. 383 genes and 8 intergenic regions containing sRNA candidates were identified as essential for growth while 288 genes and 33 intergenic regions containing sRNA candidates were specifically required for host cell colonization. Besides the role of type IV pili for colonization, we observed a metabolic reorientation towards biosynthesis of cellular components, which is consistent with greater proliferation, and identified genes important for colonization of blood vessels in vivo. Panel 29 Análisis de la maquinaria molecular implicada en multicelularidad de Bacillus cereus Joaquín Caro Astorga1, Elrike Frenzel2, Antonio de Vicente1, Oscar Kuipers2 y Diego Romero1 1 Instituto de Hortofruticultura Subtropical y Mediterránea “La Mayora”, IHSM-UMA-CSIC. Departamento de Microbiología. Universidad de Málaga. Málaga. España. 2 Department of Genetics. Groningen Biomolecular Sciences and Biotechnology Institute (GBB). Universidad de Groningen. Países Bajos. [email protected] Bacillus cereus es una bacteria Gram-positiva usualmente implicada en brotes de intoxicaciones alimentarias así como en numerosas infecciones en pacientes hospitalizados que pueden ser mortales. Estos procesos infecciosos e intoxicaciones están directa o indirectamente relacionados con el ensamblaje de un biofilm que sirve de reservorio de células o protege ante condiciones adversas, las defensas del hospedador o la quimioterapia. Son numerosos los estudios sobre los genes implicados en la formación de biofilm en diversas especies, bajo condiciones de cultivo y tiempos no estandarizados, y bajo el supuesto de que unos genes de una especie se comportan de la misma forma en otras. Hemos analizado en detalle y de forma comparativa el transcriptoma de las células planctónicas y las de biofilm a diferentes tiempos bajo condiciones estándar. Nuestros resultados desvelan un gran número de genes implicados en biofilm, muchos de ellos de función desconocida, y dan luz sobre este grupo de loci del genoma de Bacillus cereus, que además suelen estar bien conservados en Gram-positivos. En este trabajo, nos centraremos en el grupo de los metabolitos secundarios y las toxinas, un sector del metabolismo clave en la interacción de Bacillus cereus con otras bacterias y sus hospedadores. Proyecto financiado por el Plan Estatal de Investigación, Desarrollo e Innovación orientada a los retos de la sociedad. Proyecto I+D+I AGL 2013-41939-R (ó AGL201452518-C2-1R), cofinanciado con fondos FEDER (UE) y la Unión Europea (European Research Council – Starting Grant 8.06 UE/60.8003. Panel 30 La combinación de cefotaxima y ciprofloxacina como alternativa terapéutica en infecciones por Enterococcus faecalis *Laura Carrilero, *Daniel Thomas-López, Stephanie Matrat, Alfonso Santos, Andreas Hoefer, Gabriel Moyano, Cristina Martinez-Ovejero, Bruno González-Zorn * Ambas personas han contribuido de igual manera Universidad Complutense [email protected], [email protected] Enterococcus faecalis es una bacteria que forma parte de la microbiota gastrointestinal del ser humano y de diversos animales. Bajo determinadas circunstancias, puede llegar a ocasionar infecciones graves como endocarditis y septicemia. Presenta una serie de resistencias intrínsecas a antibióticos que dificulta su tratamiento, destacando entre ellas la resistencia intrínseca a cefalosporinas. En el tratamiento de infecciones por enterococos se contemplan una serie de combinaciones antibióticas. La combinación de fluoroquinolonas y cefalosporinas no se considera una opción terapeútica a priori. Hemos estudiado el efecto sinérgico de la combinación ciprofloxacina y cefotaxima, y hemos visto que en presencia de ciprofloxacina la concentración mínima inhibitoria para la cefotaxima disminuye considerablemente en la cepa JH2-2. En una librería de transposición hemos observado que un mutante para una bomba transportadora de cationes y otro para una proteína reguladora tipo HD no presentan esa sinergia antibiótica. Dicha sinergia parece así mediada por estos determinantes genéticos, cuya implicación está por esclarecer. El estudio genético de los aislados clínicos podría contribuir a describir la utilidad de esta novedosa combinación antibiótica en las infecciones enterocócicas. Fuente de financiación: Laura Carrilero: Programa Tecnologías Avanzadas en Vigilancia Sanitaria TAVS-CM. S2013/ABI-2747. Comunidad de Madrid. Daniel Thomas-Lopez: Programa FPU del MECD. Ref. AP2012-2130 Panel 31 Effectors encoded outside of the locus of enterocyte effacement of enteropathogenic E. coli are essential for attaching and effacing (A/E) lesion formation in human intestinal biopsies Massiel Cepeda1*, Cedric N. Berger2, Gad Frankel2 and Luis Ángel Fernández1 (1) Department of Microbial Biotechnology. Centro Nacional de Biotecnología, CNB-CSIC. Darwin 3, Campus UAM, Cantoblanco, 28049 Madrid, Spain; (2) MRC Centre for Molecular Bacteriology and Infection, Department of Life Sciences, Imperial College London, London, United Kingdom (*) Presenting Author. E-mail: [email protected] Although most Escherichia coli isolates are harmless commensals of the gastrointestinal tract, some strains have acquired specific virulence factors, like pathogenicity islands (PIs), insertion elements (IEs), and prophages (PPs), to become highly adapted pathogens. The enteropathogenic E. coli (EPEC) is an important category of diarrhoeagenic bacteria causing acute and chronic diarrhea in infants. The hallmark of EPEC infection is the formation of attachment and effacement (A/E) lesion in the intestinal mucosa surface, which is characterized by the intimate attachment of the bacteria to the enterocyte, microvilli effacement, and the formation of actin-pedestal-like structures underneath the attached bacteria. EPEC is endowed of a 35 kb PI called the locus of enterocyte effacement (LEE) that encodes a type III secretion system (T3SS). Through the T3SS injectisome EPEC translocates multiple effector proteins into the host cell to subvert cellular functions in benefit of the infection. Six effectors are encoded within the LEE, whereas 17 effectors are located outside the LEE, in three IEs and four PPs. We have engineered a set of effector-less EPEC strains using suicide vectors to delete the repertoire of effector genes of the EPEC prototype strain E2348/69. The deletion strategy was based on plasmid integration by homologous recombination and the marker-less resolution of co-integrants. We infected human biopsies with these EPEC mutant strains to identify the effectors necessary for the induction of the A/E lesion in human intestinal biopsies. We found that some EPEC mutant strains that were able to induce the actinpedestal formation in HeLa cells in vitro were no able to induce the A/E lesion on human intestinal mucosa. Our results indicate that effectors located outside the LEE are essential to induce efficient A/E lesion on human intestinal biopsies. Panel 32 Las islas de patogenicidad regulan genes cromosómicos de virulencia en Staphylococcus aureus Mercedes Cervera Alamar, Katerina Guzman, Migle Ziemyte, Alejandro Toledo Arana, Iñigo Lasa y Mª Ángeles Tormo Mas Instituto de Investigación Sanitaria La Fe [email protected] S. aureus es un microorganismo ubicuo que se encuentra de forma natural en piel y mucosas pero en ocasiones puede actuar como agente patogénico tanto en hombre como en animales. Esto se debe a que posee gran número de factores de virulencia lo que le permite colonizar y sobrevivir en una amplia variedad de tejidos y de hospedadores. Muchos factores de virulencia se encuentran en elementos genéticos móviles (EGMs) entre los que destacamos las islas de patogenicidad (SaPIs). Las SaPIs son fragmentos de ADN que están implicadas en virulencia ya que contienen y diseminan genes que codifican para superantígenos, toxinas, genes relacionados con la formación de biofilm o genes de resistencia antibiótica. En este trabajo se implica por primera vez a las SaPIs en la regulación de factores de virulencia cromosómicos. Mediante un Tilling array se determinó que la isla inducida producía la sobreexpresión de dos factores de virulencia como son la estafiloxantina (STX), implicada en la evasión del sistema inmune y la N-acetyltransferasa (GNAT), implicada en resistencia a antibióticos. Mediante el análisis de diferentes mutantes de la isla, demostramos que el operón I de SaPIbov1 está implicado en la regulación de la expresión de STX y GNAT y tras un análisis más detallado determinamos que la Orf7 es la principal responsable del proceso y que la Orf adyacente, la Orf6, contribuye incrementando la expresión de la Orf7. Centrándonos en el mecanismo, obtuvimos distintos mutantes cromosómicos y fusiones transcipcionales que nos ayudaron a determinar que la Orf7 no se unía al promotor de STX ni de GNAT, sino que la Orf7 actuaba mediante un mecanismo de antiterminación. Por último, generalizamos este mecanismo al determinar que proteínas similares presentes en otras islas de S. aureus y de otras especies (S. epidermidis y S. chromogenes) actuaban del mismo modo. Panel 33 A comparative genomic strategy for target discovery and evolutionary inferences in pathogenic bacteria: Stenotrophomonas sp. as a case study Oscar Conchillo-Solé1, Daniel Yero1, Aniol Valera1, Xavi Coves1, Sonia Martinez-Servat1, Isidre Gibert1, Xavier Daura1,2 1 Institut de Biotecnologia i de Biomedicina (IBB) and Dept de Genètica i de Microbiologia, Universitat Autònoma de Barcelona (UAB), 08193 Bellaterra (Cerdanyola del Vallès), Barcelona, Spain. 2Catalan Institution for Research and Advanced Studies (ICREA), 08010 Barcelona, Spain [email protected]; [email protected]; [email protected] The genus Stenotrophomonas comprises organisms with clinical and environmental importance, and it is genetically and phenotypically heterogeneous. The most predominant species, S. maltophilia, besides its biotechnological importance, is a leading multi-drug resistant (MDR) pathogen in hospitals worldwide and has been associated with serious infections in pediatric and immunocompromised patients. Here we present a comparative genomic strategy aimed to find new targets for virulence and resistance in S. maltophilia while also to unravel the complex inter- and intraspecies phylogenetic relationships in this genus. We have sequenced four new relevant strains of S. maltophilia form clinical origin with the Illuminia Miseq system. Clean reads were assembled and contigs submitted to NCBI for their annotations. In addition, all available genome sequences at NCBI for the genus Stenotrophomonas that passed the quality criteria (60 in total) were used in this study. Our comparative approach was based on all-versus-all theoretical proteome comparisons and ortholog identification was performed using a full reciprocal reverse hit algorithm identifying, this way, those proteins unique for each organism and those shared by all of them (core-proteome) or a subgroup. With this data the algorithm grouped organisms by two different methods (both represented as a Neighor Network): by how many ortholog proteins they share or by their genetic distances based on the concatenated core proteins. Both clustering methods agreed to arrange the taxa in the same clades. Interestingly, some undetermined Stenotrophomonas and the only available strain of S. pavanii clustered into the clade including all S. maltophilia strains. All members of this clade share 41 unique proteins which include several antibiotic resistance related genes, membrane proteins, ABC transporters and regulatory proteins. Moreover, the clustering strategy highlighted a defined group of phenotypically relevant S. maltophilia MDR strains from clinical origins also sharing a set of unique proteins. Overall, this comparative genomic strategy could be applied to any group of bacteria to help understand the complex evolutionary process by which an organism becomes a pathogen. In addition, the set of unique proteins shared by a group of relevant species and/or strains could rise great interest as target candidates for resistance and virulence proteins. Acknowledgments This work has been supported by Spanish MICINN/FEDER (BIO2015-66674-R) and Catalan AGAUR (2009SGR-00108). Panel 34 Nuevas herramientas para reprogramar y estudiar el flujo de fosfoinosítidos en las membranas celulares de Saccharomyces cerevisiae. Julia M. Coronas-Serna, Teresa Fernández-Acero, Víctor J. Cid, María Molina Dpto. de Microbiología II, Facultad de Farmacia, Universidad Complutense de Madrid e Instituto de Investigaciones Sanitarias Ramón y Cajal (IRyCIS). Madrid. [email protected] Los fosfatidilinositoles desempeñan un papel regulatorio como segundos mensajeros en diversas rutas de señalización celular. En mamíferos, la generación in situ de fosfatidilinositol-3,4,5-trisfosfato (PtdIns-3,4,5P3) por la fosfatidilinositol-3-quinasa (PI3K) en la membrana plasmática, regula la respuesta a factores de crecimiento mediada por la ruta de la proteín-quinasa B. Nuestro grupo ha reconstruido esta vía en Saccharomyces cerevisiae mediante expresión heteróloga, lo que permite realizar estudios funcionales de los distintos componentes que la integran. En este modelo de “levadura humanizada”, la conversión del fosfatidilinositol4,5-bisfosfato (PtdIns-4,5P2), en PtdIns-3,4,5P3 por la actividad de la PI3K heteróloga puede detectarse por inhibición del crecimiento celular. Además, la expresión de PI3K proporciona una herramienta útil para estudiar en la levadura la función del PtdIns-4,5P2, lípido esencial de la membrana plasmática. Para estudiar en profundidad la localización subcelular de la actividad PI3K in vivo, se desarrolló un plásmido capaz de expresar simultáneamente dos marcadores fluorescentes específicos para las dos especies de fosfoinosítidos, PtdIns-4,5P2 y PtdIns3,4,5P3. Dichos marcadores consisten en el dominio PH de la fosfolipasa C δ [PH(PLCδ)] en fusión a GFP y el domino PH de Akt3 en fusión a mCherry, respectivamente. Asimismo, se desarrolló un vector dual similar que permite observar en paralelo la localización del fosfatidilinositol-4-monofosfato (PtdIns-4P) y PtdIns-4,5P2, mediante fusiones entre un fragmento de la proteína de Legionella pneumophila SidC y mCherry y la citada GFP-PH(PLCδ). Con el objetivo de restringir la eliminación del PtdIns-4,5P2 en S. cerevisiae a determinados compartimentos subcelulares, se diseñó una fusión génica de la subunidad catalítica de la PI3K de mamíferos (p110α) a la septina Cdc10, que dirige la actividad PI3K a la región de septación. La fusión Cdc10-p110α se localiza de manera exclusiva en el septo, entre la célula madre y la célula hija, aunque no es capaz de complementar el fenotipo de un mutante cdc10. El uso del plásmido con el doble marcador fluorescente ha permitido determinar que, a pesar de esta localización definida, la conversión de PtdIns-4,5P2 en PtdIns-3,4,5P3 afecta a toda la membrana plasmática, causando inhibición del crecimiento de manera similar a otras señales de reclutamiento general en membranas, como una caja N-terminal de miristoilación. Panel 35 Identification of a specific epitope on the surface of virulent Haemophilus parasuis strains Florencia Correa-Fiz, Nuria Galofré-Milà, Mar Costa-Hurtado, Virginia Aragón IRTA, Centre de Recerca en Sanitat Animal (CReSA, IRTA-UAB), Campus de la Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain [email protected] Haemophilus parasuis is an early colonizer of pigs that comprises strains of different degree of virulence. Non-virulent strains can be considered components of the upper respiratory tract microbiota, while virulent strains can invade systemic organs and cause fibrinous polyserositis (Glässer’s disease). Genomic comparison of virulent and nonvirulent strains led to the identification of a family of genes differentially associated to virulence, the virulence-associated trimeric autotransporters (vtaA). This gene family was classified in three groups, where group 1 was the only associated with the virulence of the strains. Additional data supporting the role of group 1 VtaA in virulence was also reported, and VtaA8 and 9 were shown to be surface exposed and play a role in phagocytosis resistance of virulent H. parasuis. Monoclonal antibody 69C6, which was produced against VtaA8, recognized the surface of virulent strains and reacted with all VtaAs from group 1 only. Expression of six fragments, spanning the passenger domain of the of the VtaA9 autotransporter (F1 to F6), allowed the localization of the epitope in the C terminal region, F6. In agreement, a BLAST analysis showed highly conservation of F6 in group 1 VtaAs, while it was absent in VtaAs from the other two groups. In silico prediction of MHC class I and II restricted epitopes by NetMHCpan tool identified several candidates in F6. Best scored epitopes for both MHC class I and II were synthetized and the 69C6 epitope was determined. Pigs vaccinated with the passenger domain of VtaA9 developed antibodies against the 69C6 epitope. Interestingly, sera from pigs infected with H. parasuis did not recognized the same epitope. The localization of the epitope on the 3D structure model, built using Haemophilus influenza trimeric autotransporter (Protein Data Bank ID: 3emo) as template, indicates that it is surface exposed. The fact that the epitope is in the close proximity to the translocator domain, and this domain is not included in the vaccine used, could explain the different antibody response in vaccinated or infected pigs. The specificity of this epitope would allow targeting exclusively virulent strains of H. parasuis, while maintaining the non-virulent microbiota strains. Panel 36 New tools to study autophagic flux in Chlamydomonas reinhardtii Inmaculada Couso*1, M. Esther Pérez-Pérez*1 Yonghua He2, James G. Umen2 and José L. Crespo1 1 Instituto de Bioquímica Vegetal y Fotosíntesis; Consejo Superior de Investigaciones Científicas-Universidad de Sevilla; Sevilla, Spain. 2 Donald Danforth Plant Science Center. St Louis MO 63132, USA. [email protected]. Cell homeostasis is tightly controlled by the self-degradative process of autophagy. This catabolic process balances energy and recycles cell components in a way cell can survive after being subjected to different stresses. Autophagy is a dynamic process that needs to be monitored as a whole. In this sense, autophagic flux is nowadays the most reliable way of showing how active the entire process is, from the inclusion of cargo within the autophagosome to the release of the recycled molecules back into the cytosol (1). In the green alga Chlamydomonas reinhardtii, accumulation of lipidated forms of ATG8 has been used as a way of monitoring autophagy (2). However, the information given by this assay is limited, as it only provides a snapshot of the whole process. In this study, we treated Chlamydomonas cells with Concanamycin A, an inhibitor of vacuolar type H+-ATPase (3) that has been widely used to inhibit autophagy in other organisms (1). Treatment of Chlamydomonas cells with Concanamycin A led to the accumulation of ATG8 due to blockage of autophagy flux. Moreover, we found out that the ribosomal protein RPS6 is degraded in Chlamydomonas when cells are exposed to different stress conditions, and accordingly RPS6 degradation was prevented in the presence of Concanamycin A. Our results revealed a strong correlation between the activation of autophagy and the downregulation of RPS6 abundance in Chlamydomonas in a reversible manner. Thus, we concluded that RPS6 must be part of the autophagosome cargo as its abundance is regulated via autophagy and can be used to monitor autophagy flux in Chlamydomonas. 1. Klionsky et al. (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12(1):1-222. 2. Pérez-Pérez ME, Florencio FJ and Crespo JL (2010) Inhibition of target of rapamycin signaling and stress activate autophagy in Chlamydomonas reinhardtii. Plant Physiol 152(4):1874-88. 3. Drose S et al. (1993) Inhibitory effect of modified bafilomycins and concanamycins on P- and V-type adenosinetriphosphatases. Biochemistry 32(15):3902-6. Panel 37 Involvement of two paralogous EcfG σ factors in the general stress response of the tetralin degrader Sphingopyxis granuli strain TFA Rubén de Dios, Eduardo Santero and Francisca Reyes-Ramírez Departmento de Biología Molecular e Ingeniería Bioquímica, Centro Andaluz de Biología del desarrollo (CABD/CSIC), Universidad Pablo de Olavide, 41013 Seville, Spain. [email protected] In natural habitats affected by ever-changing conditions, microorganisms constantly change their expression profiles in order to increase their chances of survival. Apart from specific stress responses, bacteria can trigger the so called ‘general stress response’ (GSR), which responds to a number of unrelated stresses. Generally, in Alphaproteobacteria, the regulation pathway consists of an alternative EcfG-type σ factor, its cognate anti-σ factor, a PhyR-type response regulator and at least one sensor histidine kinase (HK). When some kind of stress appears in the environment, the HK phosphorylates the PhyR regulator. Upon phosphorylation, PhyR is able to sequester the anti-σ factor, thus releasing EcfG to activate its target genes. Sphingopyxis granuli TFA is a facultative anaerobic Alphaproteobacteria capable of growing on the organic solvent tetralin as a sole carbon source. After the analysis of the recently annotated TFA genome, two paralogs of the EcfG σ factor, the anti-σ factor and the PhyR regulator were found, along with four possible HKs involved in triggering the cascade. Once ΔecfG, ΔecfG2 and ΔecfGΔecfG2 mutant strains were constructed, a phenotype screening was performed in order to address the function of each EcfG paralog in response to several stresses. Although the ΔecfG mutant did not show significant differences with respect to the wild type, the growth of the ΔecfG2 and the ΔecfGΔecfG2 mutants was seriously affected in the presence of a variety of stressing agents. Interestingly, preliminary dRNA-seq data show that the ecfG gene was strongly overexpressed when cells are grown aerobically with tetralin as carbon source, in stationary phase, and also under anaerobic conditions using nitrate as electron acceptor. However, the expression of ecfG2 was not significantly affected in these conditions. We have quantified the level of ecfG expression under these conditions in the wild type strain, and the ΔecfG, ecfG2 and ΔecfGΔecfG2 mutant strains by measuring β-galactosidase activity from lacZ fused to the ecfG promoter. Our results indicate that ecfG expression is maximal under carbon starvation and that ecfG expression is controlled by the EcfG2 paralog. This work is a first step in understanding the GSR regulatory network in this degrader bacterium. Panel 38 Identificación y caracterización de aislados humanos, animales y ambientales portadores del gen mcr-1 procedentes de Venezuela y España Jose Francisco Delgado-Blas1, Cristina Martinez Ovejero1, Gabriel Moyano1, Manuel Ares Arroyo1, Lorena Abadia Patiño2, Maite Muniesa3 y Bruno Gonzalez-Zorn1 1 Departamento de Sanidad Animal y Centro de Vigilancia Sanitaria Veterinaria (VISAVET), Facultad de Veterinaria, Universidad Complutense de Madrid, España 2 Departamento de Biomedicina, Instituto de Investigaciones en Biomedicina y Ciencias Aplicadas (IIBCA), Universidad de Oriente, Venezuela 3 Departamento de Microbiología, Facultad de Biología, Universidad de Barcelona, España [email protected] La diseminación de bacterias extremadamente resistentes ha posicionado a la colistina como antibiótico de último recurso. Tras el descubrimiento de mcr-1, el primer mecanismo transferible de resistencia a colistina, este gen ha sido detectado en múltiples Enterobacterias de diverso origen. El objetivo de este trabajo fue la detección y caracterización de mcr-1 en 93 Enterobacterias de origen humano, animal y ambiental procedentes de Cumaná (Venezuela) y en 195 aislados de origen ambiental de Barcelona (España). La presencia de mcr-1 se determinó por PCR. La identificación bacteriana se realizó mediante secuenciación del ARNr 16S y MALDI-TOF. El nivel de resistencia fue evaluado con ensayos de microdilución en placa. La secuenciación total del genoma se realizó mediante tecnología Illumina (MiSeq). La relación epidemiológica se estableció mediante PFGE. La caracterización del plásmido portador de mcr-1 se llevó a cabo mediante su transformación y determinación del grupo de incompatibilidad. En Cumaná, se detectaron 2 E. coli multirresistentes positivos a mcr-1 (uno de origen humano, que también portaba NDM-1, y otro aislado de un cerdo). La secuenciación masiva reveló que ambos estaban relacionados con cepas patógenas humanas. En Barcelona, se detectó mcr-1 en 30 aislados (29 E. coli y 1 K. pneumoniae), procedentes de dos plantas de tratamiento de aguas residuales. Fueron resistentes a múltiples antibióticos, además de a colistina. El PFGE mostró dos pulsotipos predominantes, presentes en ambas plantas. Tanto en Cumaná como en Barcelona, el plásmido portador de mcr-1 fue IncI2 en todos los transformantes obtenidos. La presencia de mcr-1 en dos E. coli patógenos de diferente origen refleja la diseminación de este mecanismo de resistencia en Cumaná. La combinación del mismo con otros genes, como NDM-1, supone un riesgo de diseminación de Enterobacterias causantes de infecciones intratables. La presencia de aislados portadores de mcr-1 pertenecientes al mismo pulsotipo en dos plantas de tratamiento de Barcelona refleja el éxito de esta asociación. Hasta la fecha, esta es la primera descripción del gen mcr-1 en Venezuela y de su coexistencia con NDM-1 a nivel mundial. Asimismo, se trata de la primera detección de mcr-1 en muestras de agua en España y la mayor prevalencia descrita de aislados ambientales positivos a este gen. Este trabajo ha sido financiado por las Ayudas para contratos predoctorales para la formación de doctores 2015 del Ministerio de Economía y Competitividad del Gobierno de España y cofinanciado por el Fondo Social Europeo. Panel 39 Regulación de las proteínas LPXTG de superficie de Listeria monocytogenes en condiciones de crecimiento a 4 ºC Alberto Díaz-Romero1, Francisco García-del Portillo1, Taurai Tasara2 y M. Graciela Pucciarelli1,3 1 Centro Nacional de Biotecnología - Consejo Superior de Investigaciones Científicas (CSIC) 2 Institute for Food Safety and Hygiene. University of Zürich, Switzerland 3 Departamento de Biología Molecular. Universidad Autónoma de Madrid. Centro de Biología Molecular “Severo Ochoa” – CSIC [email protected] Listeria monocytogenes es una bacteria patógena que causa graves infecciones por vía alimentaria debido a su alta capacidad para crecer a temperaturas de refrigeración (4 ºC). De entre las bacterias Gram-positivas, las pertenecientes al género Listeria presentan el número más elevado conocido hasta el momento de proteínas unidas covalentemente al peptidoglicano. En un término medio, todos los genomas secuenciados contienen más de 40 genes que codifican proteínas de superficie con un motivo LPXTG, que es reconocido para la unión al peptidoglicano. La función de muchas de estas proteínas es desconocida. En este trabajo se ha analizado el subproteoma de la pared celular de bacterias crecidas en condiciones de refrigeración (4 ºC) y comparado con el de bacterias creciendo a 37 ºC. Varias proteínas de la familia LPXTG disminuyen su presencia en esta fracción de pared a 4 ºC, mientras que una de ellas, Lmo0835, se induce esencialmente a baja temperatura. Los datos de RT-PCR cuantitativa sugieren que esta regulación ocurre a nivel posttranscripcional. Se ha realizado también un estudio similar en cepas deficientes en CspA, CspB y CspD, proteínas de respuesta a choque frío presentes en L. monocytogenes. Los resultados muestran una contribución de CspB a la regulación positiva de Lmo0835 a baja temperatura. Igualmente, la ausencia de CspA produce un defecto importante en la capacidad de crecer a 4 ºC y la ausencia de varias proteínas LPXTG. Análisis de microscopía electrónica y cromatografía por HPLC confirman la contribución importante de las proteínas Csp al metabolismo de la pared celular durante la adaptación de L. monocytogenes al frío. Panel 40 Rewinding the tape of evolutionary innovation of integron integrases J. A. Escudero, A. Nivina, J. Aguilar, M. Toll-Riera, A. Babic, C. Loot, and D. Mazel Institut Pasteur and Oxford University [email protected] / [email protected] Integrons are genetic platforms that allow bacteria to evolve rapidly through the acquisition of new genes encoded in cassettes and their differential expression1. These processes are governed by integron integrases (hereafter integrases), a subfamily of tyrosine (Y)-recombinases. Integrases are a molecular example of evolutionary innovation2. They acquired a new domain (I2) within the catalytic core that allowed recombining single stranded (ss-) DNA substrates, while retaining the ancestral activity on double stranded (ds-) DNA. In modern integrases the ancestral activity is very low. We sought to obtain hyperactive integrases on ds-DNA resembling their ancestors at the moment of evolutionary divergence from the rest of Y-recombinases. To do so we have carried out a series of directed evolution experiments. Notably, in order to explore a broader protein sequence space, we recoded the integrase into two alternative yet synonymous genes3. We randomized the three alleles and selected for enhanced recombination activity using selection cycles. We found 8 independent mutations that increased integrase activity. All mutations were inserted together into the three alleles, conferring a 100-fold increase in activity. To further improve the integrase activity, these new genes were re-evolved, yielding five new beneficial mutations, with some alleles showing activity increases of 1000-fold. Throughout our experiments we observed negative epistatic interactions between mutations. In order to have the greatest increase in activity with the smaller number of mutations, we designed an epistasis purification system. We built a library containing all possible combinations of the 13 mutations and subjected it to our selective cycles. We obtained alleles showing a 10.000 fold increase in activity with only 8 mutations. Mutations found included examples of parallel and divergent evolution, hitchhiking, residue re-evolution and epistasis. Their distribution reveals the structural constraints for the recombination of ds-DNA substrates. Hyperactivity on ds-DNA came with an asymmetric trade-off with ss-DNA. Mutations specific to the alternative alleles confirmed the exploration of a larger protein sequence space. Also, mutations within the I2 domain proved its pivotal role in the evolutionary innovation of ss-DNA recognition. We have rewound the tape of evolutionary innovation in integrases and obtained a hyperactive mutant enabling further structural studies. 1, The Integron: Adaptation On Demand. Escudero JA, Loot C, Nivina A, Mazel D. Microbiol Spectr. 2015 Apr;3(2):MDNA3-0019-2014. 2, Unmasking the ancestral activity of integron integrases reveals a smooth evolutionary transition during functional innovation. Escudero JA, Loot C, Parissi V, Nivina A, Bouchier C, Mazel D. Nat Commun. 2016 Mar 10;7:10937. doi: 10.1038/ncomms10937. 3, Synonymous genes explore different evolutionary landscapes. Cambray G, Mazel D. PLoS Genet. 2008 Nov;4(11):e1000256. doi: 10.1371/journal.pgen.1000256. Panel 41 La respuesta estricta media la dispersión del biofilm en Pseudomonas putida Rocío Espinosa-Portero, Patricia Calero, Carlos Díaz-Salazar, Aroa López-Sánchez y Fernando Govantes Centro Andaluz de Biología del Desarrollo. Universidad Pablo de Olavide/Consejo Superior de Investigaciones Científicas/Junta de Andalucía [email protected] La respuesta estricta es una respuesta a estrés conservada en bacterias. En condiciones de limitación de nutrientes, se activa la síntesis de las alarmonas guanosina 5’-difosfato 3’-difosfato (ppGpp), y guanosina 5’-trifosfato 3’-difosfato (pppGpp) [conjuntamente denominadas (p)ppGpp]. El (p)ppGpp regula directa e indirectamente la expresión de numerosos genes, en ocasiones con el concurso de la proteína auxiliar DksA (1). En la naturaleza las bacterias alternan una vida planctónica con la formación de comunidades sésiles rodeadas de una matriz extracelular polimérica, denominadas biofilms. Los fenómenos de dispersión programada promueven la transición del biofilm al estado planctónico en respuesta a señales fisiológicas o ambientales. Los biofilms de la bacteria Gram-negativa de suelo Pseudomonas putida experimentan una rápida dispersión en condiciones de estrés nutricional. La dispersión es el resultado de la proteolisis de la adhesina de alto peso molecular LapA por la proteasa periplásmica LapG. La actividad de LapG está regulada por la proteína transmembrana LapD en respuesta a cambios en los niveles del segundo mensajero 3',5'-diguanilato cíclico (di-GMPc)(2). Recientemente hemos demostramos que la fosfodiesterasa BifA es responsable de la hidrólisis de di-GMPc que dispara la respuesta de dispersión (3). En esta comunicación mostramos que la respuesta estricta media la inducción de la dispersión del biofilm en P. putida. Mediante el análisis fenotípico de mutantes con defectos en respuesta estricta y análisis de expresión génica in vivo e in vitro hemos elucidado los mecanismos implicados, y hemos completando el circuito de transducción de señales que conecta la señal inductora (limitación de nutrientes) con la respuesta (proteolisis de LapA) responsable de la dispersión del biofilm en P. putida. REFERENCIAS (1) Srivatsan y Wang (2008) Curr. Op. Microbiol. 11:100-105 (2) Boyd y O'Toole (2012) Ann. Rev. Cell. Dev. Biol. 28:439-462 (3) Jiménez-Fernández et al. (2015) Environ. Microbiol. Rep. 7:78-84 AGRADECIMIENTOS Este trabajo ha sido financiado por los proyectos BIO2010-17853 (MICINN) y BIO201342073-P (MINECO) Panel 42 Implicación de los sistemas de captación y reciclaje de ácidos grasos en la infección respiratoria por Haemophilus influenzae no tipificable Ariadna Fernández-Calvet1, Irene Rodríguez-Arce1, Goizeder Almagro1, Javier Moleres1, Sara Martí2, Begoña Euba1,2, José Antonio Bengoechea3, Javier Pozueta-Romero1, Junkal Garmendia1,2 1 Instituto de Agrobiotecnología, CSIC-Universidad Pública Navarra-Gobierno Navarra, Mutilva, Navarra; 2Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES), Madrid; 3Centre for Infection and Immunity, Queen’s University Belfast, UK [email protected] El patógeno oportunista Gram negativo Haemophilus influenzae no tipificable (descapsulado, HiNT) es frecuentemente aislado del pulmón de pacientes respiratorios crónicos con patologías asociadas al tabaquismo, y es responsable de un porcentaje significativo de las exacerbaciones periódicas sufridas por este tipo de pacientes. El potencial citoprotector de varios ácidos grasos de cadena larga (LCFA) ha despertado interés terapeútico frente a patologías respiratorias crónicas por su carácter antiinflamatorio, si bien desconocemos su posible impacto en la colonización e infección crónica de las vías aéreas por HiNT. La asimetría de la membrana externa (OM) de HiNT está basada en la presencia mayoritaria de fosfolípidos (PL) en la cara interna, y en la presencia mayoritaria de lipooligosacárido (LOS) y minoritaria de PLs en la cara externa. La ruta de síntesis de ácidos grasos bacteriana genera acil-ACP y β-hidroxiacil-ACP, empleados en la síntesis de PLs y del lípido A del LOS. HiNT dispone del transportador FadL, a través del cual se produce la unión y transporte de LCFA exógenos que, convertidos en acil-CoA, son utilizados en biosíntesis de PLs o como fuente de carbono y energía mediante βoxidación. Dado que los ácidos grasos exógenos son utilizados en la biosíntesis de PLs, planteamos la existencia de una posible relación funcional entre FadL y el sistema VacJ/Mla, encargado del reciclaje de PLs a través de su transporte retrógrado al citosol para evitar su acumulación en la cara externa de la OM de HiNT. Mediante la generación de mutantes en las cepas HiNT375 y RdKW20, analizamos el papel de FadL y VacJ en el crecimiento bacteriano; la composición de PL y ácidos grasos totales, y estructura del lípido A bacteriano; la permeabilidad-resistencia bacteriana a un panel de agentes bactericidas de distinta naturaleza; la interacción de HiNT con células fagocíticas y epitelio respiratorio; la progresión de la infección pulmonar in vivo. Los resultados obtenidos permiten desacoplar funcionalmente captación y reciclaje de ácidos grasos, y asignar funciones específicas diferenciadas a los sistemas FadL y VacJ durante la infección respiratoria por H. influenzae. Panel 43 Real-Time PCR aplicada al estudio de expresión de los genes que codifican para Péptido Sintasas No Ribosomales (NRPS) en el hongo fitopatógeno Botrytis cinerea Ana Fernández-Morales, Cristina M. Ortíz-Viejo, Victoria E. González-Rodríguez, María Carbú, Carlos Garrido, Jesús M. Cantoral Universidad de Cádiz. Dpto. de Biomedicina, Biotecnología y Salud Pública, Laboratorio de Microbiología, Facultad de Ciencias de Mar y Ambientales, 11510, Puerto Real, Cádiz. [email protected]; [email protected] Botrytis cinerea es un hongo fitopatógeno causante de la enfermedad “Podredumbre gris”, que afecta a una amplia variedad de cultivos (frutales, plantas ornamentales, hortalizas, etc.), haciéndolo responsable de cuantiosas pérdidas económicas. La patogenicidad de B. cinerea, está relacionada con diferentes mecanismos de infección, siendo capaz de excretar numerosas enzimas capaces de degradar la pared celular, toxinas, y un amplio número metabolitos secundarios, algunos de ellos aún sin identificar. Entre todas estas posibilidades, existe un tipo de metabolitos secundarios, que además de B. cinerea, son sintetizados por otros muchos microorganismos: los péptidos de origen no ribosomal (NRPs). Estos péptidos, poseen un bajo peso molecular y tienen características de sideróforos. Su síntesis tiene lugar en complejos multienzimáticos denominados Péptido Sintasa No Ribosomales (NRPS). En el genoma de B. cinerea se han localizado hasta un total de 9 genes que codifican para enzimas NRPS, que deben por tanto de participar en la biosíntesis de estos compuestos sideróforos. El hierro es un ión imprescindible para el crecimiento de cualquier organismo vivo, y por ello, muchos microorganismos sintetizan sideróforos, moléculas que presentan alta afinidad por los iones de hierro. Los sideróforos son excretados al exterior formando complejos con los iones de hierro del ambiente para posteriormente ser transportados hacia el interior del microorganismo. Este tipo de metabolismo puede jugar un papel importante durante el proceso de infección desarrollado por B. cinerea, y resulta importante intentar conocer bajo qué condiciones se expresan estos genes y por tanto se sintetizan estos sideróforos. El presente trabajo, muestra los resultados del estudio de las secuencias de los 9 genes que codifican para enzimas NRPS, el diseño de “primers” específicos para cada uno de estos genes y la optimización de las condiciones de amplificación por Real-Time PCR que permitan llevar a cabo estudios de expresión en diferentes condiciones experimentales. Agradecimientos: Este trabajo ha sido financiado con ayudas del Ministerio de Economía y Competitividad (Fondos FEDER, Fondos Europeos de Desarrollo Regional), mediante los proyectos MINECO: AGL2012-39798-C02 y AGL2015-65684-C2-2-R; UCA18DGUEII02. Panel 44 Estudio de la ruta Gac-rsm en Pseudomonas syringae pv tomato DC3000 María Dolores Ferreiro, Joaquina Nogales, Gabriela A. Farias, María-Trinidad Gallegos Estación Experimental del Zaidín (CSIC), Profesor Albareda 1, 18008-Granada [email protected] Pseudomonas syringae pv. tomato DC3000 (Pto DC3000) es una bacteria fitopatógena que infecta plantas de tomate y Arabidopsis thaliana. La motilidad bacteriana juega un papel fundamental en el establecimiento de la infección y requiere la presencia de flagelos y la producción del biosurfactante siringafactina. La ruta Gac-rsm es una ruta de regulación global que controla virulencia, motilidad, producción de metabolitos secundarios, metabolismo del carbono y otros procesos en Pto DC3000 (Chaterjee et al., 2003). Sin embargo y a pesar de su importancia, esta ruta aún no ha sido caracterizada a nivel molecular. En otras Pseudomonas, la ruta se inicia con la activación del sistema de dos componentes GacS/GacA, que activa la transcripción de los ARNs no codificantes rsm, cuyo papel es secuestrar los reguladores postranscripcionales CsrA impidiendo que ejerzan su función. En nuestro grupo de investigación hemos identificado los componentes de la ruta en Pto DC3000 y estamos construyendo y caracterizando mutantes en dichos componentes. Hemos generado mutantes carentes de los genes rsmY, rsmZ, csrA-2 y csrA-3 y hemos estudiado fenotipos cruciales para la infección como la motilidad y la producción de biosurfactantes, pero también la producción de exopolisacáridos, la morfología bacteriana y su comportamiento in planta mediante ensayos de infección en plantas de tomate. Los resultados obtenidos muestran que CsrA-3 y, en menor medida, CsrA-2, pero no CsrA-1, regulan negativamente la motilidad y la producción de biosurfactante y alginato en Pto DC3000, pero su mutación no altera significativamente el progreso de la enfermedad en la planta. Los mutantes en rsmY, rsmZ y el doble mutante rsmYZ presentan un fenotipo similar a la cepa silvestre, tanto en condiciones de laboratorio como in planta, lo que sugiere que los otros ARNs tipo rsm presentes en Pto DC3000 también pueden estar contribuyendo a los fenotipos estudiados. Panel 45 Actinobacteria: una visión desde la comparación de genomas completos Abengozar D, Castejon M y Garcia MJ Facultad de Medicina. Universidad Autonoma de Madrid [email protected] La clase Actinobacteria es uno de los taxones más numerosos en cantidad y variedad de bacterias. Hasta el momento, la relación existente entre los taxones que componen esta clase, se basaba en caracteres fenotípicos o en la comparación de la secuencia del gen codificante para el RNA ribosomico 16S (Manual de Bergey). El acceso a la secuencia completa de los genomas bacterianos en las bases de datos (ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/) posibilita la comparación global del contenido genético de las Actinobacterias, pudiéndose evaluar la relación de los diferentes taxones que la componen desde una perspectiva más filogenética. Con este fin, hemos aplicado varios métodos de comparación de genomas. Cada método analiza el genoma en base a diferentes parámetros, lo que permite no solo obtener datos mas fiables, sino, además, evidenciar diferencias posibles en los mecanismos evolutivos, puestos en marcha en los distintos taxones. Los métodos utilizados han sido: 1) Firma genómica, compara la abundancia de di-tetra nucleótidos entre los genomas, calculando un valor de diferencia d entre los mismos; 2) Distancia genómica, se basa en la comparación mediante BLAST de las secuencias completas de genomas, el valor de la distancia calculado aporta información in silico del grado de similitud DNA-DNA digital (dDDH); 3) Identidad media de nucleótidos, compara secuencias de nucleótidos de genes que codifican para proteínas conservadas, que se considera son los que soportan mayor presión evolutiva; 4) Orden de genes, determina el grado de reorganización de genes en los genomas. El estudio realizado con 95 Genomas de la clase, en concreto de las familias Micrococcales, Frankiales, Propionibacteriales, Corynebacteriales y Pseudonocardiales evidencia la necesidad de revisar la clasificación aceptada de la misma en base a la información derivada de los genomas completos. Se muestran dos ejemplos mas en detalle, uno relativo a relaciones de rangos superiores, como es el orden Microccocales, en el que aparecen los géneros Kocuria y Rothia menos relacionados en base a sus genomas que lo determinado por métodos previos. El segundo ejemplo es el género Mycobacterium donde se han detectado genomas erróneamente asignados a una especie determinada y otros genomas considerados de especies diferentes, cuyo análisis muestra que no lo son. Panel 46 Does aggregation change S. aureus physiology? Pedro Dorado1, Begoña García1, Jaione Valle1, Cristina Solano1 and Iñigo Lasa1 1 Navarrabiomed - Universidad Pública de Navarra – IdiSNA [email protected] Bacterial aggregation can occur when bacteria come into contact with a surface during biofilm development and also while bacteria are freely swimming in liquid media. Bacterial aggregation usually correlates with the production of exopolysaccharides and/or specific surface proteins. In this work, we explore the concept that the contact between bacteria imposed by the aggregation process causes specific changes in bacterial physiology. The concept of sensing bacterial contact implies the existence of an apparatus that allows the perception of neighboring bacteria and provokes a biochemical cascade following the contact with other bacteria inside the aggregates. Staphylococcus aureus, a commensal and pathogenic microorganism responsible for a wide range of serious acute and chronic diseases in humans and animals, has the capacity to form aggregates when bacteria overexpresses either the exopolysaccharide PIA/PNAG or surface proteins such as SasG and Bap. Bap is a high molecular weight surface protein that connects staphylococcal cells through a self-assembly process resulting in the formation of amyloid-like aggregates. In this study we have used the Bap mediated bacterial aggregation process to identify the physiological changes undergone by S. aureus in the presence of Bap amyloid fibers. We have compared total protein extracts obtained from aggregated and planktonic bacteria using a differential proteomic analysis (label-free). Analysis of the proteomic results revealed that more than 200 proteins were differentially expressed when both conditions were compared, including global regulators and twocomponent signal transduction systems. Aggregation repressed the expression of proteins involved in peptidoglycan and cell wall synthesis, purine and pentose biosynthesis and recruitment and regulation of iron, whereas it induced the expression of proteins involved in DNA repair, genome stability, and teichoic acid biosynthesis amongst others. These results will be discussed in the presentation. Panel 47 StdE y StdF, reguladores globales de la transcripción en Salmonella enterica Lucía García-Pastor, Elena Puerta-Fernández y Josep Casadesús Departamento de Genética, Facultad de Biología, Universidad de Sevilla [email protected] Salmonella enterica es una bacteria gram-negativa patógena, causante de enfermedades gástricas y sistémicas en una gran variedad de organismos. El genoma de S. enterica serovar Typhimurium contiene 11 operones que codifican fimbrias implicadas en la colonización del intestino y tal vez de otros órganos. Uno de ellos es std, un operón adquirido por transferencia horizontal cuya expresión se encuentra reprimida en condiciones de laboratorio y se activa en el intestino de animales. El operón std está reprimido por metilación Dam, y su activación es dependiente de HdfR, un regulador transcripcional de tipo LysR. Se desconocen las señales que activan la expresión del operón y si además de la metilación Dam y HdfR existen más elementos implicados en dicha activación. En nuestro laboratorio se ha descrito previamente que los productos de los dos genes más distales (stdE y stdF) reprimen la expresión de la isla de patogenicidad-1 (SPI-1) de S. enterica. En este trabajo, con el fin de identificar otros genes regulados por las proteínas StdE y StdF, se ha realizado un análisis transcriptómico completo. Sorprendentemente se ha visto que StdE y StdF regulan múltiples genes, y el análisis fenotípico ha confirmado que StdE y StedF están involucradas en la regulación global de S. enterica, participando en virulencia, movilidad, quimiotaxis, conjugación, etc. Además, el análisis ChIP-seq sugiere que la regulación transcripcional por StdE y StdF probablemente tiene lugar de forma directa en la mayoría de los casos. Datos recientes de nuestro laboratorio parecen indicar que una pequeña subpoblación (el 2%) expresa el operón std en condiciones de laboratorio, planteando así la posibilidad de que la expresión de std esté sujeta a cambio de fase. Panel 48 Papel de la proteína Argonauta de T. thermophilus en los diferentes procesos de entrada de DNA en las células A. Blesa, N. García-Quintáns, MªL. del Pozo, M. Mencía y J. Berenguer Centro de Biología Molecular Severo Ochoa (UAM-CSIC). Facultad de Ciencias. Universidad Autónoma de Madrid. 28049 Madrid, España [email protected] Las proteínas Argonauta (Ago) están presentes en todos los eucariotas, y en algunas arqueas y bacterias, mostrando entre ellos diferentes mecanismos de actuación. En eucariotas están implicadas en la regulación de la expresión génica, mientras que en procariotas participan en la defensa del hospedador contra DNA exógeno. En Thermus thermophilus la proteína argonauta (TtAgo) está involucrada en un mecanismo de interferencia guiado por ssDNA que limita la incorporación DNA exógeno a la célula cuando éste entra por competencia natural1. Sin embargo, dicha interferencia no se observa cuando el mismo DNA es transferido por contacto desde otra bacteria del mismo género aunque utilice el mismo aparato de competencia natural, lo que implica una capacidad discriminatoria frente a la vía de entrada de DNA relacionada con algún tipo de sistema de activación. El estudio de la capacidad discriminatoria de TtAgo frente a la vía de entrada de DNA constituye un objetivo central de nuestro laboratorio. Para dilucidar los mecanismos por los que TtAgo se activa, y su papel en la fisiología de T. thermophilus, se realizaron diversos tipos de ensayos. Por un lado, purificación de la proteína recombinante y ensayos in vitro para estudiar su capacidad de corte y las condiciones de adquisición de las guías. Por otro lado, comparamos la eficiencia de transformación de T. thermophilus silvestres frente a mutantes Δago con DNA plasmídico y genómico acompañado o no por guías de ssDNA. Nuestros resultados mostraron una relación entre la eficiencia de transformación y la especificidad de las guías de ssDNA que entran en las células. Finalmente, observamos efectos pleiotrópicos en los mutantes Δago sugiriendo la participación de esta proteína en procesos de regulación celular, además de como sistema de defensa. Nuestros datos indican que la respuesta de este microorganismo a la presencia de DNA exógeno podría deberse a que la proteína forma un complejo secundario con el ssDNA-guía durante su propia síntesis. Los resultados sugieren que T. thermophilus es capaz de discriminar entre los mecanismos de entrada de DNA en las células a través de la activación de la proteína argonauta, como parte de un sistema específico de defensa. Referencias (1) Swarts, D. C., Jore, M. M., Westra, E. R., Zhu, Y., Janssen, J. H., Snijders, A. P., Wang, Y., Patel, D. J., Berenguer, J., Brouns, S. J. J. & van der Oost, J. (2014). DNAguided DNA interference by a prokaryotic Argonaute. Nature, 507 (7491): 258-261. Agradecimientos. Este trabajo ha sido financiado por los proyectos BIO2013-44963-R, y BIO2013-50068-EXP. AB disfruta de una beca FPI Panel 49 Implicación del pequeño ARN SuhB en la represión catabólica de los genes de degradación de tetralina en Sphingopyxis granuli estirpe TFA Inmaculada García-Romero, Eduardo Santero y Belén Floriano Centro Andaluz de Biología del Desarrollo, Universidad Pablo de Olavide y Departamento de Biología Molecular e Ingeniería Bioquímica. Sevilla, España. [email protected] Sphingopyxis granuli estirpe TFA es una bacteria Gram-negativa de la familia de las αproteobacterias capaz de utilizar el solvente orgánico tetralina como única fuente de carbono y energía (1). Los genes estructurales (genes thn) se organizan en dos operones divergentes (B y C) que se inducen en presencia de tetralina. Formando parte del operón C se encuentran los genes reguladores thnR, que codifica un regulador tipo LysR, y thnY, que codifica un co-activador de ThnR. En ausencia de tetralina, thnRY tienen una expresión basal desde un promotor interno constitutivo. Además de la inducción en presencia de tetralina, la expresión de los genes thn está sujeta a represión catabólica por fuente de carbono preferencial (RCC). El mecanismo de activación específica se ha estudiado en profundidad (2,3), si bien aún no se conocen los elementos implicados en la represión de los genes thn cuando en el medio, además de tetralina, está presente otra fuente de carbono preferencial, como el β-hidroxibutirato (βHB). Los únicos mutantes obtenidos hasta el momento muestran una desrepresión parcial y están afectados en la síntesis del gránulo de reserva de polihidroxibutirato (4). La comparación de la expresión génica global en células crecidas en tetralina o en β-HB puso de manifiesto la presencia de un pequeño ARN (sRNA) con expresión preferencial en células crecidas en βHB, predicho en el genoma de TFA por el software Infernal 1.1 (5) y perteneciente a la familia RF00519 (SuhB) sólo descrita en α-proteobacterias. En un mutante carente de SuhB los genes thn se encuentran parcialmente desreprimidos en condiciones de represión catabólica. La búsqueda de posibles dianas de SuhB utilizando el software IntaRNA (6) identificó, entre otros, al extremo 5’ del mRNA de thnR, localizándose la interacción de SuhB en la región de unión al ribosoma. El efecto de esta posible interacción sobre los niveles de ThnR se ha analizado mediante Western Blot en condiciones de represión catabólica, detectándose más cantidad de ThnR en la estirpe mutante que en la silvestre. Los datos disponibles hasta el momento indican que SuhB es uno de los elementos que interviene en la represión catabólica de los genes thn en Sphingopyxis granuli estirpe TFA. (1) García-Romero I, Pérez-Pulido AJ, González-Flores YE, Reyes-Ramírez F, Santero E, Floriano B. Genomic analysis of the nitrate-respiring Sphingopyxis granuli (formerly Sphingomonas macrogoltabida) strain TFA. BMC Genomics. 2016;17:93. doi:10.1186/s12864-016-2411-1. (2) López-Sánchez A, Rivas-Marín E, Martínez-Pérez O, Floriano B, Santero E. Co-ordinated regulation of two divergent promoters through higher-order complex formation by the LysR-type regulator ThnR. Molecular Microbiology. 2009;73(6):1086-1100. doi:10.1111/j.1365-2958.2009.06834.x. (3) Rivas-Marín E, Floriano B, Santero E. Genetic dissection of independent and cooperative transcriptional activation by the LysR-type activator ThnR at close divergent promoters. Scientific Reports. 2016;6:24538. doi:10.1038/srep24538. (4) Martín-Cabello G, Moreno-Ruiz E, Morales V, Floriano B, Santero E. Involvement of poly(3hydroxybutyrate) synthesis in catabolite repression of tetralin biodegradation genes in Sphingomonas macrogolitabida strain TFA. Environmental Microbiology Reports. 2011;627-631. doi: 10.1111/j.1758-2229.2011.00273.x. (5) Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933-2935. doi:10.1093/bioinformatics/btt509. (6) Busch A, Richter AS, Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008;24(24):2849-2856. doi:10.1093/bioinformatics/btn544. Panel 50 Caracterización fisiológica y molecular de Brettanomyces bruxellensis en presencia de ácidos débiles Godoy, L.1; Martinez, C1,2; Ganga, MA.1,3 1 Laboratorio de Biotecnología y Microbiología Aplicada. Facultad Tecnológica. Universidad de Santiago de Chile, 9170201, Estación Central, Santiago, Chile227184506 – 2 Centro de Estudios en Ciencia y Tecnología de los Alimentos. Universidad de Santiago de Chile. 3 Millennium Nucleus for Fungal Integrative and Synthetic Biology (MN-FISB) 120043 [email protected] Brettanomyces bruxellensis ha sido descrita como la principal levadura contaminante en vino, debido a que es capaz de convertir los ácidos hidroxicinámicos presentes naturalmente en el mosto de uva en derivados fenólicos. Estos compuestos son ácidos débiles, presentan un carácter lipofílico y se ha descrito que presentan actividad antimicrobiana inhibiendo el crecimiento de muchos microorganismos. Para evaluar el efecto de estos ácidos sobre el crecimiento de B.bruxellensis, las cepas L1359 y L2480 fueron cultivadas en medio YNB en ausencia y presencia de ácido p-cumárico (ApC). La presencia de 100 ppm de ApC en el medio de cultivo provocó un aumento en la duración de la fase lag en ambas cepas, además de una reducción en la velocidad específica de crecimiento (µ) para la cepa L2480. Sin embargo, para la cepa L1359 crecida en presencia de ApC, µ aumenta con respecto a la condición control. Asimismo se calculó el porcentaje de inhibición de crecimiento, siendo para la cepa L2480 de 9.8%, y para la cepa L1359 de -27%, lo que se traduce en que en presencia de ApC, esta última cepa mejora la cinética de crecimiento aumentando la velocidad específica de crecimiento. Por otro lado, se realizó un análisis comparativo del transcriptoma de ambas cepas, lo que nos ha permitido plantear un modelo en el cual la entrada de ApC a la célula genera un estrés generalizado en el cual se induce la expresión de bombas de protones y de eflujo de compuestos tóxicos. Estás últimas podrían estar involucradas en la salida de fuentes de nitrógeno como aminoácidos o alantoína, que al disminuir en concentración gatillarían la expresión de genes del metabolismo del nitrógeno. Este conocimiento permitirá diseñar nuevas herramientas en el control de este microorganismo causante de millonarias pérdidas económicas en la industria vitivinícola. Agradecimientos: CONICYT Postdoctorado/FONDECYT 3140083 Panel 51 Validación funcional de un sistema de expresión y secreción de antígenos heterólogos en la vacuna contra la tuberculosis MTBVAC Juan Calvet, Jesús Gonzalo-Asensio, Carlos Martín y Esther Broset* Grupo de Genética de Micobacterias, Departamento de Microbiología y Medicina Preventiva. Facultad de Medicina, Universidad de Zaragoza, 50009 Zaragoza *[email protected] El nuevo candidato a vacuna frente a la tuberculosis (TB), MTBVAC, es una bacteria viva atenuada derivada de una cepa de Mycobacterium tuberculosis. Esta vacuna se caracteriza por la inactivación de dos genes previamente descritos como implicados en la virulencia de M. tuberculosis (Arbues et al. 2013). Entre otros fenotipos, MTBVAC se caracteriza por presentar una mayor secreción de sustratos por la vía TAT (Twin Arginine Translocation). Mediante un mecanismo regulado por phoP, se transcribe un RNA no codificante denominado mcr7, que a su vez inhibe la traducción de la proteína TatC, siendo esta última un constituyente esencial del sistema TAT. La inactivación de phoP en MTBVAC conlleva un aumento de la traducción de TatC y por tanto el aumento de secreción de los sustratos por esta ruta (Solans et al. 2014). En este trabajo, y dada la capacidad adyuvante intrínseca de las micobacterias, se pretende utilizar este fenotipo de MTBVAC para expresar y secretar antígenos heterólogos. Para ello se clonaron diversos péptidos señal de sustratos de la ruta TAT (ssAg85A, ssAg85B, ssAg85C, ssβ-Lac, ssRv0203 y ssRv1987) en fase con el gen gfp y bajo el control de un promotor fuerte derivado del micobacteriofago L5. La correcta transcripción de las construcciones anteriores se confirmó mediante qRT-PCR y la expresión de GFP se verificó mediante FACS. La evaluación de la fluorescencia en la fracción secretada y en el pellet bacteriano permitió identificar que las ssAg85A y ssRv1987 presentan la mayor eficacia de secreción. Este trabajo proporciona una prueba de concepto para el potencial uso de estos péptidos señal en MTBVAC en la construcción de vacunas recombinantes multivalentes. Referencias: Arbués et al. (2013). Vaccine 31(42):4867-73 Solans et al. (2014). PLoS Pathog.10(5):e1004183. Financiación: Esther Broset es beneficiaria de una beca BES-2012-052937 del Ministerio de Economía y Competitividad de España. Este trabajo se ha financiado a cargo de los proyectos BIO2014-5258P y TBVAC2020. Panel 52 Transferencia horizontal de factores de virulencia contenidos en elementos genéticos móviles de Staphylococcus aureus en pacientes con fibrosis quística Katerina Guzman, Mercedes Cervera Alamar, Migle Ziemyte, Miguel Martí, Alicia Hernández, Amparo Solé y Mª Ángeles Tormo-Mas Grupo de Infeccion Grave. Instituto de Investigación Sanitaria La Fe, Valencia [email protected] Staphylococcus aureus es una importante bacteria patógena en pacientes con Fibrosis Quística (FQ), destacando la aparición de cepas multirresistentes a los antibióticos disponibles. La enorme versatilidad de S. aureus como bacteria patógena se debe a su habilidad para persistir y multiplicarse en diferentes ambientes junto con su capacidad para producir una gran variedad de factores de virulencia, la mayoría de las cuales están codificadas en elementos genéticos móviles (EGM), como bacteriófagos e islas de patogenicidad. Uno de los importantes factores de virulencia de Staphylococcus es su capacidad de formar biofilm, ya que le permite persistir en el foco de infección. Este trabajo se centra en la caracterización de cepas de S. aureus aisladas de los pacientes con Fibrosis Quística, así como estudiar la frecuencia y posibles cambios de elementos genéticos móviles (EGMs) y factores de virulencia. Analizamos los aislado de 118 pacientes, de los cuales, el 55,1 % obtuvimos un único aislado, el 37,3% obtuvimos aislados del mismo ST al menos dos veces a lo largo del periodo de estudio y el 7,6% obtuvimos aislados con ST diferentes. De las muestras analizadas los MLSTs más frecuentes fueron: ST-30 (12%), ST-5 (9%), ST-45, 125, 121 y otros (7%), ST-398 y ST-8 (6%). Destacar que 16% presentaron un MLST nuevo. Con respecto a los EGM, el 100% de los asilados analizados dio positivo para alguna integrasa de fago o de isla de patogenicidad. Además 11 pacientes presentaron cambios en la composición de bacteriófagos y/o islas de patogenicidad. El estudio funcional de los bacteriófagos mostró que el 70% de ellos producían lisis tras su inducción y el 24% de las SaPIs son inducidas por fagos endógenos. Referente a otros factores de virulencia, destacar que el 20% de cepas resultaron ser MRSA (SCCmecA positivas) y más del 80% son β-lactamasa positivos. Más de 90% de cepas presentaron capacidad de formación de biofilm, y 32,3% de ellas son fuertemente formadoras. En resumen, destacar la elevada presencia de SaPIs y bacteriófagos, y la capacidad de estos para actuar como vectores de transferencia horizontal y diseminar factores de virulencia. Por último, mencionar la gran capacidad para formar biofilm de estos aislados. Panel 53 Growth-phase dependent translation of the aminoglycoside resistance methylase ArmA Andreas Hoefer, Belen Gutierrez, Cristina Martinez-Ovejero, Jose Francisco DelgadoBlas, Alfonso Santos-Lopez, Daniel Thomas-Lopez, Bruno Gonzalez Zorn Departamento de Sanidad Animal y Centro de Vigilancia Sanitaria Veterinaria (VISAVET), Facultad de Veterinaria, Universidad Complutense de Madrid, España. [email protected] In the last decade an increasing number of 16S rRNA methyltransferases conferring unusually high-level pan-resistance to aminoglycosides have been rapidly disseminating amongst clinically relevant Gram-negative pathogens. Phenotypic characteristics coupled with in silico modelling have identified a putative regulatory domain in the 5’ UTR of armA, the most predominant 16S methyltransferase worldwide. By performing rapid amplification of cDNA ends (RACE) we found that the full 5’ UTR of the armA transcript is 139 nucleotides. This region along with its corresponding promoter is 100% conserved in all genetic environments of arma identified to date. Phenotypic changes upon truncation of the 5’ UTR, such as MIC reduction, demonstrate the involvement of the upstream region in the expression of the resistance determinant. To further elucidate the role of the armA upstream domain, we constructed 5’ UTR:lacZα reporter constructs. The results indicated a constitutive expression of ArmA both in the presence and absence of aminoglycosides. We then designed and performed a qRT-PCR, which also showed a constitutive expression of armA, suggesting that regulation could be dependent on a post-transcriptional mechanism. To investigate the translational frequency of active ArmA we then tagged the resistance methylase with a FlagTag in its C-terminus. The Westren Blot results revealed the differential expression of the truncations, which directly correlate to the phenotypic changes previously observed. To further characterise methylase expression throughout the life-cycle of the cell, we performed comprehensive Western blot growth curves. Under the control of the full-length 5’ UTR there is not only a significant increase in the overall expression of armA but also a significant initial peak early in the exponential phase. Our results are the first to highlight the importance of posttranscriptional regulation of acquired 16S rRNA methylases, where the presence of the active enzyme correlates with the number of ribosomes to be methylated. This project has received funding from the European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement N° 289285. Panel 54 A novel H-NS paralogue in the enteroaggregative E. coli strain 042 1 Prieto, A.1, Bernabeu, M.1, Hüttener, M.2, Juárez, A.1,2* Departamento de Genética, Microbiología y Estadística. Facultad de Biología. Universidad de Barcelona, Barcelona, España. 2 Instituto de Bioingeniería de Cataluña (IBEC), Barcelona, España. *[email protected] Bacterial nucleoid-associated proteins play a dual role. They contribute to nucleoid structure and, in addition, modulate gene expression. The H-NS protein is widesperead in γ-Proteobacteria and has been best studied in E. coli and related genera. H-NS plays a dual role, both as an architectural protein that contributes to the nucleoid structure and as a global modulator of gene expression. The E. coli hns gene encodes a 137-amino-acid protein with a molecular mass of 15.4 kDa. H-NS binds to DNA in a non-sequence-specific manner, but with a preference for intrinsically curved AT-rich regions. Genome-wide ChIPand microarray studies have identified the set of genes that are modulated by H-NS. They are mainly located in HGT DNA and include several virulence determinants. A general rule in several enterobacterial isolates as, for instance, Salmonella and E. coli strains, is the presence of a paralogue of the hns gene (the stpA gene) in their genomes . In addition, orthologues of hns are also encoded in several conjugative plasmids. The role of the StpA protein not yet well characterized. The StpA protein is overexpressed in mutants lacking HNS and, in this background, overexpression of the paralogue appears to attenuate the phenotype of the hns mutant. Otherwise, stpA deletion has only minor effects on E. coli phenotype. Enteroaggregative E. coli (EAEC) strains have been significantly associated, among others, with (i) endemic diarrhea in infants in developing and industrialized nations, (ii) persistent diarrhea in HIV-positive patients, (iii) traveller’s diarrhea, and (iv) food/waterborne outbreaks. The strain 042 is the prototypical EAEC strain for the study of virulence factors and pathogenicity. Its genome has been sequenced and the virulence factors characterized. A recent genomic analysis performed by our group has shown that, unlike many other E. coli strains, the chromosome of the strain 042 encodes a new paralogue of the hns gene: hns2. Unlike StpA, depletion of H-NS2 has significant effects on the growth rate of strain 042. Depletion of both H-NS and H-NS2 leads to a drastic reduction of its growth rate. We present in this communication information about the biological role of this new member of the H-NS family of proteins. Panel 55 Análisis funcional de la MAPK Slt2 de Saccharomyces cerevisiae sobre la dinámica e integridad del anillo de septinas Elena Jiménez, Esmeralda Alonso-Rodríguez, Pablo Fernández-Piñar, María Molina y Humberto Martín Departamento de Microbiología II, Facultad de Farmacia, UCM, e Instituto Ramón y Cajal de Investigaciones Sanitarias (IRYCIS). Plaza de Ramón y Cajal s/n, 28040, Madrid. [email protected] Las vías de señalización mediadas por MAPKs se encuentran conservadas en las células eucarióticas. En la levadura Saccharomyces cerevisiae la MAPK Slt2 es fundamental en la ruta de integridad de la pared celular (CWI), que se activa en respuesta a estrés de pared y su función es mantener la estabilidad de esta estructura fúngica esencial. Las proteín quinasas sensibles a análogos han resultado ser una herramienta muy útil para la disección de vías de señalización reguladas por fosforilación y para el esclarecimiento de las funciones celulares de las quinasas (Shokat et al. 2014). Por ello, hemos desarrollado una versión funcional de Slt2 sensible a análogos de ATP (Slt2-as), que es inhibida específicamente por el compuesto 2,3-DMBPP1 (2,3-dimetilbencil-pirazolpirimidina). Así, se ha podido estudiar el fenotipo de pérdida de actividad quinasa de Slt2 en mutantes dobles sintéticos letales con slt2Δ, como por ejemplo los afectados en la proteína adaptadora de la fosfatasa PP2C Ptc1, Nbp2, o en la β-1,3-glucanosil transferasa Gas1. La inhibición de Slt2-as en el doble mutante gas1Δ slt2-as condujo a un incremento significativo en el porcentaje de células con yema pequeña y a una deslocalización de la septina Cdc10. El mutante nbp2Δ a 37°C acumula yemas unidas a la célula madre que no desensamblan el anillo de septinas, estructura que es necesaria para un correcto crecimiento polarizado de la levadura. La adición del inhibidor al doble mutante nbp2Δ slt2-as eliminó este fenotipo. Ambas evidencias sugieren que Slt2 podría estar regulando el anillo de septinas. Aprovechando la capacidad de tiofosforilación de Slt2-as, que admite el análogo voluminoso N6-(fenetil)-ATP-γ-S, hemos analizado la posibilidad de que distintas septinas y algunos reguladores directos de éstas sean sustratos de esta MAPK. Por otro lado, estamos rastreando genes, cuya sobreexpresión, restaure la viabilidad del doble mutante gas1Δ/slt2-as en presencia del inhibidor 2,3-DMBPP1. López, M. S., Kliegman, J. I. and K. M. Shokat. 2014. “The logic and design of analogsensitive kinases and their small molecule inhibitors.” Methods Enzymol 548: 189-213. Panel 56 Protein-assisted riboregulation in the legume symbiont Sinorhizobium meliloti José I. Jiménez-Zurdo and Marta Robledo Estación Experimental del Zaidín (CSIC), Granada [email protected] Small non-coding RNAs (sRNAs) are now recognized as fundamental ubiquitous posttranscriptional regulators of virtually every aspect of bacterial physiology. Many sRNAs are transcribed from independent promoters within intergenic regions and rely on limited basepairing with trans-encoded target mRNAs to control the translation and stability of the message. Regulation mediated by trans-sRNAs is commonly assisted by RNA chaperones such as Hfq and involves cleavage of the mRNA target by cellular endoribonucleases. We are investigating riboregulation in the nitrogen-fixing α-rhizobia Sinorhizobium meliloti, the symbiont of alfalfa and related medics. S. meliloti expresses an Hfq homolog (SmHfq) and a plethora of trans-sRNAs (~550)1. Lack of SmHfq influences a diversity of free-living and symbiotic traits2. Deep-sequencing of the subpopulations of transcripts co-immunoprecipitated (CoIP-RNA) with a FLAGtagged variant of the protein mostly recovered mRNAs (1,127) and a small proportion of the annotated trans-sRNAs (85), suggesting that other proteins might assist riboregulation in this bacterium3. One candidate is the almost ubiquitous YbeY protein, which has structural homology to the MID domain of eukaryotic Ago proteins. It has been reported that deletion of S. meliloti ybeY produces a pleiotropic phenotype that resembles that of the hfq mutant, whereas the E. coli YbeY is a single-strand specific endoribonuclease4,5. S. meliloti YbeY (SmYbeY) CoIP-RNA was barely enriched in RNA species, which does not support an Hfq-like role of this protein. However, purified SmYbeY indistinctly cleaved double- and single-stranded RNA substrates, a unique ability among bacterial endoribonucleases. SmYbeY loss-of-function compromised expression of housekeeping genes, whilst promoting accumulation of motility, transport and late symbiotic mRNA transcripts. Some of the upregulated mRNAs might be SmYbeY substrates which are targeted by SmHfq-dependent trans-sRNAs. A genetic reporter assay confirmed that SmYbeY participates in the down-regulation of the amino acid ABC transporter prbA mRNA by the Hfq-binding AbcR2 sRNA. Our data thus provide a resource to dissect the regulatory mechanisms underlying Hfq and trans-sRNA activity in the post-transcriptional control of major S. meliloti stress response and symbiotic regulons. 1. 2. 3. 4. 5. Jiménez-Zurdo et al. 2013. Mol. Plant-Microbes Interact. 26, 160-167 Torres-Quesada et al. 2010. BMC Microbiol. 10, 71 Torres-Quesada et al. 2014. RNA Biol. 11, 563-579 Pandey et al. 2011. Nucleic Acids Res. 39, 4691-4708 Jacob et al. 2013. Mol. Cell 49, 427-438 This work was funded by ERDF-cofinanced grants AGL2009-07925, CSD2009-00006 and BFU2013-48282-C2-2-P from MINECO. Panel 57 La interacción entre FleQ, FleN y el diGMPc regula la expresion de genes implicados en la movilidad y la formación de biofilm en Pseudomonas putida Antonio Leal-Morales, Blanca Navarrete, Aroa López Sánchez y Fernando Govantes Area de Microbiología, Centro Andaluz de Biología del Desarrollo (CABD). Universidad Pablo de Olavide/CSIC. Sevilla [email protected] La alternancia entre un estilo de vida planctónico y la formación de estructuras complejas llamadas biofilm es un fenómeno habitual en el ciclo de vida de muchas bacterias ambientales (1). Trabajos previos llevados a cabo en nuestro grupo en la bacteria Gram-negativa de suelo Pseudomonas putida mostraron que la proteína FleQ, un activador de promotores dependientes de σ54, junto con el mensajero secundario 3',5'diguanilato cíclico (di-GMPc) participan coordinadamente en la regulación de funciones relacionadas con movilidad y colonización de superficies (2). Recientemente hemos comprobado que FleN, la proteína encargada de regular el número de flagelos en la bacteria, es también necesaria para el reconocimiento de ciertos promotores por parte de FleQ. Con el objetivo de aumentar nuestro conocimiento acerca de la red de regulación que controla esta alternancia entre estilos de vida en P. putida, se llevó a cabo un screening de promotores potencialmente involucrados en movilidad y desarrollo de biofilm fusionados a gfp y lacZ que permitió medir la expresión en diferentes fondos mutantes, incluyendo ΔfleQ y ΔfleN. Además, llevamos a cabo ensayos in vitro tales como ensayos de retardos en gel o footprint para describir la posible interacción entre los componentes en las regiones promotoras. Nuestros resultados muestran que la capacidad de unión de FleQ a sus promotores diana se encuentra modulada por la interacción con FleN, la cual actúa como proteína auxiliar a FleQ. A su vez, esta interacción se encuentra modulada de manera diferencial por los niveles de diGMPc en cada promotor. El mecanismo mediante el cual FleN interacciona con FleQ y cómo el diGMPc modifica la afinidad de esta interacción serán objeto de discusión. REFERENCIAS (1) Costerton et al. (1995). Annu. Rev. Microbiol. 49:711-45 (2) Jiménez-Fernández et al. (2016) Plos One (under review) AGRADECIMIENTOS Este trabajo ha sido financiado por los proyectos BIO2010-17853 (MICINN) y BIO201342073-P (MINECO) Panel 58 Targeted killing of bacteria using logic gates Rocío López-Igual and Didier Mazel Institut Pasteur, Unité Pasticité du Génome Bactérien, CNRS UMR3525, 75724 Paris, France [email protected] Synthetic biology aims at reprogramming bacteria to construct sophisticated novel biological systems using gene regulatory networks. This could have important applications in areas such as functional genomics, nanotechnology and biomedical therapeutics. Antibiotics are some of the most powerful drugs in modern medicine. Despite their historic relevance, the emergence of antibiotic resistance is becoming a major concern worldwide. We are implementing a deterministic model (0/1) designed to kill specific bacteria in mixed populations. This will resolve the two main problems of classical antibiotic treatments: indiscriminately killing beneficial bacteria and the emergence of resistance. Our model is based on conjugation where plasmids will act as wires (a plasmid inside a cell is 1; no plasmid is 0). Our design uses split toxins (non-functional) coupled with split intein technology. Inteins are protein sequences embedded inside a host protein (called extein) from where they can be self excised in a process called protein splicing. We designed two types of plasmids: input plasmids (A and B) and clause plasmid (CP). Only CP will be conjugative. The output in our system is cell death. Each input plasmid contains half of the toxin fused to a half intein. Separately, these plasmids do not kill the cell. Together and under the right conditions, the expression of the fusion takes place. Intein halves recognize each other in the cytoplasm and, after protein splicing, the toxin is reconstituted, killing the bacterium. Plasmid A carries a toxin-intein fusion under the control of a PBAD promoter. AraC is the activator of the PBAD promoter in the presence of arabinose. The first CP carries AraC and infects by conjugation a population (araC mutant strain) harboring the four combinations of plasmids: (A and B; only A; only B and none). CP-AraC allows the expression of the fusion in plasmid A that is otherwise repressed. CP-AraC killed specifically the bacteria harboring both plasmids, solving an AND-logic gate; one of the information units in logic systems. We are adapting our system to kill specifically pathogenic Vibrio cholerae. Currently, we are working on promoter sequences controlled by transcriptional regulators that are specific to the Vibrio cholerae genome. FUNDING: Synthetic Biology European FP7 project Project Acronym: PLASWIRES Project number: 612146 Call identifier: FP7-ICP-2013-10 Panel 59 Ni is able to induce the switch between cytochrome c6 and plastocyanin in Synechocystis sp. PCC 6803 Joaquín Giner-Lamia, Luis López-Maury, Francisco Javier Florencio Instituto de Bioquímica Vegetal y Fotosíntesis, Universidad de Sevilla- CSIC, Américo Vespucio 49, E-41092 Sevilla, Spain. [email protected] Plastocyanin (encoded by the petE gene) play an essential role in photosynthesis as electron carrier between cytochrome b6f and photosystem I (PSI) in higher plants, algae and cyanobacteria. However, cyanobacteria and some algae species are able to synthesize an additional electron carrier the heme containing protein, cytochrome c6 (encoded by the petJ gene) under low copper conditions. In presence of copper concentrations above 30 nM the expression of the petE gene is induced while petJ expression is inhibited at concentrations higher than 0.3 𝜇M while expression of both genes, and proteins, can be detected at intermediate concentrations. Here we show that nickel is able to induce expression of petE and repress petJ and not only at mRNA level but also at protein levels, showing a partial switch between cytochrome c6 and plastocyanin in low copper medium but not in copper replete medium. None of the transcriptional factors related to nickel homeostasis in Synechocystis (NrsRS and InrS) are involved, suggesting that the regulatory system(s) controlling the switch between these genes is able to respond to nickel when copper is limiting. Finally, we show that the absence of plastocyanin in a petE mutant strains, leads to nickel sensitivity and this phenotype could be rescued after petE expression even under a non-cognate promoter, suggesting that plastocyanin is able to protect against nickel toxicity. Panel 60 Biestabilidad en la expresión de genes importantes para la virulencia de Pseudomonas syringae Nieves López-Pagán, Jose S. Rufián, Diego López-Márquez, Javier Ruiz-Albert, y Carmen R. Beuzón 1 Instituto de Hortofruticultura Subtropical y Mediterránea “La Mayora” (IHSM-UMA-CSIC). Departamento de Biología Celular, Genética y Fisiología. Facultad de Ciencias. Universidad de Málaga. 29071, Málaga. [email protected] [email protected] La heterogeneidad o variación fenotípica en bacterias es un fenómeno descrito en poblaciones bacterianas desde hace décadas. Puede estar determinada por mecanismos epigenéticos, y dar lugar a linajes bacterianos con perfiles de expresión génica diferentes. Un patrón de expresión génica unimodal y heterogéneo puede volverse bimodal en ambientes homogéneos, dando lugar a la formación de linajes en poblaciones clonales. A este proceso se le conoce como bistabilidad, y ejemplos de bistabilidad se encuentran en la regulación del operon lac en E. coli, o la regulación del establecimiento de lisis o lisogénia en el fago Lambda. A pesar de ejemplos tan paradigmáticos, el estudio de muchos patógenos ha seguido analizando poblaciones enteras y definiéndolas en función de sus valores medios. La relevancia de estos procesos se ha demostrado en Salmonella entérica y otros patógenos humanos al establecerse su papel en la persistencia a antibióticos, y su implicación en la aparición de portadores crónicos. No obstante, se conoce muy poco sobre la relevancia de este tipo de procesos en el proceso de adaptación a huespedes no animales. Utilizando métodos de análisis como la microscopía confocal y la citometría hemos analizado la expresión de genes implicados en virulencia de Pseudomonas syringae, la bacteria fitopatógena de mayor relevancia académica y una de las más importantes por su impacto económico. Este análisis de expression sobre fusiones transcripcionales a gfp generadas en el cromosoma de P. syringae pv. Phaseolicolanos ha llevado a demostrar expresión biestable para distintos determinantes de virulencia. Panel 61 La regulación dependiente de fase de crecimiento de la conjugación de R27 implica el circuito regulador plasmídico TrhR/TrhY-HtdA y el regulón cAMP Marta Gibert1, Sonia Paytubi1, Sergi Beltrán2, Antonio Juárez1,3, Carlos Balsalobre1, Cristina Madrid1 1 Departament de Genètica, Microbiologia i Estadística. Secció de Microbiologia, Virologia i Biotecnologia , Universitat de Barcelona, 2Centre Nacional d’Anàlisi Genòmica (CNAG), 3 Institut de Bioenginyeria de Catalunya (IBEC). [email protected] La conjugación, proceso que permite la transferencia de material genético de forma horizontal, constituye uno de los factores más importantes en la diseminación de resistencias a antibióticos entre bacterias. Los plásmidos del grupo de incompatibilidad HI1 (IncHI1), asociados con la multiresistencia a antibióticos, se han aislado de numerosos patógenos Gram-negativos. La conjugación de estos plásmidos está finamente regulada, siendo característica su termoregulación. La conjugación está reprimida a temperaturas superiores a 30ºC, mientras que se favorece a temperaturas inferiores, lo que se ha relacionado con su transmisión en el medio ambiente, durante el tránsito de la bacteria fuera del huésped. El plásmido R27 es el prototipo de los plásmidos IncHI1. El estudio de los mecanismos de regulación de la transferencia del plásmido R27 en Escherichia coli nos ha permitido establecer que la eficiencia de la conjugación es altamente dependiente del estado fisiológico de las células donadoras. La frecuencia de conjugación es más elevada cuando las células donadoras se encuentran en fase exponencial, y disminuye drásticamente cuando se encuentran en fase estacionaria de crecimiento. Los datos del análisis transcriptómico de la expresión de los genes del plásmido R27 revelan que en fase estacionaria se produce una importante represión de la expresión de muchos de los genes del plásmido, incluyendo varios genes implicados en la transferencia (genes tra). La regulación de la conjugación por fase de crecimiento no depende de la proteína H-NS, implicada en la regulación dependiente de temperatura de la conjugación de R27 (Forns et al., 2005; Gibert et al., 2014), ni de reguladores de fase estacionaria como RpoS o ppGpp, y es estrictamente dependiente del circuito regulador TrhR/TrhY-HtdA codificado en el propio plásmido (Gibert et al., 2014). El sensor metabólico cAMP, de codificación cromosómica, también está involucrado en la regulación de la conjugación dependiendo de fase de crecimiento, mediante la modulación de la expresión de HtdA. Los datos obtenidos sugieren que existe un “crosstalk” entre el cromosoma y el plásmido para el control fisiológico de la conjugación del plásmido R27. Forns et al., 2005. Temperature-dependent conjugative transfer of R27: role of chromosome- and plasmid-encoded Hha and H-NS proteins. Journal of Bacteriology 187:3950-3959 Gibert et al., 2014. TrhR, TrhY and HtdA, a novel regulatory circuit that modulates conjugation of the IncHI plasmids. Molecular Microbiology 84:1146-1161. Panel 62 Influence of flagella motility in the targeted adhesion of bacteria to tumor cells Carmen Mañas Torres*, Eva Pico Sánchez, Carlos Piñero Lambea, and Luis Ángel Fernández Department of Microbial Biotechnology. Centro Nacional de Biotecnología, CNB-CSIC. Darwin 3, Campus UAM, Cantoblanco, 28049 Madrid, Spain (*) Presenting Author. E-mail: [email protected] The natural ability of E. coli to colonize solid tumors can be improved by expressing synthetic adhesins on its surface that specifically recognize protein receptors preferentially expressed on the tumor cell surface [1]. To characterize the factors influencing the efficient adhesion of the engineered E. coli strains to tumor cells, we have evaluated the role of bacterial flagella and motility as a potential limiting factor required by the bacterium to reach the target tumor cell. Based on an E. coli strain expressing a synthetic adhesin binding the epidermal growth factor receptor (EGFR), a tyrosine kinase receptor overexpressed in many human tumors of epithelial origin (e.g. colon and bladder carcinomas), we have constructed a set of mutants in which the assembly of the flagellar apparatus (DflhDC) or its rotation motility (DmotAB) is abrogated. The flhDC operon encodes a master regulator required for the expression of all the flagellar components. The motAB operon encodes integral inner membrane proteins that form the stator of the flagellar motor, thus DmotAB mutants express flagellar filaments unable to rotate. We tested adhesion of bacteria to Her14 cells, a mouse fibroblast cell line transfected with human EGFR, using WT, DflhDC, and DmotAB E. coli strains expressing the synthetic adhesin against EGFR. The specificity of the bacterial adhesion was demonstrated in the isogenic parental mouse fibroblast cell line 3T3 2.2, which does not express human EGFR, as well as using a control isogenic bacterial strain with a synthetic adhesin against GFP. Our results show that only the motile WT strain expressing the synthetic adhesin against EGFR was able to adhere efficiently to Her14 cells. Interestingly, non-specific bacterial adhesion to abiotic surfaces is not affected by these mutations. These results underline the crucial role of an active flagellum for the targeted adhesion of E. coli to mammalian tumor cells. 1. Piñero-Lambea, C., et al., Programming Controlled Adhesion of E. coli to Target Surfaces, Cells, and Tumors with Synthetic Adhesins. ACS Synth Biol, 2015. 4(4): p. 46373. Panel 63 Genome-wide analysis of the FleQ direct regulon in the biocontrol strain Pseudomonas fluorescens F113 Esther Blanco Romero, Daniel Garrido-Sanz, Francisco Martínez-Granero, Rafael Rivilla, Miguel Redondo-Nieto y Marta Martín Departamento de Biología. Facultad de Ciencias. Universidad Autónoma de Madrid [email protected] Bacterial motility plays a crucial role in competitiveness and colonization in the rhizosphere environment. Moreover, this trait is highly important in the biotechnological application and improvement of microbial inoculants employed in agriculture to increase yield and to avoid the use of harmful chemicals fertilizers. In this work, ChIP-seq analysis has been used to identify genes and operons regulated by the transcriptional regulatory protein FleQ. This protein was previously identified as a master regulator of flagella and exopolysaccharide synthesis. Therefore, FleQ participates in regulation of motility and biofilm formation in Pseudomonas fluorescens F113, an interesting biocontrol and plant growth promoting bacteria. ChIP-seq analysis has shown that FleQ is a global regulator and 106 FleQ putative binding sites have been identified. Genes presumably regulated by FleQ included, as expected, flagellar and motility genes and genes involved in adhesion and exopolyssacharide production. Surprisingly, the ChIP-seq analysis also identified iron uptake related genes, including the genes required for the synthesis of the main siderophore pyoverdine. Negative regulation of these genes by FleQ was shown by qRTPCR. The results also showed that FleQ shares an important part of its direct regulon with AmrZ, a global regulator implicated in environmental adaption. Although AmrZ also regulates motility and iron uptake, the overlap occurred only with the iron related genes, since both regulators control a different set of motility related genes. Funded by BIO 2015-64480-R (MINECO/FEDER EU) Panel 64 New-tagged brucellosis vaccines and associated diagnostic tests Estrella Martínez-Gómez1, Yolanda Gil-Ramírez1, José María Blasco2, Pilar Muñoz2, María Jesús de Miguel2, Amaia Zúñiga-Ripa1, Ignacio Moriyón1, Raquel Conde-Álvarez 1*and Maite Iriarte1* (*These authors contributed equally to this work) 1 Instituto de Salud Tropical. Departamento de Microbiología y Parasitología. Universidad de Navarra. España. 2 Unidad de Sanidad Animal. Centro de Investigación Tecnología Agroalimentaria (CITA) del Gobierno de Aragón. Zaragoza. España. [email protected] Brucellosis, a zoonosis caused by Brucella, infects livestock causing heavy economic losses. A complete lipopolysaccharide (LPS) is critical for its virulence. Its O-chain is a homopolymer of N-formyl-perosamine, and carries the immunodominant epitopes. The only useful vaccines to control Brucellosis are smooth (S) live attenuated: B. melitensis Rev1 (ovine), and B. abortus S19 (bovine). However, the differentiation between infected and vaccinated animals (DIVA problem) is not possible since the serological diagnostic tests are based on the immune response against the S-LPS, present in both vaccines. To solve this problem, we tagged Brucella LPS by inserting in the chromosome, wbdR that encodes an acetyltransferase that adds an acetyl group to the perosamine of E.coli O157 O-chain (Ba::Tn7wbdR). We also combined insertion of wbdR with deletion of wbkC encoding the Brucella perosamine formyltransferase (Ba::Tn7wbdRΔwbkC). Both strains carry a complete LPS, and their O-chains contain new epitopes derived of N-acetyl perosamine that are not present in Ba-parental LPS. We developed two specific serological tests (agglutination test and an iELISA) that allow the differentiation of mice infected with Ba-parental strain from those infected with Ba::Tn7wbdRΔwbkC or Ba::Tn7wbdR. These results were confirmed in a preliminary study in the natural host (sheep) using the classical vaccine Rev1 tagged with wbdR. Thus, it is tempting to speculate that introducing wbdR into Brucella vaccinal background might represent a new strategy to solve the DIVA problem associated to Brucella vaccines. Research at the laboratories of the authors is supported by grants from the Ministerio de Economía y Competitividad of Spain (AGL2011-30453-C04-04 and AGL2014-58795-C4-1R). Fellowship support for E.M.G from the Ministerio de Economía y Competitividad of Spain (FPI) is gratefully acknowledged. Panel 65 Implicación de un regulador de la familia LysR en la degradación de ectoinas en la bacteria extremófila Chromohalobacter salexigens Martínez-Martínez Lourdes, Argandoña Montserrat, Nieto Joaquín J., Vargas Carmen Departamento de Microbiología y Parasitología, Facultad de Farmacia, Universidad de Sevilla [email protected] Chromohalobacter salexigens es una bacteria extremófila productora natural de ectoínas, compuestos con un alto interés biotecnológico por sus numerosas aplicaciones en Biomedicina. Nuestro Grupo de investigación está interesado tanto en el estudio molecular de los procesos de osmoadaptación como en la Ingeniería metabólica de C. salexigens para optimizar la producción de ectoínas. Para ello es esencial el avance en el conocimiento de la regulación de las complejas rutas de síntesis y degradación de estos compuestos. Las rutas de degradación de ectoínas, a diferencia de las de síntesis, así como su regulación, no han sido estudiadas en profundidad en C. salexigens ni tampoco en bacterias extremófilas. Estudios preliminares in silico mostraron la existencia de un regulador transcripcional de tipo LysR en el cluster de los genes de degradación En el presente estudio se ha iniciado la caracterización molecular de este regulador para analizar su implicación en el control de las rutas de degradación de ectoínas. Así, se ha caracterizado el mutante CHR228, obtenido mediante deleción en fase del gen que codifica para dicho regulador (csal2729). Los resultados indican que aunque no existen diferencias entre el crecimiento de la cepa silvestre y el mutante CHR228 (ΔlysR) a diferentes salinidades (0,6 M, 1,5M y 2,5 M de NaCl) y en presencia de diferentes fuentes de C (glucosa o ectoína), sí que se observa en la cepa mutante una disminución de la expresión de los genes de degradación doeA, doeD y eutC a elevada salinidad (2,5 M) independientemente de la fuente de C. Por otro lado, los resultados también sugieren la presencia de un segundo elemento regulador que controla la expresión de dichos genes a baja y óptima salinidad (0,6 M y 1,5 M respectivamente) únicamente cuando la ectoína es la fuente de C. El análisis in silico junto con experimentos de RT-PCR ha permitido dilucidar la organización génica de la región y la localización de motivos de unión de LysR en las regiones promotoras doeAp y doeDp, sugiriendo una implicación directa de este regulador en el control de dichos genes. Financiación: BIO2015-63949-R (MINECO/FEDER, UE) y Junta de Andalucía (P11-CV17293 MO). Panel 66 La secuenciación masiva de E. coli ambientales demuestra el impacto de la actividad humana en la diversidad bacteriana y la selección de determinantes de resistencia 1 Cristina Martinez Ovejero, 1 Jose Francisco Delgado Blas, 2 William Calero-Cáceres, 1 Laura Carrilero Aguado, 1 Daniel Thomas-Lopez, 2 Maite Muniesa, 3 Fernando de la Cruz, 1 Bruno Gonzalez Zorn 1 Departamento de Sanidad Animal y Centro de Vigilancia Sanitaria Veterinaria (VISAVET), Facultad de Veterinaria, Universidad Complutense de Madrid. 2 Departamento de Microbiología, Facultad de Biología, Universidad de Barcelona. 3 Instituto de Biomedicina y Biotecnología de Cantabria, Universidad de Cantabria. [email protected] El medio ambiente desempeña un papel fundamental en la diseminación de la resistencia a antibióticos, ya que es un reservorio natural de resistencias antibióticas que se está viendo afectado por la actividad antropogénica. El objetivo de este estudio fue elucidar el efecto de la actividad humana en la ecología y evolución de los genes de resistencia y sus plataformas genéticas, comparando mediante secuenciación masiva bacterias obtenidas de ríos y de aguas residuales. Para ello, se tomaron muestras de agua y sedimentos de dos ríos (Llobregat y Cardener) y muestras de agua sin depurar de dos plantas de tratamiento de aguas residuales del área de Barcelona (Gavá y Prat). Los aislados se seleccionaron por su capacidad de crecer en agar MacConkey suplementado con aminoglucósidos. Las bacterias se identificaron por MALDI-TOF. Determinamos la susceptibilidad antimicrobiana por medio de microdilución en placa. La presencia de metilasas se analizó mediante PCR. Estudiamos la relación clonal entre los aislados mediante PFGE. Las bacterias seleccionadas se secuenciaron utilizando tecnología Illumina (MiSeq). Se obtuvieron 169 aislados, siendo E. coli la especie bacteriana predominante. Todos los aislados portaban una metilasa: armA fue la única metilasa encontrada en ríos (103 positivos), mientras que en las aguas residuales se identificó principalmente rmtB (56 positivos), seguido de lejos por armA (10 positivos). Los aislados de las aguas residuales mostraron mayor nivel de resistencia y a más antibióticos que los de los ríos. Con los resultados de los pulsotipos y los antibiotipos de los E. coli, se seleccionaron 47 cepas para su secuenciación masiva. El análisis de la secuenciación reveló la presencia de dos secuenciotipos predominantes relacionados con las aguas residuales obtenidas de ambas plantas de tratamiento, los cuales también fueron encontrados minoritariamente en los ríos, donde se observó una mayor heterogeneidad en los secuenciotipos. Por el momento, la secuenciación masiva ha revelado que los E. coli relacionados con la actividad humana presentan una menor diversidad y unos genes de resistencia concretos como rmtB. Con ello podemos concluir que la actividad antropogénica modifica y selecciona el medio ambiente favoreciendo la diseminación de clones exitosos en la población. Este trabajo está financiado por la Comisión Europea a través del proyecto EFFORT (613754-FPT7). Panel 67 Analysis of arsRB operon involved in arsenic resistance in Terribacillus sp. 2B122 Leyre Sánchez Barrionuevoa,b, Encarnación Melladoa, Almudena Escobar-Niñoa,b, David Cánovasb, a Department of Microbiology and Parasitology, University of Seville, Profesor García González s/n, 41012, Seville, Spain b Department of Genetics, University of Seville, Avda de Reina Mercedes s/n, 41012, Seville, Spain. [email protected] Arsenic is widely distributed in the environment and occurs primarily in two oxidation states, arsenate [As (V)] and arsenite [As (III)], and both are toxic to the majority of living organisms. The frequent abundance of arsenic in different environmental ecosystems has guided the evolution of detoxification systems in microorganisms. Out of these, the arsenic resistance system (ars) appears to be widely distributed among prokaryotes. It involves a set of genes that display large variations in their number and genomic organization, of which an arsenite efflux pump (ArsB), and a transcriptional repressor (ArsR)1 constituted the progenitor of the modern ars clusters. It is postulated that the arsenate reductase (ArsC) was incorporated into the ars operon during evolution upon exposure of organisms to an oxidative atmosphere, which mediated the formation of As (V). In a first stage, we analysed the resistance to heavy metals [Zn (II), Cu (II), Co (II and III) and Cd (II)] and metalloids [As (III and V) and Cr (VI)] in Terribacillus sp. AE2B 122, and found that it is moderately resistant to As, Cr and Co. We focused in arsenic resistance in this bacterium because it tolerates high concentrations of arsenite and arsenate. The homologs of arsB TA3675 and TA2733, and of arsC TA3384 and TA2854, located in the genome of Terribacillus sp. AE2B 122 were expressed in E. coli AW3110, a strain lacking the chromosomal arsRBC operon. Only TA3675 could confer high resistance to As (III) in E. coli. The arsC homologs, which contain glutathione-S-transferases (GSTs) domains, could only confer slight increased resistance to As (V) and oxidative stress. Therefore, bioinformatics, expression and functional analysis suggest that arsenic resistance in Terribacillus is mediated by the simplest and ancestral configuration of arsenic resistance genes, arsRB. References 1. Rosen, B. P. Biochemistry of arsenic detoxification. FEBS Lett. 2002, 529:86-92 Panel 68 Evolución diferencial de las 3’-UTRs en bacterias Pilar Menéndez, Carlos Caballero y Alejandro Toledo-Arana Laboratorio de Regulación Génica Bacteriana. Instituto de Agrobiotecnología, CSICUPNA. Avda. Pamplona 123, 31192-Mutilva, Navarra. [email protected] Un RNA mensajero (mRNA) está formado por tres regiones bien diferenciadas, una región central que codifica para la proteína correspondiente (CDS), la cual está rodeada por dos regiones que no son traducidas y se denominan 5’-UTR (que comprende desde el inicio de transcripción al inicio de traducción) y 3’-UTR (desde el final de la traducción al final de la transcripción). En eucariotas, las 3’-UTRs son elementos esenciales para modular la expresión de la proteína contenida en el mRNA. En esta región se encuentran los sitios de reconocimiento para diversas proteínas de unión a RNA y las zonas de interacción con los microRNAs que conjuntamente regulan la cantidad de proteína a traducir. Es interesante destacar que la complejidad y longitud de las 3’-UTRs se relaciona de manera directa con la evolución de los organismos. Es así que la longitud de las 3’-UTRs se incrementa a medida que subimos en el árbol evolutivo. Es también llamativo que la desregulación de ciertos genes causantes de algunas enfermedades como el cáncer está asociado con el acortamiento de las 3’-UTRs. En bacterias, la función de estas regiones es prácticamente desconocida. En trabajos previos de nuestro y otros grupos, se demostró que las bacterias también contienen elementos reguladores en las 3’-UTRs para modular la expresión proteica. Considerando que las 3’-UTRs han evolucionado de manera diferente según los organismos, quisimos analizar si esto también ocurría en procariotas. Así, basándonos en el mapa transcriptómico de S. aureus comparamos genes con 3’-UTRs largas con sus respectivos homólogos en diferentes especies del Género Staphylococcus. Sorprendentemente, descubrimos que la homología desaparecía a la altura del codón de parada, lo que indicaba que cada gen homólogo tiene una 3’-UTR diferente. Para comprobar si distintas 3’-UTRs provocan una expresión diferencial del mismo gen homólogo, realizamos quimeras entre una CDS y las distintas 3’-UTRs y medimos la expresión de la proteína. Los resultados mostraron que la expresión es diferente según la 3’-UTR clonada. Esto sugiere que la regulación post-transcripcional mediada por estas regiones ha evolucionado de manera diferente para un mismo gen según en la especie bacteriana que se encuentre. Agradecimientos: al Ministerio de Economía y Competitividad y al Consejo Europeo de Investigación por la financiación de los proyectos de investigación BFU2014-56698-P y ERC-2014-CoG-646869, respectivamente. A la Universidad Pública de Navarra por la financiación del contrato pre-doctoral de C.C. Panel 69 Characterization of the yebEFG locus in Salmonella enterica Ángela Mérida-Floriano and Josep Casadesús Departamento de Genética, Universidad de Sevilla [email protected] The genome of Salmonella enterica serovar Typhimurium contains >19,000 GATC sites which serve as targets for the Dam methylase. Some of these GATC sites are located within regulatory regions, affecting protein-DNA interactions that control gene transcription. Previous work of our laboratory described 176 genes showing differential expression in Dam+ and Dam– Salmonella hosts [1]. The gene cluster formed by yebG, yebF and yebE appeared among the genes that are upregulated in Dam– mutants. Little is known about the function and regulation of these genes. yebG has been described as a gene of the SOS regulon [2], yebF appears to encode a secretion protein [3], and YebE is predicted to be an inner membrane protein regulated by the Cpx stress response [4]. Interestingly, several GATC sites are found upstream of the predicted transcription start site of yebG, raising the possibility of regulation by DNA methylation. Expression of the entire yeb cluster is induced by nalidixic acid but only yebG has a putative SOS-box upstream of its transcription start site, and shows LexA-dependent expression. Single-cell analysis reveals that only 1.6% of cells express yebG in stationary phase. In contrast to other genes that have GATC sites upstream of their regulatory regions and present phenotypic heterogeneity [5], heterogeneous expression of yebG is Dam-independent. Flow cytometry analysis reveals that only a fraction of cells survives exposure to nalidixic acid, and the surviving subpopulation shows high levels of yebG expression. However, ΔyebG mutants are not more sensitive to nalidixic acid than wild type cells, suggesting that the YebG protein is not essential for nalidixic acid resistance. Despite this observation, heterogeneous yebG expression might generate a subpopulation of bacterial cells more resistant to environmental challenges. 1. 2. 3. 4. 5. Balbontín R. et al. 2006. Journal of Bacteriology 188: 8160-8168. Oh, T. J., Kim, I. G. 1999. FEMS Microbiology Letters 177: 271-277. Zhang, G., Brokx, S., Weiner, J. H. 2006. Nature Biotechnology 24: 100-104. Price, N., Raivio, T.. Journal of Bacteriology 191: 1798-1815. Cota, I. et al. 2015. Plos Genetics 11: e1005667. Panel 70 Descifrando transcriptomas en condiciones extremas: mecanismos bacterianos de resistencia a elevadas concentraciones de arsénico Mesa, Jennifer1*; Rodríguez-Llorente, Ignacio David1; Pajuelo, Eloísa1, Caviedes, Miguel Ángel1; McConkey, Brendan J2; Mooney, Steven2. 1 Departamento de Microbiología y Parasitología, Facultad de Farmacia, Universidad de Sevilla (España) 2 Department of Biology, Faculty of Science, University of Waterloo (Canada) *[email protected] El estuario conjunto de los ríos Tinto y Odiel en Huelva (España) es conocido como una de las zonas más contaminadas por metales pesados en todo el mundo. Para su restauración, se investigan actualmente estrategias de biorremediación empleando la planta autóctona hiperacumuladora Spartina maritima junto con un tratamiento de bioaumentación con bacterias promotoras del crecimiento vegetal. De las bacterias cultivables aisladas, un endófito de hoja de S. maritima identificado como Micrococcus yunnanensis SMJ12 resultó tolerar una concentración de arsénico (en forma de arsenito) de hasta 100 mM. Esta cepa se escogió para realizar un estudio de sus mecanismos de resistencia a dicho metal. La cepa fue cultivada en ausencia y presencia de arsenito, tras lo cual el ARN fue extraído y posteriormente secuenciado mediante tecnología “RNA-Seq” de Illumina®. A continuación se llevó a cabo un mapeo de las lecturas frente a una cepa de referencia y la búsqueda de genes de interés. Un número de genes apareció claramente sobreexpresado, sobre todo relacionados con genes de resistencia a arsénico (arsR), proteínas de choque térmico, proteínas de canal, transferasas y ARN polimerasas. Por el contrario, se muestra subexpresada la síntesis de proteínas de transportadores tipo ABC y otras de función aún no del todo descrita. En la actualidad se está desarrollando un profundo estudio bioinformático con el fin de esclarecer una posible ruta metabólica para el arsénico. Descifrar el mecanismo que hay detrás de elevadas resistencias es crucial para ampliar los conocimientos acerca del comportamiento de los microorganismos frente a metales pesados. Además, esto permitirá futuras aplicaciones, como conferir resistencia a otros organismos con fines de biorremediación, e incluso en algunos casos posibilita establecer paralelismos para dilucidar mecanismos de resistencia a otros compuestos, como es el caso de los antibióticos en clínica por ejemplo. Agradecimientos Junta de Andalucía, España (MARISMA P11-RNM- 7274MO) Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), España (RTA2012-00006-C03-03) Ministerio de Educación, Cultura y Deporte, España (FPU AP2012-1809 a J.M.) Panel 71 Evolución pato-adaptativa de Haemophilus influenzae en el sistema respiratorio del paciente con Enfermedad Pulmonar Obstructiva Crónica Javier Moleres1, Joshua C. Mell2, Ariadna Fernández-Calvet1, Sara Marti3,4, Lucía Caballero1, Sergio Cuesta1, Josefina Liñares3,4, Garth Elhrich2, Junkal Garmendia1,4 1 Instituto de Agrobiotecnología, CSIC-UPNa-Gob. Navarra, Mutilva, Navarra; 2Department of Microbiology and Immunology, Drexel University College of Medicine, Philadelphia, USA; 3Departamento de Microbiología, Hospital Bellvitge, IDIBELL, Universidad de Barcelona, Barcelona; 4CIBERES, Centro Investigación Biomédica en Red en Enfermedades Respiratorias, Madrid [email protected] Haemophilus influenzae (Hi) es un patógeno oportunista causante de infección respiratoria crónica y de exacerbación periódica en pacientes con patologías respiratorias crónicas asociadas al tabaquismo como la Enfermedad Pulmonar Obstructiva Crónica (EPOC). El pulmón EPOC es un nicho ecológico complejo, con niveles elevados de inflamación, mucosidad, enfisema y fibrosis, expuesto a humo de tabaco y a fármacos. Todo ello genera una presión selectiva que modula la evolución, adaptación y persistencia pulmonar de Hi. Para entender la dinámica de la evolución pato-adaptativa de Hi en el pulmón EPOC, se dispuso de una colección prospectiva longitudinal de 86 aislados de Hi procedentes de muestras de esputo de 12 pacientes EPOC en exacerbación, generada entre los años 2005 y 2014. Para identificar rasgos fenotípicos y genotípicos de pato-adaptación, se analizaron crecimiento bacteriano, auto-agregación y resistencia a la muerte por péptidos antimicrobianos, y se realizó secuenciación genómica de la colección de aislados. El análisis bioinformático del material secuenciado estableció la existencia 20 series de aislados clonales. El análisis de polimorfismos en ORFs en las 20 series clonales identificó 231 SNPs no sinónimos en 180 genes, y reveló la existencia de 15 genes que presentan rasgos de evolución paralela (SNPs en varias series clonales aisladas de distintos pacientes). Entre ellos, 6 series clonales mostraron SNPs en el gen fadL (transiciones e inserciones), generando 12 variantes alélicas con cambios en loops extracelulares y 9 variantes alélicas truncadas. FadL es una proteína con función dual, (i) transportador de ácidos grasos exógenos de cadena larga (LCFA), y (ii) ligando del receptor eucariota Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 (hCEACAM1) que facilita la invasión epitelial por Hi. El significado biológico de la variabilidad alélica de FadL en ambas categorías funcionales se analizó mediante el diseño y utilización de (i) un medio mínimo con ácido oleico como fuente de carbono, y (ii) un ensayo de interacción con CEACAM1 mediante línea celular HeLa transfectada con hCEACAM1. En conjunto, este trabajo constituye el primer estudio evolutivo de un patógeno bacteriano adaptado al pulmón EPOC, e identifica rasgos de pato-adaptación y potenciales dianas que serán explotadas en el desarrollo de nuevos antimicrobianos. Panel 72 Discovery of a novel gluten-degrading prolyl endopeptidase: potential application in enzymatic celiac therapy María de Lourdes Moreno Amador1, Miguel Arévalo Rodríguez2, Juan Carlos Martínez Reyes2, Ángel Cebolla Ramírez2 y Carolina Sousa Martín1 1 Departamento de Microbiología y Parasitología, Facultad de Farmacia, Universidad de Sevilla, C/ Profesor García González S/N, 41012-Sevilla, Spain; 2Biomedal S.L., C/ Américo Vespucio 5, 41092- Sevilla, Spain [email protected], [email protected], [email protected], [email protected], [email protected] Celiac disease (CD) is an autoimmune disorder characterized by mucosal damage of the small intestine and triggered by ingestion of proline-rich, digestion-resistant proteins present in gluten from different cereals, including wheat, rye and, in some cases, oats. Prolyl endopeptidases (PEP), a family of serine proteases with the ability to hydrolyze the peptide bond on the carboxyl side of an internal proline residue, are able to degrade gluten immunotoxic peptides (GIP) responsible for CD. The use of PEP has also been proposed for gluten detoxification in food as well as oral enzyme supplementation. The aim of this study was to identify novel gluten-digesting microbial enzymes with the potential to reduce GIP toxicity by neutralizing their antigenic epitopes. In this work, microorganisms with gluten-degrading capacity were isolated by following a selective plating strategy using gluten agar. A bacterial isolate, 2RA3, displaying a high level of glutenase activity was identified initially by 16S rDNA gene sequencing as 99,1% similar to Chryseobacterium taeanense. Gel electrophoresis and gliadin zymography revealed the presence of a ~65 kDa gluten-degrading enzyme in the culture medium of 2RA3. Fingerprinting by MALDI-TOF and ion-trap MS allowed identification in the C. taeanense 2RA3 genome of the coding sequence for this glutenase, designed as PEP 2RA3. Based on sequence similarity, the new enzyme belongs to the S9A family of the SC clan of serine peptidases, with high similarity to prolyl endopeptidase from Flavobacterium sp. B17. The PEP 2RA3 coding sequence was PCR-amplified from C. taeanense 2RA3, cloned and expressed in Escherichia coli as C-terminally His-tagged recombinant protein that was purified by Ni-NTA affinity chromatography. The recombinant protein, with a predicted molecular mass and isoelectric point of 80.29 kDa and 7.5, respectively, exhibited prolyl endopeptidase activity with standard chromogenic substrates, showing more hydrolysis efficiency for Suc-Ala-Pro-pNa than for Z-Gly-Pro-pNa. The optimal pH for the activity PEP 2RA3 was found to be pH 8.0. Panel 73 Efectos del transporte de glucosa en la cianobacteria marina Prochlorococcus José Ángel Moreno-Cabezuelo, Antonio López-Lozano, María Agustina DomínguezMartín, Jesús Diez, José Manuel García-Fernández Departamento de Bioquímica y Biología Molecular. Universidad de Córdoba [email protected] La picocianobacteria marina Prochlorococcus domina la biomasa fotosintética en latitudes medias de los océanos oligotróficos, siendo responsable de una parte importante de la producción primaria. Además Prochlorococcus es el organismo fotosintético más abundante del planeta, y se ha convertido en un modelo de estudio en ecología marina 1,2. Resultados previos de nuestro grupo demostraron que Prochlorococcus puede transportar glucosa, observando un incremento en la expresión de genes que participan en su metabolización 3. Posteriormente se caracterizó el gen Pro1404, que codifica un transportador de glucosa de cinética bifásica en Prochlorococcus sp. SS120. Además se detectó transporte de glucosa en poblaciones naturales de Prochlorococcus en el Atlántico 4. En éste trabajo se han utilizado las estirpes de Prochlorococcus sp. SS120 (ecotipo de profundidad), y MED4 (ecotipo de superficie). En ambas se ha determinado la expresión de melB en presencia de glucosa, observando un incremento significativo con diferentes concentraciones de glucosa al cabo de 24 horas. Por otra parte, se está estudiando la implicación de diversos aminoácidos en la cinética de transporte de glucosa. Para ello, se han identificado algunos residuos específicos conservados en este transportador. Actualmente estamos trabajando en la obtención de mutantes en los mismos, para analizar a continuación el impacto de los cambios sobre la cinética del transporte. Financiado por los proyectos BFU-2013-44767-P (Ministerio de Economía y Competitividad, cofinanciado por el European Social Fund de la Unión Europea), P12BIO-2141 (Proyectos de Excelencia, Junta de Andalucía) y la Universidad de Córdoba (Programa Propio de Investigación). JAMC y ALL han recibido contratos de personal de investigación de la Junta de Andalucía. 1 Partensky, F., Hess, W. & Vaulot, D. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 63, 106-127 (1999). 2 Biller, S., Berube, P., Lindell, D. & Chisholm, S. Prochlorococcus: the structure and function of collective diversity. Nat. Rev. Microbiol. 13, 13-27, doi:10.1038/nrmicro3378 (2015). 3 Gómez-Baena, G. et al. Glucose uptake and its effect on gene expression in Prochlorococcus. PLOS One 3, e3416 (2008). 4 Muñoz-Marín, M. C. et al. Prochlorococcus can use the Pro1404 transporter to take up glucose at nanomolar concentrations in the Atlantic Ocean. Proc. Natl. Acad. Sci. USA 110, 8597-8602 (2013). Panel 74 Identification of glycoproteins involved in virulence in Ustilago maydis Ismael Moreno-Sánchez and Jose I. Ibeas Centro Andaluz de Biología del Desarrollo. Universidad Pablo de Olavide, de SevillaCSIC-Junta de Andalucía, Sevilla, Spain. [email protected] Ustilago maydis has raised as an excellent model for the study of plant-pathogen interactions, and its relation with maize plant is one of the systems in which studies can be tackled from both plant and pathogen perspective (Djamei & Kahmann, 2012). U. maydis genome contains more than 500 putative secreted proteins, of which more than 50% doesn`t have known functional domains and many of these proteins have been related to infection process (Kämper et al., 2006). Protein N- and O- glycosylation are critical processes in host-pathogen relations, in fact mutations in genes pmt4, gls1 and gas2 compromise U. maydis virulence on maize, affecting different steps in the infection process (Fernandez-Alvarez et al., 2009; Fernandez-Alvarez et al., 2013). We have now performed a secretome analysis in order to identify proteins glycosylated by Pmt4 and/or Gls1, produced only when the virulence program is activated by over-expressing the transcription factor Biz1. Up today we have identified by mass spectrometry and MASCOT analysis two proteins glycosylated by Pmt4 and seven proteins glycosylated by Gls1, all dependent of the virulence program activation. These proteins are actually being characterized. Moreover, we have also isolated cell wall proteins from wild-type, Δpmt4 and Δgls1 mutants that will be analysed in a similar way. This work was funded by BIO2013-48858-P from MEC Spain. References: Djamei A, Kahmann R. (2012). Ustilago maydis: dissecting the molecular interface between pathogen and plant. PLoS Pathog. 8(11):e1002955. Fernández-Álvarez A, Elías-Villalobos A, Ibeas JI. (2009). The O-mannosyltransferase PMT4 is essential for normal appressorium formation and penetration in Ustilago maydis. Plant Cell. 21(10):3397-412. Fernández-Álvarez A, Elías-Villalobos A, Jiménez-Martín A, Marín-Menguiano M, Ibeas JI. (2013). Endoplasmic reticulum glucosidases and protein quality control factors cooperate to establish biotrophy in Ustilago maydis. Plant Cell. doi:10.1105/tpc.113.115691 Kämper J, Kahmann R, Bolker M, Ma LJ, Brefort T, et al. (2006). Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444: 97–101. Panel 75 EFFORT: Ecología y transmisión de las resistencias a antibióticos de la granja a la mesa Gabriel Moyano, Alfonso Santos-Lopez, Cristina Bernabe-Balas, Daniel Thomas-Lopez, Jose Francisco Delgado-Blas, Natalia Montero y Bruno Gonzalez-Zorn. Departamento de Sanidad Animal y Centro de Vigilancia Sanitaria Veterinaria (VISAVET), Facultad de Veterinaria, Universidad Complutense de Madrid, España. [email protected] Ecology from Farm to Fork Of microbial drug Resistance and Transmission (EFFORT) se centra en estudiar el complejo mundo de la epidemiología, la ecología de la resistencia a antimicrobianos (RAM) y las interacciones entre las comunidades bacterianas (comensales y patógenas) en los animales, la cadena alimentaria y el medio ambiente. Los principales objetivos son: comprender la epidemiología de la RAM en la cadena alimentaria, estudiar la ecología de la RAM en las comunidades microbianas, determinar la contribución relativa de las vías de exposición de los seres humanos a determinantes de resistencia a antimicrobianos con origen en los animales y comprender el impacto económico y sobre el bienestar animal de la RAM en la cadena alimentaria. La recogida de muestras y datos más amplia y armonizada realizada en la Unión Europea combinada con las más novedosas tecnologías micromoleculares y bioinformáticas permitirán profundizar más allá que ningún proyecto realizado hasta la fecha. La secuenciación masiva de más de 20.000 muestras obtenidas a lo largo de toda la cadena alimentaria: cerdos, pollos, vacas, pavos, truchas, animales salvajes, animales de compañía, medio ambiente (aire, agua y polvo), granjeros, técnicos de mataderos y carne comercializada, combinada con técnicas moleculares clásicas (qPCR y antibiogramas), proporcionarán a la comunidad científica el primer microbioma, plasmidoma y resistoma de la cadena alimentaria. Los resultados obtenidos se comprobarán mediantes estudios in vitro e in vivo. Además, se realizarán estudios de intervención a tiempo real con el objetivo de validar diferentes medidas que permitan reducir el uso de antimicrobianos en la práctica veterinaria. Finalmente, toda la información obtenida será utilizada para el desarrollo de modelos predictivos con el objetivo principal de limitar el desarrollo de resistencias bacterianas a los antibióticos de importancia crítica para los seres humanos. En último término, los resultados obtenidos por EFFORT servirán para apoyar políticas basadas en la evidencia y la priorización sobre la prevención y la correcta gestión del riesgo al que están expuestos los seres humanos a lo largo de la cadena alimentaria. EFFORT (Ecology from Farm to Fork Of microbial drug Resistance and Transmission) is supported by the European Commission under the Food, Agriculture and Fisheries, and Biotechnologies theme of the 7th Framework Programme for Research and Technological Development (Grant Agreement no 613754). Panel 76 Importancia del hierro y de los reguladores globales Fur en el control de la síntesis de ectoínas en la bacteria de interés biomédico Chromohalobacter salexigens Emilia Naranjo, Montserrat Argandoña, Joaquín J. Nieto y Carmen Vargas Departamento de Microbiología y Parasitología, Facultad de Farmacia, Universidad de Sevilla [email protected] Chromohalobacter salexigens es una bacteria halófila modelo de máximo interés en Biotecnología, ya que sintetiza los solutos compatibles ectoína e hidroxiectoína, compuestos bioestabilizadores que destacan por sus potenciales aplicaciones en el campo de la Biomedicina. Se sabe que la homeostasis del hierro en esta bacteria está directamente relacionada con su capacidad de adaptación al estrés osmótico y por lo tanto, con la síntesis de ectoinas, a través del regulador global Fur. En el genoma de C. salexigens hemos encontrado dos parálogos pertenecientes a la superfamilia Fur, Fur1, descrito anteriormente como activador de los genes de síntesis de ectoína (1), y un segundo regulador denominado Fur2, que por resultados anteriores se sabe que interviene en la osmoadaptación. Ambos intervienen en la homeostasis del hierro mediante el control de la síntesis de sideróforos. En este estudio se ha profundizado en el papel que juegan ambos reguladores Fur en el control transcripcional de los genes implicados en la síntesis de ectoína (ectABC) e hidroxiectoína (ectD y ectE), en condiciones limitantes y con aporte de hierro, y a diferentes salinidades. Para ello se realizaron estudios de expresión mediante qPCR y mapeo de promotores mediante fluorescencia (FLOE) de diferentes mutantes. Además se cuantificó el contenido intra y extracelular de ectoínas mediante HPLC-MS así como el contenido intracelular de hierro de las diferentes cepas mediante ICP-OES. Los resultados obtenidos muestran la implicación de ambos reguladores en el control de la expresión de los genes de síntesis tanto de ectoína como de hidroxiectoína, aunque tienen un papel diferente (activador o inhibidor) dependiendo de las condiciones ambientales. Además sugieren la existencia de un mecanismo de control adicional dependiente de hierro pero independiente de Fur. Por otro lado, cabe destacar la influencia del hierro tanto en la expresión de los genes de síntesis de ectoínas como en el contenido intracelular de las mismas. Estos resultados confirman la compleja regulación de los genes de síntesis de ectoinas y la importancia del hierro en la osmoadaptación de esta bacteria. Financiación: BIO2015-63949-R (MINECO/FEDER, UE) y Junta de Andalucía (P11-CV17293 MO) Referencias: (1) Argandoña et al. (2010).Appl Environ Microbiol.76(11):3575-89. Panel 77 Nuevos elementos reguladores de la movilidad y la adhesión en Pseudomonas putida Blanca Navarrete, Laura Serrano, Aroa López-Sánchez and Fernando Govantes Área de Microbiología, Centro Andaluz de Biología del Desarrollo (CABD). Universidad Pablo de Olavide/CSIC/Junta de Andalucía [email protected] Pseudomonas putida es una bacteria de suelo que puede encontrarse en la naturaleza como células móviles individuales o como parte de comunidades sésiles adheridas a sustratos denominadas biofilm. La transición entre ambos estilos de vida está regulado por la proteína FleQ y los niveles intracelulares de di-GMPc (2). El aislamiento de mutantes de inserción en el gen flhF, defectivos en la formación de biofilm, sugieren la implicación de elementos adicionales en la regulación de este proceso. flhF codifica una GTPasa necesaria para la correcta localización del flagelo, y se encuentra localizado aguas arriba de fleN, que regula el número de flagelos, y fliA, que codifica un factor sigma específico. Con el fin de estudiar el posible papel regulador de estos elementos, se han caracterizado mutantes de deleción en flhF, fleN y un doble mutante flhFfleN. Para ello se han llevado a cabo curvas de crecimiento planctónico y formación de biofilm, ensayos de adhesión y movilidad tipo swimming y swarming. También se han elaborado tinciones de flagelo de todos los mutantes para su posterior visualización al microscopio óptico. Además, se han realizado RT-PCR para el estudio de la organización transcripcional de los genes flhF y fleN, Para estudiar la implicación de estos elementos en regulación de la movilidad y el desarrollo del biofilm, se han realizado ensayos β-galactosidasa de fusiones transcripcionales al gen lacZ de promotores relacionados tanto con la síntesis de flagelo como la formación de biofilm. Los resutados indican que flhA, flhF, fleN y fliA están organizados en un único operón. FleN y FlhF están involucrados en movilidad tipo swimming, pero solo FleN es necesario para la adhesión y formación de biofilm. Por otro lado, ambos genes son necesarios para la movilidad tipo swarming. El análisis de la expresión génica indica que FleN está implicado en la regulación de genes relacionados con movilidad y formación de biofilm. Además, flhF parece estar involucrado en la regulación de, al menos, uno de los promotores flagelares estudiados. Todo ello, sugiere que FleN es un elemento importante en la regulación de la movilidad flagelar y de la adhesión en P. putida. REFERENCIAS (1) Costerton et al. (1995). Annu. Rev. Microbiol. 49:711-45. (2) Jiménez-Fernández et al. (2016).Plos One (under review) AGRADECIMIENTOS Este trabajo ha sido financiado por los proyectos BIO2010-17853 (MICINN) y BIO201342073-P (MINECO) Panel 78 Influencia del sistema de dos componentes EupK/EupR en la adaptación metabólica y la producción de ectoínas en la bacteria halófila Chromohalobacter salexigens Rosa García-Valero1, Montserrat Argandoña1, José Mª Pastor2, Manuel Cánovas2, Carmen Vargas1, Joaquín J. Nieto1 1 Departamento de Microbiología y Parasitología, Universidad de Sevilla, 41012 Sevilla 2 Departamento de Bioquímica y Biología Molecular B e Inmunología, Universidad de Murcia, Campus de Espinardo 30100, Murcia [email protected] La bacteria halófila Chromohalobacter salexigens es un productor natural de ectoínas, compuestos osmoprotectores de gran interés biotecnológico en los campos de la Biomedicina, la Biología Molecular y la Dermofarmacia, entre otros. La síntesis de las ectoínas como osmolitos se produce en respuesta a estrés por elevada salinidad y/o temperatura y está ligada al metabolismo central, de forma que se produce una adaptación metabólica para asegurar la producción de los mismos. Nuestro objetivo es el desarrollo de un modelo de regulación transcripcional que conecte las señales ambientales, que desencadenan la acumulación de ectoínas, con los sistemas de transducción de señales que regulan la respuesta metabólica. Previamente demostramos la existencia en C. salexigens del sistema de dos componentes EupK/EupR implicado en el metabolismo de las ectoínas (síntesis, degradación y transporte) y, posiblemente, en el metabolismo de la glucosa a través de un sistema de regulación cruzada. En este estudio se ha profundizado en su caracterización, centrándonos principalmente en el catabolismo de la glucosa y su implicación en la acumulación de ectoínas. Para ello se cuantificó mediante HPLC la producción de ectoínas y se analizó el perfil metabólico, monitorizando el consumo de glucosa y los niveles extracelulares de ácidos orgánicos. La pérdida del regulador EupR originó una mayor acumulación de ectoínas a baja salinidad, junto con una alteración en el perfil de excreción de ácidos orgánicos y en el consumo de glucosa, indicando su posible papel en el metabolismo central. Posteriormente, se obtuvo el regulón de EupR mediante RNA-seq (secuenciación masiva por Illumina). Un análisis preliminar de los datos mostró 634 y 772 genes diferencialmente expresados a baja y elevada salinidad, respectivamente, con respecto a la cepa silvestre, de los cuales la mayoría estaban relacionados con el metabolismo, la regulación y el transporte. También sugieren que EupR actuaría principalmente como activador, sobre todo a elevada salinidad. Nuestros resultados revelan la importancia del regulador EupR en la adaptación metabólica de C. salexigens en respuesta a elevada salinidad, siendo éste un elemento clave en la red de regulación que controla la producción de ectoínas. Financiación:BIO2015-63949-R (MINECO/FEDER,UE), Junta de Andalucía P11-CV17293MO Panel 79 Glucoside transporters that influence function and structure of septal junctions in the heterocyst-forming cyanobacterium Anabaena Mercedes Nieves-Morión1, Conrad W. Mullineaux2, C. Peter Wolk3, and Enrique Flores1 1 Instituto de Bioquímica Vegetal y Fotosíntesis, CSIC and Universidad de Sevilla, E-41092 Seville, Spain; 2School of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London E1 4NS, United Kingdom; 3MSU-DOE Plant Research Laboratory, Michigan State University, East Lansing, Michigan 48824, USA. [email protected] Heterocyst-forming cyanobacteria perform oxygenic photosynthesis and, in absence of combined nitrogen, can fix atmospheric nitrogen in specialized cells called heterocysts. The vegetative cells provide the heterocysts with reduced carbon and the heterocysts provide the vegetative cells with fixed nitrogen. Thus, an intercellular molecular exchange takes place in the diazotrophic filament, which behaves as a multicellular organism. In the model heterocyst-forming cyanobacterium Anabaena sp. PCC 7120, proteins—including SepJ—have been identified that are located at the cell poles in the intercellular septa of the filaments and that appear to contribute to the formation of cell-cell joining structures termed septal junctions. Intercellular molecular exchange can be assessed with fluorescent tracers including calcein (1), 5-carboxyfluorescein (2), and the sucrose analog esculin (3). We investigated the effect of temperature on the intercellular transfer of these tracers and found kinetics consistent with transfer by diffusion, implying that the septal junctions—like the metazoan gap junctions—contain conduits that join adjacent cells. We have recently identified components of ABC transporters for glucosides that mediate esculin uptake into the cells of Anabaena. MalK is an ATP-binding protein subunit and GlsP is a permease subunit of two different ABC transporters. Mutant strains bearing inactivated malK and/or glsP were impaired in intercellular molecular exchange tested with the three fluorescent tracers. The effect of these mutations on intercellular transfer of calcein was greater than on transfer of esculin or 5-CF. This result is reminiscent of the effect of inactivating sepJ. MalK was found to be necessary for the normal subcellular localization of SepJ and for the formation of a normal number of septal peptidoglycan nanopores (which are related to SepJ). On the other hand, bacterial two-hybrid analysis showed a possible interaction of GlsP with SepJ. Thus, whereas MalK is involved in the formation of septal structures that mediate intercellular communication, GlsP might influence SepJ function through a physical interaction between the two proteins. We do not know, however, whether the glucoside transporters are needed only for synthesis and functionality of septal junctions or may also be involved in regulation of these cell-cell joining structures. Work in Seville was supported by grant no. BFU2014-56757-P from Plan Nacional de Investigación, Spain, co-financed by the European Regional Development Fund. (1) (2) (3) Mullineaux et al. (2008) EMBO J 27:1299-1308. Merino-Puerto et al. (2011) Mol Microbiol 82:87-98. Nürnberg et al. (2015) mBio 6(2):e02109-14. Panel 80 NsiR8, un pequeño RNA que se transcribe específicamente en los heterocistos Elvira Olmedo-Verd, Alicia M. Muro-Pastor, Agustín Vioque Instituto de Bioquímica Vegetal y Fotosíntesis, Universidad de Sevilla-CSIC [email protected] Las cianobacterias constituyen un grupo de bacterias capaces de llevar a cabo la fotosíntesis oxigénica, un proceso similar al que realizan las plantas superiores. En respuesta a la carencia de nitrógeno combinado, algunas cianobacterias filamentosas, como Nostoc (Anabaena) sp. PCC 7120, desarrollan un tipo celular especializado en la fijación de nitrógeno atmosférico, los heterocistos. El programa transcripcional que culmina en la diferenciación en heterocistos de un 10-15% de las células del filamento está estrictamente regulado y es en gran parte exclusivo de las células en diferenciación. La identificación y caracterización en los últimos años de múltiples RNAs pequeños pone de manifiesto la implicación de estas moléculas en la regulación de distintos aspectos de la fisiología bacteriana, principalmente a nivel post-transcripcional y, en muchos casos, como un nivel adicional a la regulación mediada por factores transcripcionales. Con el objeto de caracterizar los mecanismos reguladores orquestados por RNAs no codificantes en cianobacterias, en nuestro laboratorio se identificaron por primera vez, mediante secuenciación de RNA total (RNA-seq), posibles RNAs pequeños cuya expresión está regulada por la disponibilidad de nitrógeno combinado en Nostoc sp. PCC 7120 (1). NsiR8 (Nitrogen stress-induced RNA 8), es un RNA pequeño de 171 nucleótidos que aparece codificado exclusivamente en genomas de cianobacterias formadoras de heterocistos. Utilizando una estirpe derivada de Nostoc sp. PCC 7120 que porta una fusión entre el promotor de NsiR8 y el gen testigo gfp, hemos podido demostrar que la transcripción de NsiR8 tiene lugar en condiciones de estrés por nitrógeno y exclusivamente en las células del filamento que se están diferenciando como heterocistos (2). El algoritmo CopraRNA identifica con alta probabilidad una posible interacción entre NsiR8 y el mRNA del gen alr1041, que codifica la fructosa-1,6bisfosfatasa/sedoheptulosa-1,7-bisfosfatasa, una enzima esencial para la asimilación fotosintética de CO2 en el ciclo de Calvin y cuya abundancia en el heterocisto está reducida con respecto a las células vegetativas (3). La interacción entre NsiR8 y la región 5´UTR de alr1041 ha sido validada en un sistema heterólogo establecido en E. coli. Actualmente estamos analizando los efectos de la alteración en los niveles de NsiR8 (deleción, sobreexpresión) sobre la actividad fructosa-1,6-bisfosfatasa en Nostoc, al objeto de determinar la posible contribución de NsiR8 a los mecanismos que regulan la reducción en la cantidad de este enzima en los heterocistos. 1. Mitschke J, Vioque A, Haas F, Hess WR, and Muro-Pastor AM (2011). Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc. Natl. Acad. Sci. USA 108, 20130-20135. 2. Brenes-Álvarez M, Olmedo-Verd E, Vioque A and Muro-Pastor AM (2016). Identification of conserved and potentially regulatory small RNAs in heterocystous cyanobacteria. Front. Microbiol. 7:48. 3. Sandh, G, Ramström, M and Stensjö, K (2014). Analysis of the early heterocyst Cys-proteome in the multicellular cyanobacterium Nostoc punctiforme reveals novel insights into the division of labor within diazotrophic filaments. BMC Genomics 15, 1064. Financiación: MICINN (BFU2010-14821), MINECO (BFU2013-48282-C2-1) Panel 81 La bEBPs altamente homólogas RedR1 y RedR2 utilizan mecanismos diferentes de activación y cooperan para inducir la ruta de degradación de 1,3-dihidroxibenceno Daniel Pacheco, Patricia Marín, Oscar González y Silvia Marqués Departamento de Microbiología Ambiental y Biodegradación, Estación Experimental del Zaidin, CSIC, Granada [email protected] Azoarcus anaerobius es una bacteria anaerobia estricta que degrada 1,3dihidroxibenceno utilizando el nitrato como aceptor de electrones, mediante una ruta oxidativa independiente de oxigeno1. La agrupación génica que codifica dicha ruta incluye dos reguladores de la familia de bEBP, redR1 y redR2. En respuesta a la presencia del sustrato, estos reguladores, que presentan más del 97% de identidad entre sí, activan la expresión de 3 operones regulados por promotores dependiente de σ54. Ambos reguladores son necesarios para obtener una inducción máxima de la ruta. RedR1 y RedR2 presentan un dominio sensor (NTD), un dominio central AAA+ y un dominio HTH de unión a ADN. El modelo estructural del extremo N-terminal basado en el cristal de DhaR2 ha puesto de manifiesto la existencia de dominios GAF y PAS conectados por un linker. En un mutante btdS, que codifica una proteína integral de membrana, la actividad de los tres promotores es constitutiva y depende exclusivamente de RedR2. El análisis de los reguladores truncados desprovisto del dominio NTD reveló que RedR2 pertenece al grupo de bEBPs reguladas positivamente (inactivas cuando se elimina el dominio sensor) y presentan actividad parcial en ausencia del efector, mientras que RedR1 está regulada negativamente por su dominio NTD y presenta actividad constitutiva cuando este es eliminado. Hemos establecido que BtdS interacciona con RedR2 a través de una cisteína presente en el dominio GAF, y lo mantiene secuestrada en la membrana. Dado que ambas proteínas son necesarias para activar los mismos promotores hemos planteado la hipótesis de que ambos reguladores formen un heterohexámero para conseguir una máxima actividad como ya se ha descrito en otro miembro de la familia3. Para determinar la existencia de interacción entre ambos reguladores hemos expresando ambas proteínas en un sistema heterológo observando una disminución de la actividad de la versión activa de RedR1 en presencia de la versión inactiva de RedR2 indicio de un posible secuestro del regulador activo por interacción entre ambos. Mediante ensayos de doble híbridos hemos corroborado la existencia de interacción entre ambos reguladores, donde el extremo NTD no juega ningún papel. Estas interacciones se confirmarán mediante ensayos de pull-down y cross-linking in vivo. Referencias: 1. Darley PI, Hellsten JA, Medina-Bellver JI, Marqués S, Schink B, Philipp B (2007) J Bacteriol 189,3824-33. 2. Shi R, McDonal L, Cygler M, Ekiel I (2014) Structure 22, 478-87. 3. Hutcheson S W, Bretz J, Sussan T, Jin S, Pak K (2001) J Bacteriol 183, 5589-98. Agradecimientos: Trabajo financiado con fondos FEDER a través de los proyectos BIO2011-23615 del MICINN y BIO2014-54261-R del MINECO Panel 82 Detección molecular de factores de virulencia y colonización en Campylobacter jejuni mediante PCR multiplex Sònia Paytubi, Sergio Huertas, Cristina Madrid, Carlos Balsalobre Departament de Genètica, Microbiologia i Estadística. Secció de Microbiologia, Virologia i Biotecnologia. Universitat de Barcelona. [email protected] Campylobacter es el agente causal de la la campilobacteriosis, una de las enfermedades bacterianas de transmisión alimentaria más común en todo el mundo. Se estima que el número de casos de campilobacteriosis es de 9 millones por año. Campylobacter jejuni, bacteria comensal de aves y algunos mamíferos, es la especie de Campylobacter más frecuentemente asociada con la campilobacteriosis. La infección por Campylobacter causa diarrea aguda, generalmente autolimitante. Las infecciones extraintestinales son poco frecuentes e incluyen bacteremia, meningitis, y otras infecciones invasivas. Además, una pequeña proporción de infectados por Campylobacter pueden desarrollar el síndrome de Guillian-Barré, una complicación neurodegenerativa grave. A pesar de su importancia como patógeno, se conoce relativamente poco sobre los mecanismos por los que Campylobacter causa enfermedad. Durante la patogénesis se ha constatado la importancia de la motilidad y la producción de la toxina citoletal distensiva (Cdt), así como de estructuras de superficie relacionadas con la adherencia e invasión. En el marco de un proyecto que pretende caracterizar una colección de cepas de Campylobacter jejuni aisladas de pollos de engorde, aves silvestres y humanos, hemos puesto a punto una PCR multiplex que permite detectar la presencia de distintos factores de virulencia y colonización: toxina citoletal distensiva (CdtA, CdtB, CdtC), flagelo (FlaA), proteína de membrana externa de unión a fibronectina (CadF), antígeno invasivo (CiaB), sistema de secreción de tipo VI (Hcp), proteasa/chaperona periplasmática (HtrA) y plásmido pVir (virB11). Tras el diseño de los cebadores marcados con fluorescencia y el desarrollo de la PCR multiplex, se ha procedido al análisis de los resultados obtenidos mediante electroforesis capilar en un secuenciador Sanger. Mediante el software GeneMapper, el secuenciador automático ofrece un análisis sencillo y preciso de PCRs múltiples, pues en una misma electroforesis se pueden separar diferentes fragmentos combinado tamaños y colores. Esta técnica de análisis ofrece una serie de ventajas sobre los geles de agarosa como son rapidez, resolución, sensibilidad y manejo de los datos. De esta manera se ha caracterizado la presencia de los factores estudiados en las distintas colecciones de aislados de Campylobacter de las que disponemos. Panel 83 Biofilm oxygen gradients and ribonucleotide synthesis: Anaerobic ribonucleotide reductases are essential for proper biofilm formation Anna Crespo, Lucas Pedraz, Josep Astola, Eduard Torrents Instituto de Bioingeniería de Cataluña (IBEC), Barcelona [email protected], [email protected] Pseudomonas aeruginosa is well known as a ubiquitous and extremely adaptable pathogen, able to live free in soil and water, but also causing infections in plants and animals, including humans. As an opportunistic pathogen, it is especially dangerous in immunocompromised individuals, where it usually causes extremely resistant chronic infections. These chronic infections correlate with the formation of a biofilm, which increases resistance to physical stress and antibiotic therapies, while enhancing cell-to-cell communication. During biofilm formation, due to the impeded oxygen diffusion, together with the oxygen consumption by the different cell strata, an oxygen gradient is established, from a completely aerobic top part to microaerophilic or even fully anaerobic environments in the lower layers. While usually listed as an obligate aerobe, P. aeruginosa is able to grow in the absence of oxygen via anaerobic respiration through dissimilatory nitrate reduction. To adapt to the different oxygenation conditions, this bacterial species displays a whole range of changes in gene expression. One of the genetic pathways that need to be tightly regulated depending on the situations confronted by the bacteria is the ribonucleotide reduction, performed by a sophisticated family of enzymes, the ribonucleotide reductases (RNRs). They constitute the only de novo pathway for the formation of the deoxyribonucleotides, the building blocks needed for DNA synthesis and repair. P. aeruginosa is one of the few bacteria encoding all three known RNR classes (Ia, II, and III). In this work we explored the role of the different RNR classes in biofilm formation. We demonstrated the importance of class II and III RNR for proper cell division in biofilm development and maturation. We also determined that these classes are transcriptionally induced during biofilm formation and under anaerobic conditions, through a specific activation by the Anr/Dnr system. These data can be integrated with previous knowledge about biofilms in a model where these structures are understood as a set of layers determined by oxygen concentration and contain cells with different RNR expression profiles, bringing us a step closer to the understanding of this complex growth pattern, essential for P. aeruginosa chronic infections. This work was supported in part through grants to ET from the Ministerio de Economia y Competitividad (BFU2011-24066 and BIO2015-63557-R), Generalitat de Catalunya (2014 SGR01260). LP is thankful to the Generalitat de Catalunya for its financial support through the FI programme (2015-FI-B-00817). Referencias: 1. Crespo, A., Pedraz, L., Astola, J., Torrents, E. (2016). Pseudomonas aeruginosa exhibits deficient biofilm formation in the absence of class II and class III ribonucleotide reductases due to hindered anaerobic growth. Frontiers in Microbiology. 7:688. 2. Crespo, A., Pedraz, L., Torrents, E. (2015). Function of the Pseudomonas aeruginosa NrdR transcriptional factor: global transcriptomic analysis and its role on the ribonucleotide reductase gene expression. PLoS One. 10(4):e0123571. 3. Torrents, E. (2014). Ribonucleotide reductases: essential enzymes for bacterial life. Frontiers in Cellular and infection microbiology. 4:52. doi: 10.3389/fcimb.2014.00052. Panel 84 Carbonic anhydrase is essential for Brucella growth at ambient CO2 concentrations. Lara Pérez-Etayo1, María Jesús De Miguel2, Raquel Conde-Álvarez1, Ignacio Moriyón1, Maite Iriarte1 and Amaia Zúñiga-Ripa1. 1 Departamento de Microbiología y Parasitología e Instituto de Salud Tropical, Universidad de Navarra, 2 Unidad de Sanidad Animal, Centro de Investigación y Tecnología Agroalimentaria (CITA). [email protected] Brucellosis is an important zoonosis caused by bacteria of the genus Brucella. Animal vaccination is the main way to prevent this disease, and the main vaccines available are B. abortus S19 and B. melitensis Rev1. However, there is no specific vaccine against B. ovis and protection is achieved using B. melitensis Rev1. Stopping vaccination with Rev1 when B. melitensis is eradicated leads to a clear increase in the number of infections caused by B. ovis and thus, research on specific vaccines is an area of increasing interest. Nevertheless, one of the main difficulties to develop a B. ovis-vaccine is the requirement of a high-CO2 atmosphere to grow. Another important issue concerns the antigens used for the serological diagnosis of B. ovis infections, which are obtained from the CO2independent strain B. ovis REO198. This strain carries a defect in the lipopolysaccharide, a molecule of major importance as diagnostic antigen (Soler-Lloréns 2014, PhD, Universidad de Navarra), and thus, this defect may result in reduced sensitivity and/or specificity. In this work, we have analyzed the mechanisms underlying CO2 dependence in Brucella and demonstrated that the incapability of B. ovis and two B. abortus strains (292 and 544) to grow under atmospheric CO2 conditions is due to the lack of an active carbonic anhydrase (CA). In the case of B. ovis, both genes encoding CAI and CAII are disrupted. In contrast, although CAI is conserved in the two B. abortus strains, the lack of a functional CAII seems to be responsible for the requirement of a high-CO2 atmosphere. Consequently with these results, the introduction of an active CAII is enough to allow Brucella growth in atmospheric conditions, while the role of CAI remains to be unveiled. Interestingly, the introduction of a functional CAII into B. ovis does not affect the virulence of this strain, and therefore, it is an excellent background for the development of specific vaccines. Finally, this B. ovis strain able to grow under ambient air is a useful tool for the production of B. ovis antigens for diagnosis. This research was supported by a grant from the Ministerio de Economía y Competitividad of Spain (AGL2014-58795-C4). L.P-E has a grant from the Instituto de Salud Tropical (Fundación Empresa Universidad de Navarra). Panel 85 Novel c-di-GMP binding domain involved in production a mixed-linkage β-Glucan in Sinorhizobium meliloti Daniel Pérez-Mendoza1, Daniela Bertinetti2, Oliver Bertinetti2, Friedrich W. Herberg and Juan Sanjuán1 1 Dpto. Microbiología del Suelo y Sistemas Simbióticos, Estación Experimental del Zaidín, CSIC. Granada, Spain. 2 Department of Biochemistry, University of Kassel, 34132 Kassel, Germany. [email protected] An artificial increase of c-di-GMP levels in Sinorhizobium meliloti 8530 leads to overproduction of a mixed-linkage (1→3)(1→4)-β-d-glucan (MLG) that resembles ML βglucans found in cereals and lichens1. In contrast to lichenan and barley glucans, this unique bacterial exopolysaccharide has a distinctive primary structure with a perfect alternation of β(1→3) and β(1→4) bounds, which may give new interesting biotech properties to this biopolymer2. Moreover, the MLG participates in bacterial aggregation and biofilm formation, and is required for efficient attachment to the roots of a host plant, resembling the biological role of cellulose in other plant associated bacteria1. A two-gene operon bgsBA required for production of this MLG, is conserved amongst several genera within the order Rhizobiales, where bgsA encodes a glycosyl transferase (GT) with domain resemblance and phylogenetic relationship to bacterial cellulose synthases (CS). Furthermore, MLG synthesis is also subjected to both transcriptional and posttranslational regulation, but in contrast to cellulose: (i) bgsBA transcription is dependent on the ExpR/SinI quorum sensing regulatory system and (ii) a novel c-di-GMP binding domain, different to PilZ, is involved in the posttranslational regulation1. Different Biomolecular Interaction Analysis (BIA), including Fluorescence Polarisation (FP) and Surface Plasmon Resonance (SPR)3, have been used in this study to characterise the binding of BgsA to c-di-GMP. Various site directed BgsA mutants has been assayed to identify the residues involved in the c-di-GMP binding and activation of MLG production. Preliminary results obtained so far suggest a different activation mechanism from other cdi-GMP-activated GTs like cellulose synthase. References: 1 Pérez-Mendoza, D. et al. Novel mixed-linkage beta-glucan activated by c-di-GMP in Sinorhizobium meliloti. Proc Natl Acad Sci U S A 112, E757-765, doi:10.1073/pnas.1421748112 (2015). 2 Pérez-Mendoza, D. & Sanjuán, J. Exploiting the commons: cyclic diguanylate regulation of bacterial exopolysaccharide production. Curr Opin Microbiol 30, 36-43, doi:10.1016/j.mib.2015.12.004 (2016). 3 Moll, D. et al. Biomolecular interaction analysis in functional proteomics. J Neural Transm 113, 1015-1032, doi:10.1007/s00702-006-0515-5 (2006). Acknowledgments: DPM is supported by Andalucía Talent Hub Program launched by the Andalusian Knowledge Agency, co-funded by the European Union’s Seventh Framework Program, Marie Skłodowska-Curie actions (COFUND – Grant Agreement nº 291780) and the Ministry of Economy, Innovation, Science and Employment of the Junta de Andalucía. Panel 86 Autophagy as an adaptive response to stress in Chlamydomonas reinhardtii M. Esther Pérez-Pérez1, Stéphane Lemaire2 and José L. Crespo1 Instituto de Bioquímica Vegetal y Fotosíntesis, Consejo Superior de Investigaciones Científicas (CSIC)-Universidad de Sevilla. 41092 Sevilla, Spain. 2 Centre National de la Recherche Scientifique; UMR8226; Laboratoire de Biologie Moleculaire et Cellulaire des Eucaryotes; Institut de Biologie Physico-Chimique; Paris, France. [email protected] 1 Autophagy is a membrane-trafficking process by which unnecessary or damaged cellular components are degraded and recycled to maintain the cellular homeostasis or to cope with stress. This degradative process is characterized by the formation of double membrane vesicles, called autophagosomes, which engulf the cellular material that will be finally degraded in the vacuole. ATG8 protein plays an essential role in autophagosome formation and has been widely used as autophagy marker [1]. We have described the autophagy process in the model green alga Chlamydomonas reinhardtii [2]. Using ATG8 as a specific autophagy marker, we have demonstrated that autophagy is induced in Chlamydomonas by a wide range of stress conditions including nutrient limitation, endoplasmic reticulum stress, oxidative stress or photo-oxidative damage [2, 3]. Therefore, autophagy can be considered as an adaptive response to stress in Chlamydomonas. Our results also revealed a link between the production of reactive oxygen species (ROS) and autophagy activation in Chlamydomonas [3, 4]. Mounting evidence suggests that ROS may play a role in the control of autophagy by regulating ATG4 activity [5, 6]. ATG4 is a cysteine protease with an essential function in ATG8 maturation and thus in autophagosome biogenesis [1]. In this study we have unraveled the molecular mechanism for the redox regulation of Chlamydomonas ATG4. We demonstrate that ATG4 activity depends on the redox potential and is regulated by the formation of a disulfide bond controlled by thioredoxin. In addition, we found that ROS led to the oxidation and consequent inactivation and aggregation of ATG4. Thus, we propose that the fine-tuning of ATG4 activity and conformation by the intracellular redox state regulates autophagy in Chlamydomonas. [1] Ichimura et al. (2000) Nature 408:488-492. [2] Pérez-Pérez ME, Florencio FJ and Crespo JL (2010) Plant Physiol 152(4):1874-1888. [3] Pérez-Pérez ME, Couso I and Crespo JL (2012) Autophagy 8(3):376-388. [4] Pérez-Martín M, Pérez-Pérez ME, Lemaire SD and Crespo JL (2014) Plant Physiol 166(2):997-100. [5] Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L and Elazar Z (2007) EMBO J 26(7):1749-1760. [6] Pérez-Pérez ME, Zaffagnini M, Marchand CH, Crespo JL and Lemaire SD (2014). Autophagy, 10(11):1953-1964. Panel 87 Construction and characterization of TB vaccines based on MTBVAC in modern lineages of Mycobacterium tuberculosis Irene Pérez1, Carlos Martín1,2, Jesús Gonzalo-Asensio1,2 1 Grupo de Genética de Micobacterias, Universidad de Zaragoza, Zaragoza, Spain 2 CIBER enfermedades respiratorias. Instituto de Salud Carlos III, Madrid, Spain [email protected], [email protected], [email protected] Tuberculosis (TB) is caused by Mycobacterium tuberculosis (MTB) and is one of the infectious diseases that cause more mortality (WHO, 2015). BCG is the only actual vaccine against TB, but its efficacy against pulmonary diseases in adults is variable. To overcome this problem of BCG, it has been constructed a new live attenuated vaccine candidate, MTBVAC, which is in clinical trials (phase Ib, NCT02013245). MTBVAC is based on the deletion in phoP and fadD26 genes, both related with virulence factors (Arbués, 2013). MTBVAC belongs to lineage 4 (Euro-American-Asian) of Mycobacterium tuberculosis (Mtb). The principal objective of this work is to construct the same deletions of MTBVAC in two strains of linages 2 (Asian Beijing) and 3 (African-Indian). These are modern lineages, which together with lineage 4, are highly distributed and responsible for the current transmission of MTB in humans. We have selected clinical strains from lineages 2 and 3 using RFLP and spoligotyping techniques. Vaccines will be constructed in these lineages using the same method that was used to construct MTBVAC. Suicide plasmids which contains the gene (phoP or fadD26) disrupted by an antibiotic resistance gene and a counter-selectable (negative) marker will be used (Arbués, 2013). Mutants will be constructed through a two-step process involving a positive/negative double selection. The antibiotic marker will be eliminated by using a γδ-resolvase which acts on res sites flanking the resistance marker since these constructions will be used as potential vaccine candidates. MTBVAC has been extensively characterized. It has been studied the transcriptome of the transcription factor PhoP, its lipidomics profile by thin-layer chromatography in which it was observed the devoid of cell-wall lipids phthiocerol dimycocerosates (DIM), diacyltrehaloses and polyacyltrehaloses (DAT/PAT) as direct consequence of fadD26 and phoP deletions, respectively (Arbués, 2013). It has also been studied the expression profile of the principal PhoP-regulated genes (pks2, pks3, espA…) by qRT-PCR (Gonzalo-Asensio, 2008; Solans, 2014) and the inability to secrete ESAT-6 (Gonzalo-Asensio, 2014) by Western blot. To study whether these newly constructed vaccines show the same properties as MTBVAC, we plan to deeply characterize these phenotypes prior to conduct preclinical studies. References Arbues, A., et al., Construction, characterization and preclinical evaluation of MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials. Vaccine, 2013. 31(42): p. 4867-73. Gonzalo-Asensio, J., et al., PhoP: a missing piece in the intricate puzzle of Mycobacterium tuberculosis virulence. PLoS One, 2008. 3(10): p. e3496. Gonzalo-Asensio, J., et al., Evolutionary history of tuberculosis shaped by conserved mutations in the PhoPR virulence regulator. Proc Natl Acad Sci U S A, 2014. 111(31): p. 11491-6. Solans, L., et al., The PhoP-dependent ncRNA Mcr7 modulates the TAT secretion system in Mycobacterium tuberculosis. PLoS Pathog, 2014. 10(5): p. e1004183. WHO (2015). Global Tuberculosis Report 2015. Funded by: Diputación General de Aragón (DGA). European Project TBVAC2020. Panel 88 iFA762: Modelo metabólico a escala genómica con capacidad para simular adaptaciones metabólicas dependientes de salinidad en Chromohalobacter salexigens Francine Piubeli1, Montserrat Argandoña1, Manuel Salvador2, Carmen Vargas1 y Joaquín Nieto1 1 Departamento de Microbiología y Parasitología, Facultad de Farmacia, Universidad de Sevilla 2 Faculty of Health and Medical Sciences, University of Surrey, Guildford, UK [email protected] La bacteria halófila Chromohalobacter salexigens es capaz de crecer en un amplio rango de salinidades (0,1-4 M NaCl, uno de los más amplios descritos en la naturaleza) gracias, principalmente, a complejas adaptaciones de su metabolismo en función de la salinidad permitiendo generar los intermediarios metabólicos necesarios para la síntesis y acumulación de ectoínas en cada condición. Con objeto de profundizar en el conocimiento del complejo metabolismo de C. salexigens y la adaptación de éste al estrés osmótico, así como su relación con la producción de ectoínas, hemos desarrollado un modelo metabólico a escala genómica (iFA762) con capacidad para simular la respuesta metabólica a baja y alta salinidad (0,6 M vs 2,5 M NaCl). Este modelo refleja con bastante precisión el sistema biológico ya que las rutas bioquímicas incluidas y genes asociados han sido manualmente revisados en base a estudios multiómicos, bioquímicos y fisiológicos previos. Además se han incluido dos reacciones de biomasa (reacción asociada al crecimiento) específicas para cada salinidad, modificando los coeficientes de determinados metabolitos dependientes de salinidad basándonos en datos experimentales de C. salexigens. Las simulaciones se realizaron limitando el sistema utilizando datos metabólicos obtenidos previamente, como las tasas de consumo de fuente de C y excreción de ácidos orgánicos a diferentes salinidades, permitiendo así aumentar la capacidad de predicción del modelo. Las distribuciones de flujos obtenidas por el modelo en condiciones de alta y baja salinidad se compararon de forma cualitativa con datos de transcriptómica diferencial (RNA-seq, SOLID) obtenidos en las mismas condiciones. Los resultados mostraron una correlación entre la expresión diferencial de algunos grupos de genes y su distribución de flujos en función de la salinidad. Entre ellos destacan la síntesis de ectoína a alta salinidad y la oxidación fosforilativa, así como la síntesis del grupo hemo a baja salinidad, entre otros. Estos resultados muestran la capacidad del modelo iFA762 para simular en distintas salinidades permitiéndonos conocer los principales mecanismos metabólicos implicados en la adaptación al estrés osmótico. Esta información nos ayudará a desarrollar nuevas estrategias de ingeniería metabólica para la optimización de cepas productoras de ectoínas. Financiación: MINECO/FEDER (BIO2015-63949-R) y Junta de Andalucía (P11-CV1-7293 MO). Panel 89 A different slant in anti-biofilm drug discovery Juana María Prieto Mariscal1, Iñigo Lasa2 and Cristina Latasa1 1- Recombina SL. Calle Nueva 8 local 10, 31192 Mutilva, Navarra, España. 2- Navarrabiomed-Universidad Pública de Navarra, IdiSNA, Pamplona-31008, Navarra, Spain [email protected] After 100 years of extensive research, it may be said that we are capable of keep acute infections at bay. Though, we are witnessing a rising concern about the dramatic spread of antibiotic-resistant strains and chronic infections caused by biofim-forming bacteria, against whom traditional therapies are not sufficiently effective. Specially worrying are the matrix-enclosed communities assembled by the pathogens Staphyloccocus aureus and epidermidis, the most frequent causes of nosocomial infections and infections on indwelling medical devices The polysaccharide PIA/PNAG is a major component of staphylococcal biofilms. The production of this high molecular weight polymer depends on the proteins encoded in the icaADBC intercellular adhesion locus, an operon that is subjected to strict regulation, both at transcriptional and post-transcriptional levels. Contrary to what it might seem, production of PIA/PNAG does not always confer a selective advantage, proof of which are the on-off mechanisms like phase-variation that regulate its expression (Arciola, Campoccia, Ravaioli, & Montanaro, 2015). Our work hypothesis is based on the fact that production or overproduction of PIA/PNAG entails a significant fitness cost for the bacteria and such process might be turned toxic, and thus become an Achilles heel. We have designed a High Throughput Screening assay in which several PIA/PNAG overproducer strains and their icaADBC mutant counterparts have been treated with different concentrations of a collection of marine extracts and compounds. Up to date, we have selected two compounds, previously proved to possess antibiotic and antibiotic-cytotoxic activities respectively, and five crude extracts proceeding from the fermentation of marine Actinomyces and fungal species that specifically inhibit, or do it in significant higher levels, the growth of those strains capable of producing a PIA/PNAG-dependent biofilm in comparison to their PIA/PNAG-defective derivatives. Further studies are being undertaken in order to characterize the molecular mechanisms undelaying the action of these compounds/extracts and their potential synergies whether in combination between them or with classical antibiotics. Arciola, C. R., Campoccia, D., Ravaioli, S., & Montanaro, L. (2015). Polysaccharide intercellular adhesin in biofilm: structural and regulatory aspects. Frontiers in Cellular and Infection Microbiology, 5(114), 7. http://doi.org/10.3389/fcimb.2015.00007 This project is supported by the Spanish Ministry of Economy and Competitiveness. (Retos-colaboración 2015 call) Panel 90 Killing effect of R-pyocins on susceptible and multiresistant Pseudomonas aeruginosa clinical isolates Mar Redero1, Carla López-Causapé2, Jana Basas3, Joan Gavaldà3, Antonio Oliver2, Jesús Blázquez1,4 and Anabel Prieto1 Instituto de Biomedicina de Sevilla, Sevilla, Spain; 2Servicio de Microbiología y Parasitología, Hospital Universitario Son Espases, Palma de Mallorca, Spain; 3 Institut de Recerca-Hospital Vall d`Hebron, Barcelona, Spain; 4 Centro Nacional de Biotecnología, Madrid, Spain. [email protected] The recent appearance and dissemination of multidrug resistance (MDR) among bacteria have created a new threat for which traditional antibiotics offer little protection. After decades of neglect, the phage-therapy is beginning to be considered as a possible reemerging alternative. R-type pyocins are high-molecular-weight bacteriocins encoded in the chromosome of some bacterial species, such as Pseudomonas aeruginosa, supposedly to defend themselves against other strains of the same species. R-pyocins resemble the tails of lysogenic Myoviridae bacteriophages from which probably they evolved. The development of pulmonary MDR-P. aeruginosa infections is one of the most serious complications in the course of disease in patients with cystic fibrosis (CF), which often have very few antibiotic options . The analysis of approximately 200 P. aeruginosa clinical isolates shows that CF P. aeruginosa strains are hyper-susceptible to R-pyocins compared to isolates from other sources (bronchial aspirates, bloodstream, skin, sputum, abdominal, urine). The efficacy of R-pyocins in the eradication of P. aeruginosa infections has been tested through in vitro biofilms experiments and animal models. Treatments with R1, R2, or R5 pyocins prevent biofilm formation and also displayed a strong antimicrobial activity against established biofilms. In addiction, R-pyocins were also effective in ex vivo pig lung infection experiments, showing a 1000-fold decrease in the number of infecting bacteria. Moreover, administration of R2 pyocins in an in vivo murine model of ventilator-associated pneumonia (VAP) with a MDR clinical isolate (Pa1016) also showed a reduction of 100fold in the number of bacteria recovered from lung after 24h of infection. Altogether, these results strongly suggest that R-pyocins could be a possible alternative to be used as antimicrobial agent alone or in combination with antibiotic therapy for the treatment of P. aeruginosa infections. Panel 91 Mutational analysis of the permease domain of the cell-cell joining protein SepJ in Anabaena Félix Ramos-León, Vicente Mariscal, Antonia Herrero, Enrique Flores Instituto de Bioquímia Vegetal y Fotosíntesis, CSIC and Universidad de Sevilla, Sevilla [email protected] Anabaena sp. PCC 7120 is a filamentous cyanobacterium that grows as chains of cells. Under nitrogen deprivation, cells specialized in N2 fixation, known as heterocysts, differentiate from vegetative cells. Heterocysts are found in the filament with a semiregular pattern, separated by 10-20 vegetative cells. Heterocyst patterning is governed by positive transcription factors (NtcA and HetR) and by PatS- and HetN-related diffusible regulators, which inhibit heterocyst differentiation in cells away from those that already started differentiation. The cells in the filament are joined by septal junctions, structures analogous to metazoan gap junctions through which the diffusible regulators may be transferred between cells. Several proteins, including SepJ, that are located at the intercellular septa and may be components of the septal junctions have been identified in Anabaena. They appear to play a dual role, since they are required for filament integrity and are involved in intercellular molecular transfer, which can be probed with fluorescent tracers. Predictions of SepJ secondary structure show three well-defined domains: an Nterminal coiled-coil domain important for cell-cell binding, a linker domain required for septal localization and oligomerization, and a C-terminal permease domain that is similar to Drug and Metabolite Export (Dme) transporters. With the aim of gaining information on the transfer function of SepJ, Anabaena mutant strains expressing SepJ with alterations in the permease domain were constructed. Mutations included single amino acid substitutions and deletions of internal peptides and of C-terminal fragments resulting in truncated versions of the protein. The mutant strains were analyzed for growth on different nitrogen sources, filament length, expression of nitrogenase activity, heterocyst pattern and transfer of fluorescent tracers. Four mutants bearing amino acid substitutions in SepJ that produce long filaments but show an Mch (Multiple contiguous heterocysts) phenotype were identified. This phenotype was previously known from mutants lacking the diffusible regulators of heterocyst differentiation and may indicate an alteration in the intercellular transfer of the regulators in those SepJ site-directed mutants. Our results permit to separate the filamentation (cell-cell binding) and intercellular molecular transfer functions of SepJ, opening the way to a better understanding of the septal junctions. Work supported by grant no. BFU2014-56757-P from Plan Nacional de Investigación, Spain, co-financed by the European Regional Development Fund. Panel 92 Cambios transcripcionales inducidos por el efector SseK1 de Salmonella enterica en células humanas Fernando Baisón Olmo, Francisco Ramos Morales Departamento de Genética, Universidad de Sevilla [email protected] Salmonella enterica serovar Typhimurium posee dos sistemas de secreción tipo III, T3SS1 y T3SS2, codificados en las islas de patogenicidad 1 y 2, respectivamente. Estos sistemas se encargan de translocar proteínas, denominadas efectores, desde la bacteria al citosol de la célula hospedadora eucariótica. El T3SS1 transloca efectores a través de la membrana citosólica hospedadora y es importante para la invasión de células no fagocíticas. El T3SS2 se expresa después de la invasión y es necesario para el establecimiento del nicho intracelular típico de este patógeno, un fagosoma modificado denominado SCV (por Salmonella containing vacuole). SseK1 es uno de los efectores de los dos T3SS de Salmonella (1). Aunque puede secretarse a través de ambos sistemas, los patrones y cinéticas de translocación difieren según el tipo celular donde se ensayen (epiteliales, macrófagos o fibroblastos) (2). Se trata de un factor relevante para la virulencia en el modelo de ratón ya que la estirpe mutante nula para sseK1 es significativamente menos virulenta que la silvestre en ratones BALB/c, tanto inoculados por vía oral como intraperitoneal (2). Se desconoce, sin embargo, la función concreta que realiza SseK1 una vez translocado. Como primer paso para tratar de entender los efectos de SseK1 en la célula hospedadora hemos analizado el transcriptoma de células epiteliales humanas HeLa en diferentes condiciones. Hemos utilizado dos aproximaciones. La primera ha consistido en expresar de manera constitutiva el efector SseK1 en células HeLa mediante transfección estable y comparar el perfil transcriptómico de estas células con el de células transfectadas con el vector vacío usando micromatrices. En la segunda se han comparado los transcriptomas de células HeLa infectadas, bien con la estirpe silvestre de S. enterica serovar Typhimurium, bien con una estirpe mutante para sseK1. La integración de los resultados de ambos análisis ha permitido detectar 18 genes humanos que aumentan su expresión de manera significativa en respuesta a SseK1 y 14 que disminuyen su expresión debido a este efector. Los datos globales obtenidos se han sometido a análisis de ontología génica y se han validado mediante PCR cuantitativa. 1-Kujat Choy SL, Boyle EC, Gal-Mor O, Goode DL, Valdez Y, Vallance BA, Finlay BB. 2004. SseK1 and SseK2 are novel translocated proteins of Salmonella enterica serovar typhimurium. Infect Immun 72:5115–25. 2-Baisón-Olmo F, Galindo-Moreno M, Ramos-Morales F. 2015. Host cell type-dependent translocation and PhoP-mediated positive regulation of the effector SseK1 of Salmonella enterica. Front Microbiol 6:396. Financiación: MINECO SAF2013-46229-R Panel 93 Caracterización global del subproteoma regulado por cada sistema de doscomponentes en Staphylococcus aureus Beatriz Rapún1, Joaquín Fernández-Irigoyen2, Xabier Martínez2, Enrique Santamaría2, Jaione Valle1, Cristina Solano1, Begoña García1 e Iñigo Lasa1 1 Centro de Investigación Biomédica. Navarrabiomed. Universidad Pública de Navarra. Pamplona 2 Unidad de Proteómica. Centro de Investigación Biomédica. Navarrabiomed-Fundación Miguel Servet. Pamplona [email protected] Introducción: Las bacterias cuentan con diferentes sistemas de transducción de señales para establecer conexiones funcionales entre las señales ambientales y la fisiología bacteriana. Los sistemas más utilizados son los basados en procesos de fosforilación asociados a los sistemas de dos componentes (Two-component system, TCS). Los TCS están formados por un sensor de membrana o histidine kinase (HK) y un regulador de respuesta citoplasmático (RR). El número de TCS que contiene una bacteria varía en función del número de ambientes en los que es capaz de crecer y la complejidad de su diferenciación celular. En estudios previos, se generó en el laboratorio una cepa de Staphylococcus aureus deficiente en todos los TCS no esenciales (15) y un conjunto de cepas derivadas, cada una de las cuales conteniendo un único TCS. Objetivo: Identificación del subproteoma regulado por cada TCS de S. aureus. Material y Métodos: Para cada una de las 15 cepas de S. aureus que contienen un único TCS se prepararon extractos de proteínas totales en una condición de crecimiento (TSB, 37ºC, DO 0,8) y se llevó a cabo su caracterización mediante proteómica diferencial (label-free). Los resultados obtenidos se compararon con el extracto de proteínas totales de la cepas salvaje y del mutante deficiente en todos los TCS no esenciales, crecidos en las mismas condiciones. Resultados: En aquellos casos en los que se conoce un fenotipo asociado al TCS se confirmó que la expresión ectópica de dicho TCS era capaz de restaurar el fenotipo. Este control se realizó para los sistemas srr, vra, nre, arl y gra. Los análisis bioinformáticos permitieron caracterizar las proteínas específicamente afectadas por la expresión de cada TCS. Estas, están implicadas en determinadas funciones fisiológicas, algunas previamente descritas y otras identificadas por primera vez. Conclusiones: Hemos caracterizado el proteoma dependiente de cada TCS sin las posibles interferencias causadas por la presencia de otros TCS. Globalmente, los resultados indican que el número de proteínas específicas para cada TCS es bajo, sin grandes solapamientos entre los diferentes proteomas caracterizados y sugieren que existe una especificidad funcional de cada TCS en la regulación de determinadas funciones fisiológicas. Panel 94 Pseudomonas fluorescens F113 can produce a second flagellar apparatus, which is important for rhizosphere colonization Emma Barahona, Ana Navazo, Daniel Garrido-Sanz, Esther Blanco-Romero, Candela Muriel, Francisco Martínez-Granero, Miguel Redondo-Nieto, Marta Martín and Rafael Rivilla Departamento de Biología. Universidad Autónoma de Madrid [email protected] The genomic sequence of Pseudomonas fluorescens F113 has shown the presence of a 41 kb cluster of genes that encode the production of a second flagellar apparatus. Among 2535 pseudomonads strains with sequenced genomes, these genes are only present in the genomes of F113 and other six strains, all but one belonging to the P. fluorescens cluster of species, in the form of a genetic island. The genes are homologous to the flagellar genes of the soil bacterium Azotobacter vinelandii. Regulation of these genes is mediated by the flhDC master operon, instead of the typical regulation in pseudomonads, which is through fleQ. Under laboratory conditions, F113 does not produce this flagellum and the flhDC operon is not expressed. However, ectopic expression of the flhDC operon is enough for its production, resulting in a hypermotile strain. This flagellum is also produced under laboratory conditions by the kinB and algU mutants. Genetic analysis has shown that kinB strongly represses the expression of the flhDC operon. This operon is activated by the Vfr protein probably in a c-AMP dependent way. The strains producing this second flagellum are all hypermotile and present a tuft of polar flagella instead of the single polar flagellum produced by the wild-type strain. Phenotypic variants isolated from the rhizosphere produce this flagellum and mutation of the genes encoding it, results in a defect in competitive colonization, showing its importance for rhizosphere colonization. Funded by BIO2015-64480-R (MINECO/FEDER EU) Panel 95 La inactivación de la timidilato sintetasa ThyA modula la resistencia antibiótica y la interacción de Haemophilus influenzae con el aparato respiratorio humano Irene Rodríguez-Arce1, Sara Martí2,3, Begoña Euba1,2, Nahikari López-López1, Ariadna Fernández-Calvet1, José E. Yuste2,4, José Ramos-Vivas5,6, Carmen Ardanuy2,3, Josefina Liñares2,3, Junkal Garmendia1,2 1 Instituto de Agrobiotecnología, CSIC-Universidad Pública Navarra-Gobierno Navarra, Mutilva, Spain; 2Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES), Madrid, Spain; 3Departmento Microbiología, Hospital Universitari Bellvitge, University of Barcelona, IDIBELL, Barcelona, Spain; 4Centro Nacional de Microbiología, Instituto de Salud Carlos III (ISCIII), Madrid, Spain; 5Servicio Microbiología, Hospital Universitario Marqués de Valdecilla and Instituto de Investigación Marqués de Valdecilla (IDIVAL), Santander, Spain; 6Red Española de Investigación en Patología Infecciosa (REIPI), ISCIII, Madrid, Spain [email protected] Haemophilus influenzae (Hi) es un miembro habitual del microbioma respiratorio y el patógeno oportunista más frecuentemente aislado en muestras de esputo recogidas durante la exacerbación de pacientes con Enfermedad Pulmonar Obstructiva Crónica (EPOC). El tratamiento de las exacerbaciones implica la administración de antibióticos como Cotrimoxazol (SMX), y la consiguiente adquisición de resistencias. Entre los mecanismos de resistencia a SMX, se encuentra la auxotrofía a timidina asociada a mutaciones en el gen thyA, que codifica una timidilato sintetasa. En un estudio observacional de 2,542 aislados clínicos EPOC de Hi obtenidos entre 2010 y 2014 en el HU Bellvitge (Barcelona), 119 formaron colonias pequeñas con crecimiento lento en placas de Mueller Hinton Fastidioso (MHF), un medio con baja concentración de timidina. El análisis de estos 119 aislados identificó Hi8233, una cepa de un paciente que recibió SMX, auxótrofa natural a timidina, resistente a SMX (SMXR), con una inserción de seis nucleótidos en thyA. Para entender la relación entre auxotrofía a timidina, SMXR e interacción hospedador-patógeno durante la infección respiratoria por Hi, se generaron y caracterizaron los mutantes HiRdKW20ΔthyA y Hi375ΔthyA, en relación a auxotrofía a timidina, SMXR, morfología, crecimiento, auto-agregación, expresión génica, interacción con inmunidad innata, e infección pulmonar. La inactivación del gen thyA genera cepas Hi SMXR y produce auxotrofía a timidina. Esta auxotrofía se revierte en placas de MHF y cultivo líquido por adición de timidina exógena, y genera cambios morfológicos asociados a menor tasa de división, lo que aumenta el tamaño y facilita la auto-agregación bacteriana. La expresión del transportador de nucleósidos nupC aumenta en cepas HiΔthyA, lo que sugiere que la reversión fenotípica por adición de timidina exógena está relacionada con su captación. Asimismo, la auxotrofía a timidina disminuye la adhesióninvasión epitelial, y la carga bacteriana en pulmón y lavado broncoalveolar murino, si bien aumenta el fitness del patógeno en animales sometidos a un régimen terapéutico consistente en la administración oral de SMX. En conjunto, la auxotrofía a timidina en Hi es un mecanismo de SMXR que, concomitantemente, disminuye su capacidad infectiva. Este equilibrio resistencia antibiótica-virulencia debe ser clínicamente considerado en el tratamiento de las exacerbaciones EPOC. Panel 96 Estudio del papel de las proteínas ribosómicas en el ensamblaje de los ribosomas de Saccharomyces cerevisiae Francisco J. Espinar-Marchena1, José Fernández-Fernández1, Sara Martín Villanueva1, Julia Contreras, Olga Rodríguez-Galán1, Eduardo Villalobo2 y Jesús de la Cruz1 1 Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla y Departamento de Genética, Universidad de Sevilla. 2 Departamento de Microbiología, Universidad de Sevilla. [email protected] La mayoría de las proteínas ribosómicas juegan un papel esencial en la sintesis y función de los ribosomas. Nuestro grupo está interesado en conocer la función precisa de algunas de éstas proteínas en los procesos de síntesis del ribosoma y de traducción proteica, y su relación con otros procesos celulares como el ciclo celular. En esta comunicación nos centramos en los resultados obtenidos en el análisis las proteínas ribosómicas L16 y L14. Ambas proteínas son esenciales, su depleción in vivo resulta en un déficit de subunidades 60S y en la aparición de polisomas tipo “half-mer”. Este fenotipo es probablemente debido a la inestabilidad y rápida degradación de partículas pre-60S tempranas e intermedias, como lo demuestran la disminución en los niveles estacionarios de los RNAs pre-ribosómicos 27SBS y 7SL/S detectados por northern blot y primer extension y las pequeñas cantidades de 27S pre-rRNA y 25S rRNA detectados en experimentos de pulso y caza. Además, la depleción de L14 ó L16 bloquea el transporte nucleo-citoplásmico de las partículas pre-60S. Igualmente, mostramos que ambas proteínas se ensamblan en el nucleolo de manera temprana en partículas pre-ribosómicas 90S. Muchas proteínas ribosómicas están conservadas evolutivamente, presentando una estructura similar en bacterias, arqueas y eucariotas, sin embargo, muchas otras son más complejas y presentan extensiones extra amino- o carboxilo-terminales y/o lazos internos específicos de eucariotas. En esta presentación, también se discute la función de la extensión C-terminal, específica de eucariotas, de L16. Panel 97 A DNA methylation-dependent switch for the construction of biological sensors David R. Olivenza, Ignacio Cota, and Josep Casadesús Departamento de Genética, Universidad de Sevilla [email protected] The Salmonella enterica ovpAB operon encodes inner membrane proteins that control Oantigen chain length (1). Expression of opvAB shows phase variation under transcriptional control by Dam methylation and OxyR (2, 3). The OpvAB phase variation system can be used to construct artificial toggle switches with a variety of potential uses. An example is the use of OpvAB as a sensor of Dam methylase inhibition. Searches for DNA methylase inhibitors that might serve as antibacterial drugs have been sought for more than a decade, and have been hampered by lack of suitable sensors. A strain carrying a gfp fusion downstream of opvAB provides a highly sensitive sensor because Dam methylase inhibition yields an amplifiable signal: the number of cells in the ON state provides quantitative assessment of the inhibition (measured by fluorescence intensity). The OpvAB switch can also serve as a sensor of the presence of bacteriophages. A variety of bacterial surface structures serve as receptors for bacteriophages, including the O-antigen of the lipopolysaccharide. OpvABOFF cells are sensitive to bacteriophages that target the Oantigen while OpvABON cells are resistant. Because the OpvABOFF subpopulation is predominant (in LB, 99.8%), a Salmonella strain carrying an opvAB::gfp fusion shows very low fluorescence. In the presence of a bacteriophage, only the OpvABON subpopulation survives, and the presence of a bacteriophage can be detected by an increase in fluorescence. Sensors of this kind can be useful to assist in bacteriophage isolation trials, and to monitor the presence of bacteriophages (and hence of host bacteria) in food, water, environmental samples, etc. A ~700 bp DNA fragment containing the opvAB control region and the promoter can be used as a module that confers phase variable-expression to a lac operon cloned immediately downstream. This construction is active in E. coli and provides a proof of concept to imagine additional applications of the OpvAB switch. 1. Cota I, Blanc-Potard AB, Casadesús J. PLoS One 7:e36863, 2012 2. Cota I, Sánchez-Romero MA, Hernández SB, Pucciarelli MG, García-Del Portillo F, Casadesús J. PLOS Genetics 11:e1005667, 2015 3. Cota I, Bunk B, Spröer C, Overmann J, König C, Casadesús J.Nucleic Acids Research doi: 10.1093/nar/gkv1483, 2015 Panel 98 Un sistema de dos componentes homólogo a RstAB regula la expresión de tres hemolisinas en el patógeno Photobacterium damselae subsp. damselae Mateus S. Terceti, Amable J. Rivas, Manuel Noia, Carlos R. Osorio Instituto de Acuicultura, Universidade de Santiago de Compostela [email protected] Photobacterium damselae subsp. damselae es una bacteria marina de la familia Vibrionaceae que causa septicemia en peces y otros animales marinos, y también puede causar infecciones oportunistas mortales en humanos. Las cepas fuertemente hemolíticas contienen el plásmido pPHDD1, que codifica las hemolisinas Damselisina (Dly) y Phobalisina (PhlyP) [1,2]. Ambas, conjuntamente con la hemolisina HlyAch codificada en el cromosoma I, constituyen factores de virulencia claves. Las tres hemolisinas ejercen un efecto sinérgico tanto en la hemólisis como en la virulencia para ratones y peces [3]. Si bien conocemos su papel en la virulencia, los mecanismos mediante los cuales se regula la expresión de estas tres hemolisinas son desconocidos. En este trabajo hemos aislado un mutante por transposición con mini-Tn10, que mostró una fuerte afectación en su actividad hemolítica. La identificación del punto de inserción reveló que se trataba de un mutante en el gen rstB, homólogo a uno de los genes del sistema regulador de dos componentes RstAB descrito previamente en Enterobacterias. La reconstrucción del mutante por intercambio alélico demostró la implicación de rstAB en la hemólisis, y los ensayos de expresión mediante fusiones transcripcionales con el gen lacZ demostraron que la transcripción de los promotores de los genes de las tres hemolisinas Dly, PhlyP y HlyAch se veía fuertemente disminuida en el mutante rstB. Además, demostramos que la mutación de rstB produce efectos pleiotrópicos en el crecimiento y la división celular en condiciones de baja salinidad. La mutación de rstB causó una fuerte disminución de la virulencia de P. damselae subsp. damselae en peces utilizando la lubina como modelo animal. Este estudio representa la primera evidencia del papel de un sistema de dos componentes homólogo a RstAB implicado en la regulación de toxinas en bacterias. [1]-Rivas, AJ., Balado, M., Lemos, ML., & Osorio C.R. Infect. Immun. 79: 4617-4627. 2011. [2]-Rivas, AJ., Von Hoven, G., Neukirch, C., Meyenburg, M., Qin, Q., Füser, S., Boller, K., Lemos, ML., Osorio C.R., & Husmann, M. Infect. Immun. 83(11): 4335-4348. 2015. [3]-Rivas, AJ., Balado, M., Lemos, ML., & Osorio C.R. Infect. Immun. 81(9): 3287-3299. 2013. Panel 99 Effect of N-acetyl-cysteine on antibiotic mediated bacterial mutagenesis Ana I. Rodríguez-Rosado1#, Estela-Ynes Valencia1,2,3,#, Coloma Costas1,2 , Rodrigo Galhardo3, Jesús Blázquez1,2* and Jerónimo Rodríguez-Beltrán1,2, Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain Centro Nacional de Biotecnología (CNB-CSIC), Madrid, Spain Department of Microbiology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, SP 05508-900, Brazil. [email protected] Bactericidal antibiotics increase bacterial mutagenesis mediated by induction of the SOS response and production of Reactive Oxygen Species (ROS) promoting the emergence of antibiotic resistance variants. Interestingly, high levels of ROS can additionally induce SOS. Therefore, antibiotics may produce cumulative mutagenic effect (ROS plus SOS), although the relative contribution of each factor has not been yet clarified. Inhibition of SOS and/or ROS could be useful strategies to diminish the mutagenic effect of antibiotics. Among other molecules, antioxidants have been suggested as possible adjuvants in antibiotic therapy. Nonetheless, ROS production has been linked to lethality of bactericidal antibiotics in bacteria. Thus, antioxidants could have an antagonistic effect on this lethality. Our research is aimed at investigating i) whether antioxidants can affect the killing activity of antibiotics, and ii) the relative contribution of SOS and ROS in bacterial mutagenesis. N-Acetyl-Cysteine, a widely used FDA approved antioxidant, was able to decrease the mutagenesis promoted by ciprofloxacin, a bactericidal antibiotic and well-known SOS inducer without affecting viability of Escherichia coli. The relative contribution of ROS and SOS on mutagenesis will also be presented. Panel 100 Manipulación de la formación de biofilm y del metabolismo de Pseudomonas putida para procesos de bioconversión Laura Claret Fernández, Álvaro Escobar Doncel, José Martín Gómez, Gabriel Ruiz Romero, Fernando Govantes Romero y Rafael Rodríguez Daga Centro Andaluz de Biología del Desarrollo [email protected] Los biofilms bacterianos son biocatalizadores robustos ante cambios en las condiciones de operación, autoinmovilizados y autorregenerantes, especialmente adecuados para procesos que operan durante periodos prolongados de tiempo o aquellos en los que sustrato y/o productos pueden afectar a la viabilidad del organismo (1). Pseudomonas putida es una bacteria Gram-negativa formadora de biofilms que presenta numerosas ventajas como hospedador para procesos biotecnológicos (2). En el proyecto iGEM-UPO 2016 se pretenden utilizar métodos de ingeniería genética, ingeniería metabólica y biología sintética para implementar el uso de P. putida como plataforma para producción biotecnológica basada en biofilms. Para ello se ha realizado un diseño modular basado en dos líneas de actuación: (i) la generación de un sistema de control de la formación y dispersión del biofilm de P. putida que permita inducir la formación de un biofilm robusto o la dispersión del mismo a voluntad del usuario, y (ii) la generación de una ruta metabólica sintética que permita la bioconversión del glicerol, un subproducto de la industria del biodiésel, en un producto de valor añadido, el propionato. Para controlar el ciclo del desarrollo del biofilm pretendemos insertar de forma estable construcciones sintéticas que produzcan de forma alternativa factores que promuevan la formación del biofilm, como la diguanilato ciclasa PleD* o componentes del sistema de secreción de la adhesina LapA, o bien factores que promuevan la dispersión del biofilm, como la fosfodiesterasa YhjH o LapG. Para conseguir un derivado de P. putida que produzca propionato a partir de glicerol, se expresarán de forma regulada los genes glpF, implicado en el transporte de glicerol, y scpAB, argK y scpC, implicados en la conversión de succinato en propionato. La funcionalidad de los dos módulos se probará por separado en las estirpes indicadoras adecuadas, y finalmente se incorporarán todas las modificaciones en una única estirpe. Nuestros resultados actuales indican que nuestra estrategia de acción sobre el ciclo de desarrollo del biofilm permite controlar de forma estable y eficaz los procesos de formación y dispersión, y que la producción de GlpF promueve un crecimiento más rápido y robusto usando glicerol como sustrato. BIBLIOGRAFÍA 1. Rosche et al. (2009) Trends Biotechnol. 27(11):636-43 2. Poblete-Castro et al. (2012) Appl. Microbiol. Biotechnol. 93:2279-2290 Panel 101 Estudio de una nueva arquitectura de transcripción de genes en Staphylococcus aureus Sonia Sáenz1, Nerea Bitarte1, Begoña García1, Maite Villanueva1, Igor Ruiz de los Mozos1, Alejandro Toledo-Arana1, Íñigo Lasa1 1 Navarrabiomed-Fundación Miguel Servet [email protected] En un estudio previo caracterizamos el transcriptoma de Staphylococcus aureus y observamos que un elevado porcentaje del genoma se transcribe desde ambas hebras simultáneamente generando transcritos solapantes (1). Este solapamiento puede ocurrir en las regiones 5´ y 3´ de genes adyacentes, por la presencia de RNAs antisentido o por la existencia de operones solapantes. Los últimos se refieren a genes que encontrándose en la mitad del operón se transcriben en dirección opuesta. En todos los casos RNasaIII reconoce las regiones de solapamiento y lleva a cabo un procesamiento dando lugar a RNAs cortos de 20 nucleótidos (1,2). Los RNAs cortos son el producto de la degradación de los transcritos solapantes que están presentes simultáneamente en la misma bacteria ya que RNasaIII digiere únicamente RNA de cadena doble. El objetivo de nuestro estudio es estudiar la importancia biológica de esta arquitectura de transcripción utilizando como modelo el operón SA_01912-SA_01913-menCE cuya expresión solapa con la del gen SAOUHSC_01914. Para estudiar detenidamente esta arquitectura se generaron mutantes en los que los promotores de ambos RNAs fueron delecionados o sustituidos por promotores constitutivos para posteriormente analizar mediante Western y Northern-blot el efecto que uno de los RNAs solapantes tiene sobre el otro. El Northern-blot confirmó que la arquitectura objeto de estudio es un operón solapante. El Western-blot reveló que la deleción o expresión constitutiva del promotor de SAOUHSC_01914 influye en los niveles de expresión de proteínas SA_01912-SA_01913-menCE y viceversa lo que nos llevó a pensar que esta arquitectura de operón solapante conlleva una regulación de su expresión. Los resultados revelan que el operón SA_01912-SA_01913-menCE presenta un mecanismo de regulación post-transcripcional que coordina su expresión con la expresión de genes adyacentes basado en el solapamiento de transcritos que posteriormente serán procesados por RNasaIII. 1. Lasa I, et al. (2011). Genome-wide antisense transcription drives mRNA processing in bacteria. PNAS 108:20172-20177. 2. Lybecker M, et al. (2014). The double-stranded transcriptome of Escherichia coli. PNAS 111: 3134-3139. Panel 102 Identification of WadD, a new glycosyltransferase acting on Brucella LPS core synthesis Salvador-Bescós M., Moriyón I., Iriarte M., Conde-Álvarez R. Universidad de Navarra, Departamento de Microbiología y Parasitología [email protected] Brucellosis is a zoonotic disease caused by Brucella. The lipopolysaccharide (LPS) of Brucella plays a major role in its virulence as impairs normal recognition by the immune system, which delays the immune response. The LPS core of Brucella has an important role in the recognition by the immune system and in the resistance to the lytic action of complement and polycationic peptides. Mutants in glycosyltransferases involved in the LPS core synthesis are attenuated and good vaccine candidates against brucellosis. The chemical structure of the Brucella core LPS suggests that, in addition to the already identified WadB and WadC (Conde-Álvarez et al., 2012; Gil-Ramírez et al., 2014), other glycosyltransferases should also be implicated in its biosynthesis (Fontana et al., 2016). Thus, the study of genes putatively encoding enzymes with this function could help to improve the generation of new vaccines against brucellosis. Using bioinformatics tools (Carbohydrate-Active enZYmes Database) we identified 7 new ORFs putatively encoding core glycosyltransferases in B. abortus. We constructed mutants in these ORFs and observed that all of them kept the O-chain in their LPS. Interestingly, a mutant in ORF BAB1_0953 (named wadD) lost reactivity against polyclonal and monoclonal antibodies that recognize the core section. These results suggest that WadD is a new glycosyltransferase probably adding one or more sugars to the core ramification of Brucella LPS that is not linked to the O-chain. A mutant in WadD was more sensitive than the parental strain to the killing action of complement present in bovine serum and to bactericidal peptides. In vivo studies are being carried out in order to confirm its role in virulence. References: Conde-Álvarez, R., Arce-Gorvel, V., Iriarte, M., Manček-Keber, M., Barquero-Calvo, E., Palacios-Chaves, L., … Gorvel, J.-P. (2012). The Lipopolysaccharide Core of Brucella abortus Acts as a Shield Against Innate Immunity Recognition. PLoS Pathogens, 8(5), e1002675. http://doi.org/10.1371/journal.ppat.1002675 Fontana, C., Conde-Álvarez, R., Ståhle, J., Holst, O., Iriarte, M., Zhao, Y., … Widmalm, G. (2016). Structural studies of lipopolysaccharide defective mutants from Brucella melitensis identify a core oligosaccharide critical in virulence. Journal of Biological Chemistry, 291(14), 7727–7741. http://doi.org/10.1074/jbc.M115.701540 Gil-Ramírez, Y., Conde-Álvarez, R., Palacios-Chaves, L., Zúñiga-Ripa, A., Grilló, M.-J., Arce-Gorvel, V., … Iriarte, M. (2014). The identification of wadB, a new glycosyltransferase gene, confirms the branched structure and the role in virulence of the lipopolysaccharide core of Brucella abortus. Microbial Pathogenesis, 73, 53–59. http://doi.org/10.1016/j.micpath.2014.06.002 Research at the laboratories of the authors is supported by grants from the Ministerio de Economía y Competitividad of Spain (AGL2014-58795-C4-1-R). Fellowship support for M.S-B from the Asociación de Amigos de la Universidad de Navarra is gratefully acknowledged. Panel 103 Diversidad de elementos genéticos que codifican para las proteínas de las fimbrias F17 en Gallibacterium anatis y Avibacterium paragallinarum Ma. Patricia Georgina Sánchez Alonso1, María Elena Cobos Justo1, Jaqueline López Ochoa1, Erasmo Negrete Abascal2, Gloria Paniagua2, Sergio Vaca2, Norma Elena Rojas Ruiz1, Armando Tapia1, Teresita Jiménez1, Candelario Vázquez Cruz1. 1 Instituto de Ciencias, Centro de Investigaciones en Ciencias Microbiológicas. Benemérita Universidad Autónoma de Puebla. Edificio IC11, Ciudad Universitaria, Colonia San Manuel, CP. 72570, Puebla México. 2Carrera de Biología, Facultad de Estudios Superiores-Iztacala, UNAM, Avenida de los Barrios # 1, Los Reyes Iztacala, Tlalnepantla, Edo de México, 54090, México. [email protected] Introducción: Las bacterias Gallibacterium anatis y Avibacterium paragallinarum son patógenos de importancia veterinaria y económica, porque disminuyen la producción de huevo y carne de pollo para consumo humano. Se ha propuesto que uno de los factores de virulencia en estas bacterias son las fimbrias del tipo F17. Por lo que es importante conocer a nivel funcional y genético la participación de las fimbrias F17 en el proceso infeccioso e inmunogénico. Objetivos: Identificar in silico y clonar en E. coli los genes que constituyen los operones fimbriales en el genoma de G. anatis y A. paragallinarum. Material y Métodos: Por medios bioinformáticos se identificaron los posibles operones que codifican proteínas fimbriales. Y se sintetizaron oligonuclétidos para amplificar los segmentos de DNA que codifican para las proteínas de las chaperonas, proteínas estructurales y adhesinas de fimbrias F17 en G. anatis y A. paragallinarum. Los fragmentos de DNA obtenidos por PCR, se ligaron con el vector de clonación pBluescriptII KS (-). La selección de clonas candidatas fueron comprobadas mediante PCR, digestiones con enzimas HindIII y KpnI y secuenciación. Los datos se analizaron bioinformáticamente para establecer la relación filogenética de las secuencias. Resultados: Se identificaron tres potenciales operones que codifican fimbrias tipo F17 en G. anatis y en A paragallinarum, la organización genética de estos operones es muy conservada entre estas dos bacterias, pero hay diversidad de secuencias de genes que codifican adhesinas y proteínas estructurales. No obstante, el estudio predictivo estructural muestra una conservación funcional importante. Discusión: Las secuencias identificadas en estas bacterias son relativamente importantes para estudiarlas como medio para el diseño de inmunógenos y la generación de vacunas. Habrá que estimar experimentalmente la utilidad de las proteínas fimbriales como blancos del sistema inmune ya que siendo frecuentes estas proteínas en las bacterias se reportan poco como blancos para vacunas. Conclusiones: Para continuar el proyecto se requieren evaluar los aspectos inmunogénicos de cada uno de los componentes de las estructurales fimbriales, entre las que están la proteína estructural y la adhesina, especialmente para determinar la posibilidad de cruce antigénico en la misma cepa o entre cepas o especies. - RED-MODELOS MICROBIANOS DE IMPORTANCIA AGROBIOTECNOLOGICA - Panel 104 A regioselective synthesis of acylated carbohydrates and polyphenols with a novel acetylesterase (AE-6L) from Bacillus sp. HR21-6 Leyre Sánchez Barrionuevo,a,b Alejandro González-Benjumea,c María Guzmán,c David Cánovas,b Almudena Escobar-Niño,a,b María Teresa García,a José G. FernándezBolaños,c Inés Maya,c Óscar López,c Encarnación Mellado,a a Department of Microbiology and Parasitology, University of Seville, Profesor García González s/n, 41012, Seville, Spain b Department of Genetics, University of Seville, Avda de Reina Mercedes s/n, 41012, Seville, Spain. c Department of Organic Chemistry, University of Seville, Profesor García González 1, E-41012, Seville Spain. [email protected] Chemo-, regio- and stereoselective transformations constitute one of the most widely used strategies in organic synthesis, being especially valuable in the synthesis of complex and polyfunctional molecules such as carbohydrates and polyphenols1. In this context, chemoenzymatic synthesis is a powerful tool for accessing valuable derivatives and fine chemicals in a non-expensive and green fashion. Esterases are ubiquitous enzymes with important biotechnological roles in the synthesis or hydrolysis of ester-containing compounds. Herein, we describe the selection of a lipolytic microorganism, Bacillus sp. HR21-6, from food industries wastes, and its use for tailoring a new set of compounds with relevant pharmacological and biotechnological interest. In the first stage we used the cell-free lyophilized supernatant obtained from the microbial culture directly as a biocatalyst in the chemoselective and regioselective synthesis of partially acetylated compounds. Recently we have identified a novel acetylesterase (AE-6L) from the supernantant of Bacillus sp. HR21-6 sp. as the protein responsible of such activity described above. This activity appears to be unique of AE-6L, when compared with the activities of commercially available lipases from Candida antarctica (Novozyme® 435), since no reaction in these substrates was observed with the commercial enzymes. Expression and purification of this protein is the main objective of the present work. This enzyme displays broad substrate specificity and is active toward different p-nitrophenyl compounds. Therefore, the chemoenzymatic route proposed herein gives access to modify raw materials, and produce compounds of biological and pharmacological interest, in a green and inexpensive way compared to classical chemical synthesis that would involve timeconsuming protecting/deprotecting steps. References 1. González-Sabín, J.; Morán-Ramallal, R.; Rebolledo, F. Chem. Soc. Rev., 2011, 40, 5321. Panel 105 Efecto de las proteínas Hfq y Crc sobre el transcriptoma de P. putida Sánchez-Hevia, D.L., Moreno, R. and Rojo, F. CNB-CSIC [email protected] Pseudomonas putida es una bacteria metabólicamente muy versátil cuyo metabolismo está optimizado mediante un proceso de represión catabólica (RC) que le permite asimilar de forma preferente unas fuentes de carbono frente a otras. Los reguladores centrales de este sistema son las proteínas Hfq y Crc, que inhiben la expresión de genes implicados en la asimilación de sustratos no preferentes. Hfq, ayudada por Crc, se une a los mRNAs de estos genes, inhibiendo su traducción. La acción de Crc/Hfq está regulada por dos RNAs pequeños (sRNAs), CrcZ y CrcY, que pueden unirse a estas proteínas y secuestrarlas. Los niveles de CrcZ/CrcY varían mucho en función de la situación metabólica de la célula. La inactivación del gen hfq tiene un fenotipo mucho más acusado que la inactivación de crc, presumiblemente porque Hfq necesita a Crc para regular a algunos genes, pero puede regular otros de forma independiente. En otras bacterias, Hfq está implicada en muchos procesos de regulación post-transcripcional. Ensayos previos han indicado que, aunque Crc y Hfq son reguladores posttranscripcionales, su presencia o ausencia afecta mucho al transcriptoma de la célula. Para comprender mejor cuál es la influencia global estos reguladores en el metabolismo de la bacteria hemos realizado un análisis de RNA-Seq, comparando el transcriptoma de la estirpe parental con el de derivados mutantes en los que se han inactivado los genes crc, hfq, o crc y hfq. Un análisis preliminar de los datos ha confirmado que el efecto de la ausencia de Hfq es mayor que la falta de Crc. Además, el efecto de la doble mutación crchfq es bastante mayor que el de cada una de ellas por separado, lo que sugiere que el funcionamiento de Crc y Hfq es más complejo de lo que se piensa. Uno de los procesos alterados en el mutante hfq, pero no en el mutante crc, es el metabolismo del hierro, el cual está regulado por los sRNAs PrrF1 y PrrF2. Actualmente estamos estudiando este proceso regulador en más detalle. Panel 106 Comparative genomic analysis of Aquisalimonas species Carmen Infante-Domínguez, Rafael R. de la Haba, Antonio Ventosa and Cristina Sánchez-Porro Department of Microbiology and Parasitology, Faculty of Pharmacy, University of Sevilla, Spain [email protected] The genus Aquisalimonas comprises three moderately halophilic species, growing at 5 to 20% NaCl and optimally at 7.5-10% NaCl. They belong to the family Ectothiorhodospiraceae and class Gammaproteobacteria. They were isolated from different hypersaline habitats and geographical areas: Aquisalimonas asiatica from an alkaline, saline lake in Inner Mongolia Autonomous Region, China, Aquisalimonas halophila from a soil sample of a salt mine in Yunnan, China, and Aquisalimonas lutea was recently described and isolated from Santa Pola saltern, Spain (Infante-Domínguez et al., 2015). This moderately halophilic species of Aquisalimonas was isolated on the basis of the recent metagenomics studies carried out by our research group, which indicated that members of the family Ectothiorhodospiraceae constitute an abundant cluster of the prokaryotic fraction of hypersaline habitats. The purpose of this study was the comparative analysis of the genomes of these three Aquisalimonas species, specially focused on their adaptation mechanisms to the stress conditions, in comparison with other phylogenetically related non-halophilic species, as well as the determination of their abundance/presence in hypersaline environments. The genomes of the type strains of the three species of Aquisalimonas were fully sequenced (Ilumina), assembled and annotated. Their size was comprised between 3.6 and 4.1 Mb and their GC content is relatively high (65-67%). The genomes had three rRNA operons and showed a high level of synteny of A. halophila and A. lutea but not between these species and A. asiatica. This feature is in correlation with the low Average Nucleotide Identity (ANI) between A. asiatica and the other two species of Aquisalimonas (85.0 %), while the ANI between A. halophila and A. lutea was 94.5%. The calculated in silico DDH between A. lutea and A. halophila and A. asiatica was 63% and 31%, respectively. A remarkable feature is the presence of six genomic islands, related to a large urease cluster, phosphonate metabolism, phage recognition target, other genes related with different metabolic functions or without specific function. Finally, we have studied in more detail the genes involved on the carbon and nitrogen metabolism, osmoregulatory mechanisms and the recruitment of the genomes from metagenomes obtained previously from hypersaline habitats with different salinities. The data indicate that they are not as abundant as members of the phylogenetically related genus Spiribacter, but constitute a proportion of the prokaryotic diversity on saline habitats with 13 to 21% NaCl. Reference Infante-Domínguez, C., Sánchez-Porro, C. and Ventosa, A. (2015). Aquisalimonas lutea sp. nov., a moderately halophilic bacterium from a saltern. Int J Syst Evol Microbiol 65, 1354-1359. This research was funded by the Spanish Ministry of Economy and Competitiveness (MINECO) through project CGL2013-46941-P, and by the Junta de Andalucía Excellence project P10-CVI-6226, both including FEDER funds. Panel 107 La importancia de la biestabilidad de SPI-1 en Salmonella enterica: el papel activo de la subpoblación OFF María Antonia Sánchez-Romero & Josep Casadesús Departamento de Genética, Facultad de Biología, Universidad de Sevilla, Sevilla, 41012 [email protected] La isla de patogenicidad 1 (SPI-1) de Salmonella enterica contiene genes que codifican para el aparato de secreción tipo 3 y para efectores que están implicados en la invasión de células epiteliales. Uno de los rasgos más característicos de SPI-1 es su expresión biestable: se generan dos subpoblaciones, una que expresa la SPI-1 (subpoblación SPI-1 ON) y la otra que no lo hace (subpoblación SPI-1 OFF). El significado biológico de esta biestabilidad ha sido abordado con anterioridad por estudios detallados y exhaustivos. Entre ellos, cabe destacar la biestabilidad considerada como una división de labores que implica un altruismo autodestructivo por la subpoblación SPI-1 OFF (Ackermann et al. Nature 454, 987-90, 2008). En otro estudio, sin embargo, se ha observado cómo la subpoblación SPI-1 OFF podría beneficiarse de la inflamación desencadenada por la subpoblación SPI-1 ON (Stecher et al. PLOS Biology 5:2177-89, 2007). Además, también se ha detectado un incremento en la tolerancia a antibióticos en células creciendo lentamente de la subpoblación SPI-1 ON (Arnoldini et al. PLOS Biology e1001928, 2014). En esta comunicación se describe un aspecto complementario e inesperado de la biestabilidad de SPI-1 en Salmonella enterica. Se muestra como una subpoblación SPI-1 ON, obtenida tras la separación de un cultivo celular con ambas subpoblaciones, no invade las células epiteliales. Esto sugiere un papel activo de la subpoblación SPI-1 OFF en la invasión. A favor de esta conclusión, también se revela que los defectos en invasión asociados a una expresión unimodal de SPI-1 pueden ser suprimidos por mutaciones que permitan la formación de una subpoblación SPI-1 OFF. Panel 108 El operón trn de Anabaena sp. PCC 7120: regulación y aspectos funcionales Javier Santamaría-Gómez, Miguel Ángel Rubio, Mauro Napolitano e Ignacio Luque Instituto de Bioquímica Vegetal y Fotosíntesis. CSIC y Universidad de Sevilla [email protected] En bacterias Gram-positivas son frecuentes los operones de genes de tRNA (trn). En Gram-negativas en cambio, los genes de tRNAs se encuentran generalmente dispersos en el genoma y se transcriben de forma independiente. No obstante, en algunas especies de proteobacterias y cianobacterias existen agrupaciones largas de genes trn probablemente adquiridos por transferencia horizontal. La cianobacteria Anabaena sp. PCC 7120 cuenta con una dotación de 48 genes trn, distribuidos de forma dispersa en el cromosoma, que se transcriben a un alto nivel y son suficientes para descodificar todos los codones del código genético. Además cuenta en su plásmido delta con un agrupamiento de 26 genes trn (7 de ellos son posiblemente pseudogenes) que se expresan como un operón, generando un único transcrito que se procesa para dar lugar a tRNAs individuales [1]. En condiciones estándar de cultivo, el nivel de expresión de este operón es muy bajo. Ello, junto al hecho de que los tRNAs del cromosoma son suficientes para sostener la traducción plantea la cuestión de si los tRNAs del operón cumplen alguna función o constituyen un vestigio evolutivo afuncional. Hemos determinado que la expresión del opéron trn del plásmido delta se induce considerablemente en condiciones diversas de estrés. Se ha observado la coexpresión del operón con la ORF vecina situada aguas arriba (all8564), que codifica una posible nucleasa. Mediante 5'-RACE se han identificado dos promotores, uno delante del gen all8564 y otro delante del primer tRNA del agrupamiento. La secuencia de este último se ajusta a la secuencia consenso del promotor dependiente de sigma 70 en cianobacterias, de lo que se deduce que la expresión del operón se regula probablemente por represión. La expresión regulada del operón sugiere que los tRNAs codificados en él cumplen una función en las condiciones de inducción. Actualmente estamos investigando si estos tRNAs participan en la síntesis de proteínas en el ribosoma mediante el análisis de su presencia en fracciones polisómicas. Se ha comprobado que en condiciones normales de crecimiento varios de estos tRNAs se aminoacilan [1] y actualmente analizamos si también lo hacen en condiciones de inducción. Dada la notable desviación de su secuencia respecto a los tRNAs isoaceptores codificados en el cromosoma, se contempla la hipótesis de que algunos tRNAs codificados en el operón puedan cargarse con aminoácidos que no corresponden a su anticodón, lo que daría lugar a que algunos codones de los mRNAs se tradujesen con un cierto grado de ambigüedad. [1] Puerto-Galán L & Vioque A (2012) FEMS Microbiol Lett 337(1):10-17. Panel 109 Adaptaciones compensatorias facilitan la coexistencia de plásmidos ColE1 Alfonso Santos-Lopez, Cristina Bernabe-Balas, Alvaro San Millan, Rafael Ortega-Huedo, Manuel Ares-Arroyo, Natalia Montero y Bruno Gonzalez-Zorn Departamento de Sanidad Animal y Centro de Vigilancia Sanitaria Veterinaria (VISAVET), Facultad de Veterinaria, Universidad Complutense de Madrid, Spain. [email protected] La coexistencia de plásmidos pequeños es un fenómeno muy común en la naturaleza: aproximadamente el 50% de los plásmidos ColE1 descritos en las bases de datos, cohabitan con otro plásmido tipo-ColE1, y el 99% de ellos cohabitan con al menos otro plásmido no ColE1. Sin embargo, puesto que la adquisición de plásmidos conlleva un coste biológico para la bacteria, es razonable pensar que la acumulación de varios replicones disminuirá significativamente el fitness bacteriano y por tanto, las bacterias portadoras de varios plásmidos se perderán en ausencia de presión selectiva. En este trabajo estudiamos dos aproximaciones que puedan explicar la cohabitación sin presión selectiva: i) cando los plásmidos son adquiridos inicialmente por las bacterias existen interacciones epistáticas positivas entre ellos que disminuyen su coste biológico, o ii) una vez que la bacteria ha compensado el coste biológico de un replicón, la adquisición de otro plásmido no conlleva ninguna disminución del fitness bacteriano. Para probar ambas hipótesis, utilizamos como modelo la cepa Haemophilus influenzae Rd y los plásmidos pB1000, pB1005 y pB1006. Cuando los replicones son adquiridos por la bacteria, cada plásmido confiere el mismo fenotipo, mantiene su propio número de copias y produce el mismo coste biológico independientemente de si están habitando la célula solos o cohabitando con otros ColE1. Por tanto no parece que la cohabitación de los replicones pueda ser explicada por interacciones epistáticas entre los replicones. Sin embargo, utilizando evolución experimental, demostramos como H. influenzae Rd era capaz de compensar completamente el coste biológico producido por pB1000. Interesantemente, una vez que H. influenzae Rd ha compensado el coste biológico de pB1000, la adquisición de nuevos replicones (pB1005, pB1006 o ambos) no produce un coste biológico extra. Por tanto proponemos que las adaptaciones compensatorias facilitan la adquisición y el mantenimiento de múltiples plásmidos ColE1 en un mismo hospedador. Este trabajo ha sido financiado por la Comisión Europea a través del proyecto EFFORT (613754-FP7). Panel 110 Prevalence of multi drug resistant Acinetobacter baumannii clinical isolates in a spanish hospital over five years Dahdouh E.1, Gómez-Gil R.2, Mingorance J.2, Pacho, S.1, Daoud Z.3, Suárez, M.1 1-Faculty of Veterinary, Department of Animal Health, Complutense University of Madrid, Madrid, Spain 2-Servicio de Microbiología, Hospital Universitario La Paz, IdiPAZ, Madrid, Spain. 3-Faculty of Medicine, Department of Clinical Microbiology, University of Balamand, Balamand, Lebanon. [email protected] Objectives. Acinetobacter baumannii is a nosocomial pathogen that is infamous and relevant for its ability to cause a wide range of infections and acquire resistance to many antibiotics. Three International Clones (ICs) have been described for this organism and have been implicated with certain carbapenemases. In this study, we aim at identifying the most prevalent IC as well as determining the antibiotic susceptibility profiles and carbapenemases among A. baumannii bloodstream isolates. Methods. 59 bloodstream isolates were collected from Hospital la Paz, Madrid between 2009 and 2013. The Vitek-2 system was used for the determination of the MICs. Multiplex PCRs for the identification of the international clones and the detection of the blaOXA-51-like, blaOXA-23-like, blaOXA-24-like, blaOXA-58-like, blaOXA-48, blaNDM, and blaKPC genes was done. Results. The majority of the tested isolates belonged to IC II and harbored blaOXA-24-like. More than 80% of the isolates were non-sensitive to ticarcillin, piperacillin, ampicillin/sulbactam, piperacillin/tazobactam, ceftazidime, ciprofloxacin, levofloxacin, and trimethoprim/sulfamethoxazole. 84.75% of the isolates were non-susceptible to carbapenems, 79.66% to cefepime and 16.95% to minocycline. Non-susceptibility to aminoglycosides ranged from 18.64% for amikacin to 54.24% for gentamycin and only 2 strains (3.39%) were resistant to colistin. This data shows that although alarmingly high resistance rates were detected for a wide range of antimicrobials, including carbapenems, most isolates remain susceptible to some aminoglycosides and to colistin. blaOXA-51-like was detected in 94.92%, blaOXA-24-like in 64.41%, blaOXA-58-like in 13.56%, and blaOXA-23-like in 11.86% of the isolates. blaOXA-48, blaNDM, and blaKPC weren’t detected in any isolate. 8.47% of the strains pertained to IC I, 71.19% to IC II, 6.78% to IC III, and 13.57% did not belong to any IC. This agrees with other studies that show that IC II is the predominant clone in Spain and that blaOXA-24-like is the most common carbapenemase among A. baumannii isolates. Conclusion. High rates of antimicrobial resistance was detected and non-susceptibility to carbapenems was as high as 84.75%. Nonetheless, the tested isolates remained susceptible to aminoglycosides and colistin. The high resistance rates detected highlight the need for intervention and the implementation of antimicrobial stewardship programs for safeguarding these valuable antibiotics for future use. Keywords. Acinetobacter baumannii, International Clone, Carbapenemases, Multi-Drug Resistance Panel 111 Caracterización de un citocromo de tipo c exclusivo de cianobacterias filamentosas formadoras de heterocistos Alejandro Torrado1, Mercedes Roncel1, José M. Ortega1, Manuel Hervás1, José A. Navarro1, Adrián Velázquez-Campoy2 y Fernando P. Molina-Heredia1 1 Instituto de Bioquímica Vegetal y Fotosíntesis, Universidad de Sevilla y CSIC. 2 Instituto de Biocomputación y Física de Sistema Complejos. Universidad de Zaragoza. [email protected] En cianobacterias, las cadenas de transporte de electrones fotosintética y respiratoria se encuentran situadas en el mismo sistema de membranas, compartiendo algunos elementos, como son el complejo de citocromos b6f y algunos mediadores móviles de naturaleza quinónica o proteica. Entre estos últimos encontramos el citocromo c6 que transfiere electrones desde el complejo b6f al fotosistema I y a las oxidasas terminales. El citocromo c6 es una hemoproteína que se encuentra en todas las cianobacterias. En algunas de ellas se ha encontrado una segunda isoforma, el citocromo c6-2, que es capaz de reducir al fotosistema I, pero no a las oxidasas terminales, ni es capaz de oxidar al complejo b6f. Además, en cianobacterias filamentosas formadoras de heterocistos hemos encontrado una tercera isoforma, el citocromo c6-3. En este trabajo describimos el análisis físico-químico, estructural y funcional del citocromo c6-3. Mediante estudios de interacción por calorimetría (ITC) y biocomputación, se ha determinado que esta proteína puede interaccionar con el complejo b6f y, por su potencial redox, podría captar electrones de éste. Sin embargo, el citocromo c6-3 no es capaz de donar electrones de manera eficiente al fotosistema I, aunque sí es capaz de donarlos a las oxidasas terminales que se expresan específicamente en heterocistos. También, mediante inmunofluorescencia se ha demostrado que la proteína se expresa de forma constitutiva en células cultivadas con nitrógeno combinado, aunque su expresión disminuye ligeramente en condiciones diazotróficas. Estos resultados nos inducen a pensar que la función del citocromo c6-3 podría estar relacionada con el desarrollo o funcionalidad del heterocisto. Panel 112 Disassembling bacterial extracellular matrix with DNase-coated nanoparticles to enhance antibiotic delivery in biofilm infections Aida Baelo1,, Riccardo Levato2,, Esther Julián3, Anna Crespo1, José Astola1, Joan Gavaldà4, Elisabeth Engel2,3, Miguel Angel Mateos-Timoneda2, Eduard Torrents1,* 1 Bacterial infections and antimicrobial therapies and 2Biomaterials for regenerative therapies. Institute for Bioengineering of Catalonia (IBEC), Barcelona, Spain. 3 Departament de Genètica i de Microbiologia, Facultat de Biociències, Universitat Autònoma de Barcelona, 08193, Bellaterra, Spain. 4Infectious Diseases Research Laboratory, Infectious Diseases Department, Vall d’Hebron Research Institute VHIR, Hospital Universitari Vall d'Hebron, Barcelona, Spain. *[email protected] Infections caused by biofilm-forming bacteria are a major threat to hospitalized patients and the main cause of chronic obstructive pulmonary disease and cystic fibrosis. There is an urgent necessity for novel therapeutic approaches, since current antibiotic delivery fails to eliminate biofilm-protected bacteria. In this study, ciprofloxacin-loaded poly(lactic-coglycolic acid) nanoparticles which were functionalized with DNase I, were fabricated using a green-solvent based method and their antibiofilm activity was assessed against P. aeruginosa biofilms. Such nanoparticles constitute a paradigm shift in biofilm treatment, since, besides releasing ciprofloxacin in a controlled fashion, they are able to target and disassemble the biofilm by degrading the extracellular DNA that stabilize the biofilm matrix. These carriers were compared with free-soluble ciprofloxacin, and ciprofloxacin encapsulated in untreated and poly(lysine)-coated nanoparticles. DNase I-activated nanoparticles were not only able to prevent biofilm formation from planktonic bacteria, but they also successfully reduced established biofilm mass, size and living cells density, as observed in a dynamic environment in a flow cell biofilm assay. Moreover, repeated administration over three days of DNase I-coated nanoparticles encapsulating ciprofloxacin was able to reduce by 95% and then eradicate more than 99.8% of established biofilm, outperforming all the other nanoparticles formulations and the freedrug tested in this study. These promising results, together with minimal cytotoxicity as tested on J774 macrophages, allow obtaining novel antimicrobial nanoparticles, as well as provide clues to design the next generation drug delivery devices to treat persistent bacterial infections. Referencias: 1 Baelo, A., Levato, R., Julian, E., Crespo, A., Astola, J., Gavaldà, J., Engel, E., Mateos-Timoneda, MA., Torrents, E. (2015). Degrading bacterial ECM with DNase-coated nanoparticles to enhance antibiotic delivery in biofilm infections. Journal of Controlled Release. 209:150-158. This work was supported in part through grants to ET from the Ministerio de Economia y Competitividad (BFU2011-24066 and BIO2015-63557-R), Generalitat de Catalunya (2014 SGR01260) and La Caixa (CAIXAIMPULSE). Panel 113 Caracterización de aislados de Salmonella enterica resistentes a bilis procedentes de la vesícula biliar del ratón Verónica Urdaneta y Josep Casadesús Departamento de Genética, Universidad de Sevilla [email protected] La bilis es una secreción digestiva con propiedades antimicrobianas. Las sales biliares actúan como detergentes en las membranas lipídicas, desnaturalizan proteínas y causan daños en el ADN. Un fenómeno relevante en la resistencia de Salmonella enterica a la bilis es la adaptación, que consiste en un aumento del grado de resistencia a sales biliares tras la exposición a una dosis subletal. Dicha resistencia, que permite resistir concentraciones letales de sales biliares, es reversible y desaparece en ausencia de bilis [1]. La adaptación implica cambios en la expresión génica. Una alternativa a la adaptación es la adquisición de resistencia a bilis por mutación, y se ha especulado con la posibilidad de que las propias sales biliares aumenten la tasa de mutación ya que son agentes oxidantes que causan lesiones en el DNA [2]. Esta comunicación describe estudios cuyo objeto era determinar si la adaptación de Salmonella a la vesícula biliar es mutacional o no mutacional. Para ello se infectaron ratones BALB/c y 129Sv5, y se rescataron células de Salmonella presentes en la vesícula biliar. Dichos aislados fueron clasificados en dos tipos: mutantes (con resistencia a bilis estable) y no mutantes ("adaptados", con resistencia a bilis inestable). La gran mayoría de aislados de la vesícula eran inestables, indicando que Salmonella sobrevive en la vesícula biliar por adaptación, y sugiriendo que la mutación es infrecuente pese a la alta concentración de sales biliares. Sin embargo, también se obtuvieron mutantes resistentes a bilis. La secuenciación del genoma completo de algunos de dichos mutantes confirmó que la envoltura celular de S. enterica tiene un papel relevante en la resistencia a bilis: la mayoría de aislados presentaban mutaciones en genes relacionados con membranas bacterianas, con la división celular, con el transporte, y con la síntesis de LPS. [1] S. B. Hernández, I. Cota, A. Ducret, L. Aussel, J. Casadesús. Adaptation and preadaptation of Salmonella enterica to bile. PloS Genetics 8(1): e1002459, 2012 [2] A. I. Prieto, F. Ramos-Morales, J. Casadesús. Bile-induced DNA damage in Salmonella enterica. Genetics 168: 1787-1794, 2004 Panel 114 Identificación de nuevos genes implicados en el crecimiento de Saccharomyces cerevisiae en condiciones de estrés osmótico Ramón González1, Pilar Morales1, Jordi Tronchoni1, Gustavo Cordero-Bueso2, Enrico Vaudano3, Rafael Torres-Pérez1, Eva Valero2 1 Instituto de Ciencias de la Vid y del Vino (CSIC-Universidad de La Rioja-Gobierno de La Rioja) 2 Universidad Pablo de Olavide 3 Consiglio per la Ricerca e Sperimentazione in Agricoltura, Centro di Ricerca per l’Enologia [email protected] Las altas concentraciones de azúcar del mosto de uva hacen que el estrés osmótico sea uno de los factores limitantes del crecimiento de S. cerevisiae durante las primeras etapas de fermentación. Los mecanismos de adaptación de las levaduras a dicho estrés han sido ampliamente estudiados, conociéndose los genes implicados en las principales cascadas de transducción de señal que desencadenan la respuesta de la célula, sin embargo constantemente se descubren nuevos aspectos fisiológicos de las células bajo estas condiciones. Este trabajo pretende identificar nuevos genes implicados en la tolerancia de la levadura al estrés osmótico mediante una aproximación a escala genómica, no sesgada; con experimentos de competición de colecciones de cepas de levaduras con una representación de todas las deleciones posibles de genes individuales y marcadas con código de barras. Los experimentos se realizaron por triplicado en cultivo continuo durante diez generaciones, equivalente a un crecimiento exponencial, en dos medios diferentes, con glucosa o sorbitol como agente estresante. La abundancia relativa de cada cepa al inicio y final del experimento se estimó mediante secuenciación masiva, tras amplificar la región correspondiente a los dos códigos de barras de cada cepa. Para la identificación de genes cuya deleción tiene un efecto negativo y específico sobre la tolerancia al estrés, se tuvieron en cuenta los resultados del experimento control, en condiciones no estresantes. El experimento permitió también identificar deleciones que confieren una desventaja selectiva en medio basal, pero que pueden ser rescatadas, elevando la presión osmótica del medio. Los resultados obtenidos son diferentes entre los experimentos con glucosa o sorbitol, aunque hay coincidencias, como el gen HOG1. Además, se revela la importancia de funciones no relacionadas hasta ahora con la tolerancia al estrés osmótico. Así, se aprecia que las células son muy dependientes del buen funcionamiento mitocondrial para tolerar el estrés por glucosa, o del control de la gluconeogénesis y funciones del peroxisoma, en el caso del sorbitol. También se observa el rescate por alta presión osmótica de cepas defectivas en algunos procesos de control transcripcional o en el sistema de membranas internas (Golgi, endosoma, retrómero, o los complejos EXCRT-I, II, y III). Agradecimientos Ministerio de Economía y Competitividad: proyecto AGL2015-63629-R, Junta de Andalucía: proyecto P10-AGR6544. Panel 115 Reconstrucción de una red de regulación transcripcional en respuesta al estrés osmótico en Chromohalobacter salexigens Ali Tahrioui1, Montserrat Argandoña1, Manuel Salvador2, Joaquín J. Nieto1, Carmen Vargas1 1 Departamento de Microbiología y Parasitología, Facultad de Farmacia, Universidad de Sevilla 2 Faculty of Health and Medical Sciences, University of Surrey, Guildford, UK [email protected] La regulación transcripcional es esencial para la adaptación de las bacterias a cambios en las condiciones ambientales. En Chromohalobacter salexigens una bacteria halófila de amplio rango salino de crecimiento, los mecanismos de osmoadaptación incluyen la adaptación de las membranas, el transporte y acumulación de osmoprotectores, como las ectoínas, y la adaptación de su metabolismo central. Esta respuesta necesita de complejos mecanismos de regulación, en su mayoría desconocidos. El objetivo principal de este estudio consiste en la reconstrucción de una red de osmorregulación en C. salexigens mediante un enfoque integrado que incluye el análisis de datos moleculares genómicos y transcriptómicos. Para ello se analizaron los datos de RNA-seq obtenidos a partir de células de C. salexigens crecidas a baja (0,6 M NaCl) y alta salinidad (NaCl 2,5 M), observándose que al menos 60 factores de transcripción (FTs), pertenecientes a diferentes familias, se expresaron diferencialmente en función de la salinidad. Posteriormente, se realizó un análisis in silico de dichos FTs, para su reanotación y asignación funcional, mediante el uso combinado de herramientas bioinformáticas e información procedente de diferentes bases de datos. Así, se infirieron interacciones regulatorias de los FTs reanotados con posibles genes dianas mediante predicción in silico de regulones, utilizándose redes de regulación caracterizadas en otros microorganismos como molde para inferir interacciones regulatorias adicionales. Finalmente, las interacciones obtenidas sirvieron de base para realizar un enriquecimiento por operones incorporando así nuevas interacciones. Los resultados obtenidos nos permitieron establecer una red de regulación transcripcional preliminar compuesta por 598 genes y 1192 interacciones regulatorias. Un análisis posterior de subredes de los FTs sobreexpresados a baja y alta salinidad mostraron que éstos controlan genes dianas asociados a una variedad de funciones celulares, como la obtención y conversión de energía, el transporte y metabolismo de amino ácidos, carbohidratos, nucleótidos, e iones inorgánicos, además de otros procesos biológicos. Estos resultados servirán como base para ampliar la red de regulación en C. salexigens, en la cual se podrán integrar otros elementos regulatorios relacionados con la osmoadaptación, como por ejemplo, sistemas de dos componentes o reguladores globales, entre otros. Financiación:BIO2015-63949-R(MINECO/FEDER,UE), Junta de Andalucía (P11-CV17293MO) Panel 116 El genoma de Bacillus thuringiensis DNG102III un potencial controlador biológico de actividad tóxica intermedia contra Aedes aegipty Candelario Vázquez Cruz1, María Patricia Georgina Sánchez Alonso1, Elena Cobos Justo1, Ana Jaqueline López Ochoa1, Erasmo Negrete Abascal2, Sergio Vaca Pacheco2, Moisés Carcaño Montiel1, Lucía López Reyes1. 1 Centro de Investigaciones en Ciencias Microbiológicas, Instituto de Ciencias, Benemérita Universidad Autónoma de Puebla, Edificio IC11, Ciudad Universitaria, Colonia San Manuel, CP 72570 Puebla México. 2Carrera de Biología, Facultad de Estudios Superiores, UNAM, Av. de los Barrios # 1, Los Reyes Iztacala, Tlalnepantla, Edo de México, 54090, México. [email protected] Introducción. Los mosquitos hembras de Aedes aegipty transmiten varias enfermedades virales al ser humano, como la fiebre amarilla, la del Oeste del Nilo, el dengue, chikungunya y zika. La alternativa de control integrado de vectores por medio de antagonistas biológicos no debe cesar como vía de control de estas enfermedades, dado que han repuntado, afectando la salud de la población y recientemente han aparecido enfermedades desconocidas en la América Latina (chikungunya y zika). Como medio de control biológico esta, caracterizar nuevas cepas de Bacillus thuringiensis con actividad mosquitocida. Objetivo. Identificar los genes toxigénicos cry albergados en la cepa DNG102III con actividad tóxica en larvas de Aedes aegypti. Material y Métodos. En el Laboratorio de Bioquímica y Genética Microbiana del CICMBUAP se describió la cepa de Bacillus thuringiensis DNG102III como toxigénica para Aedes aegipty que prolifera exitosamente a 70 kilómetros (Izúcar de Matamoros, 1283 m. s. n. m.) de la Ciudad de Puebla en México (1980 m. s. n. m. y coordenadas 19°03′05″N 98°13′04″O). El genoma de esta cepa fue secuenciado por medio de la tecnología 454 y ensamblado con MIRA4. Resultados y Discusión. La cepa DNG102III tiene limitada actividad contra Aedes aegipty, pero buena actividad contra Culex spp. La utilidad mayor de DNG102III se observó al hacer mezclas con el prototipo BTI, observándose sinergia. Para conocer cómo se logra esta se hizo la secuenciación del genoma de BT DNG102III, este genoma está ensamblado en 115 contigs y se estima su tamaño en 6.1 MB. En este genoma faltan genes cry para tener la combinación del prototipo BTI. Los genes identificados tienen la siguiente identidad: 76% con cry4B, 86% con cry10Aa, 60% con cry11A, 53% con etx_mtx2). Conclusión. No obstante que faltan genes importantes en DNG102III para igualar la actividad mosquitocida de BTI, la cepa es una herramienta útil para elaborar cocteles que quizás contribuyan a disminuir la incidencia de enfermedades virales transmitidas por mosquitos vectores y se puede convertir en una alternativa para remediar la aparición de resistencia hacia los biopesticidas de Bacillus thuringiensis. - RED-MODELOS MICROBIANOS DE IMPORTANCIA AGROBIOTECNOLOGICA - Panel 117 Chimeric Csl2 enzybiotic: towards the tailor-made anti-infectives Vázquez, R., García, P. Departamento de Microbiología Molecular y Biología de las Infecciones, Centro de Investigaciones Biológicas (CIB-CSIC) [email protected] Bacterial resistance to common antimicrobial therapies (i.e., antibiotics) is reaching alarming levels worldwide. This situation is pushing for the development of alternative antimicrobials, such as bacteriophage-encoded endolysins, also called enzybiotics. This antibacterial approach could pose a new and valuable adding to the antimicrobial arsenal, with increased specificity and much lower chances of enabling bacterial resistance mechanisms. Both database sequence search and protein engineering have proved to be effective approaches towards the discovery and improvement of phage endolysins for therapeutic uses, leading to the development in our lab of two highly efficient enzybiotics derived from Streptococcus pneumoniae phage lysins: the broad-acting Cpl-7S and the chimeric enzybiotic Cpl-711, the most potent enzybiotic described to date specifically against S. pneumoniae. We have recently added two more lysins to our current armamentarium: a natural endolysin (Sil73) from a Streptococcus ictaluri prophage active against a relevant fish pathogen (Streptococcus iniae) and a chimeric lysin (Csl2) built upon the fusion of the catalytic domain of S. pneumoniae phage Cp-7 endolysin and the cell wall binding domain from Streptococcus suis SMP phage endolysin, which resulted to have a rather specific bactericide activity against S. suis and some other closely related streptococci. Csl2 was able to kill the 99% of a S. suis culture with a 15 µg/ml treatment and showed also lethal activity against S. suis biofilms, so it is proposed as a specific therapeutic and prophylactic agent against S. suis With these results herein we are confident that enzybiotics are nowadays much more an immediate reality than a future promise and the possibilities that they present might lead in a not so distant future to the construction of case-specific tailor-made anti-infectives. Panel 118 Genomics of Spiribacter, new successful bacteria in hypersaline environments María José León, Cristina Sánchez-Porro and Antonio Ventosa Department of Microbiology and Parasitology, Faculty of Pharmacy, University of Sevilla, Spain [email protected] Hypersaline ecosystems are widely distributed habitats on Earth in which the main lifelimiting factor is their high salt concentration. Studies focused on culture-independent techniques in natural environments have shown that the most abundant, slow growing and well adapted to these extreme conditions microbes, are currently still extremely difficult to retrieve in pure culture. As a consequence, the physiology of the microorganisms that are present in abundance in hypersaline ecosystems is still mostly indirectly assessed through metagenomic approaches, or at best, studied through a few isolated species. Metagenomics-based studies in salterns, have recently permitted the isolation of new Gammaproteobacteria that appeared to be representative of the dominant bacterial group at intermediate salinities (10-20 % salts). It was taxonomically described as a new genus, Spiribacter. Altogether we have isolated eight new bacterial strains in pure culture in the laboratory. Their complete genomes have been sequenced and a detailed inter-species and inter-strains genomic analysis is being performed. Representatives of Spiribacter are widespread worldwide in habitats with intermediate salinity and the isolated strains represent the first ecologically defined moderate halophiles (León et al., 2014; 2015). Spiribacter species could be considered as interesting model organisms for understanding stress management mechanisms allowing microbes to cope with these extreme habitats. Inspection of the type species Spiribacter salinus M19-40 genome sequence revealed that it adheres to the “salt out” type of osmotic adjustment strategy. In this sense we combined physiological approaches and genome mining to derive a comprehensive picture of the molecular and cellular events that allow to the members of the genus Spiribacter to be successful bacteria in hypersaline environments. Collectively, our data show that newly synthesized ectoine and imported glycine betaine play key roles in the cellular defense of S. salinus M19-40 against the detrimental effects of high salinity on cellular physiology and growth. References León, M. J., Fernández, A. B., Ghai, R., Sánchez-Porro, C., Rodriguez-Valera, F. and Ventosa, A. (2014). From metagenomics to pure culture: Isolation and characterization of the moderately halophilic bacterium Spiribacter salinus gen. nov., sp. nov. Appl and Environ Microbiol 80, 3850-3857. León, M. J., Rodríguez-Olmos, A., Sánchez-Porro, C., López-Pérez, M., RodríguezValera, F., Soliveri, J., Ventosa, A. and Copa-Patiño, J. L. (2015). Spiribacter curvatus sp. nov., a new moderately halophilic bacterium from Santa Pola saltern in Spain. Int J Syst Evol Microbiol 65, 4638-4643. This research was funded by the Spanish Ministry of Economy and Competitiveness (MINECO) through project CGL2013-46941-P, and by the Junta de Andalucía Excellence project P10-CVI-6226, both including FEDER funds. Panel 119 Metabolic diversity of hypersaline environments Blanca Vera-Gargallo, Cristina Sánchez-Porro, Antonio Ventosa Department of Microbiology and Parasitology, Faculty of Pharmacy, University of Sevilla, Spain. [email protected] The advent of omics technologies has enabled the access to the metabolic potential of uncultured organisms, the so called “dark matter”, and it has brought to light the most abundant functional categories in diverse habitats. Recent metagenomic studies on hypersaline habitats have permitted to describe in detail the phylogenomic diversity of these habitats. The goal of this study is to explore the key prokaryotic metabolic pathways in hypersaline environments, comprising terrestrial and aquatic habitats with different salinities, making use of metagenomic databases. Salinity exerts a primary limitation on water availability and osmotic potential and has been shown to be an important microbial stressor in most environments. It can also lead to high internal levels of ions that are toxic to metabolic activities and can affect to the stability of the extracellular enzymes necessary for carbon and nutrient acquisition. Thus, increased salinity represents a biological stress that requires evolutionary or energetically expensive solutions. This leads to functional categories being differentially represented compared to non-saline environments. Furthermore, although several molecular studies have been carried out concerning aquatic hypersaline settings such as marine salterns, hypersaline lakes and the Dead Sea, hypersaline soils have received much less attention, resulting in a scarce knowledge about their microbial inhabitants and their capabilities. Previous studies focusing on saline soils have traditionally made use of techniques lacking the ability to access the vast diversity harbored by terrestrial systems. Hence, these communities represent a pool of rare forms of biodiversity containing likely important genomic resources that remain unexplored, as denoted by the extent of unannotated genes present in saline soil metagenomes. Comparison of terrestrial and aquatic representative genes involved in pathways related to osmotic stress resistance permit the assessment of how bacteria and archaea dwelling in such different hypersaline habitats cope with salinity. Acknowledgements This work was supported by grants from MINECO (CGL2013-46941-P) and Junta de Andalucía (P10-CVI-6226), both including FEDER funds. Blanca Vera-Gargallo was supported by a FPU scholarship from the Spanish Ministry of Education, Culture and Sports. Panel 120 Rifampicin resistant rpoB alleles, or multicopy thioredoxin/thioredoxin reductase, suppress the lethality of the global stress regulator spx in Staphylococcus aureus Maite Villanueva, Ambre Jousselin, Kristoffer T. Baek, Julien Prados, Diego O. Andrey, Adriana Renzoni, Hanne Ingmer, Dorte Frees, and William L. Kelley Department of Microbiology and Molecular Medicine, University Hospital and Medical School of Geneva, Geneva, Switzerland [email protected] Staphylococcus aureus is capable of a remarkable disease spectrum ranging from mild skin eruptions to life threatening infections. Part of S. aureus’ survival and pathogenic potential depends on its ability to sense and respond to changes in its environment. Spx is a thiol/oxidative stress sensor that interacts with the RNA polymerase RpoA subunit Cterminal domain leading to changes in gene expression to help sustain viability in various conditions. Using genetic and deep sequencing methods, we show that spx is essential in S. aureus and that a previously reported Δspx strain harbored suppressor mutations allowing S. aureus to grow without spx. One of these mutations is a single missense mutation in rpoB (P519L) conferring high-level resistance to rifampicin. This mutation alone was found to be sufficient to bypass the requirement for spx. The generation of rifampicin resistant libraries led to discovery of an additional rpoB mutation, R484H, which supported spx disruption. Other rifampicin resistant mutations either failed to support Δspx, or were recovered with unexpectedly low frequency in genetic transduction experiments. Both rpoB P519L and R484H map in close spatial proximity and comprise a highly conserved region of RpoB. We also discovered that multicopy expression either of trxA (thioredoxin), or trxB (thioredoxin reductase) driven by a T-box riboswitch glyS tRNA synthetase promoter supports the deletion of spx supporting the link between transcription, redox homeostasis, and at least one Spx known function: regulation of trxB. Follow-on experiments using deep sequencing methods demonstrated a surprising global impact of rifampicin-resistant rpoB alleles on transcription regulation. Our results reveal intriguing properties, especially of RNA polymerase, that compensate for the loss of an essential gene that is a key mediator of diverse processes in S. aureus including redox and thiol homeostasis, antibiotic resistance, growth and metabolism. Panel 121 Role of operon rmlBACD of Stenotrophomonas maltophilia in lipopolysaccharide biosynthesis, antibiotic resistance and virulence-associated phenotypes Daniel Yero1, Uwe Mamat2, Sonia Martinez-Servat1, Roser Marquez1, Xavier Daura1,3, Isidre Gibert1 1 Institut de Biotecnologia i de Biomedicina (IBB) and Dept de Genètica i de Microbiologia, Universitat Autònoma de Barcelona (UAB), 08193 Bellaterra (Cerdanyola del Vallès), Barcelona, Spain. 2Division of Structural Biochemistry, Research Center Borstel, LeibnizCenter for Medicine and Biosciences, Parkallee 1-40, D-23845, Borstel, Germany. 3 Catalan Institution for Research and Advanced Studies (ICREA), 08010 Barcelona, Spain [email protected]; [email protected]; [email protected] Stenotrophomonas maltophilia is an environmental multi-drug resistant organism that is increasingly associated with nosocomial respiratory infections in humans. Most of the Oantigen repeating units of the lipopolysaccharide (LPS) of S. maltophilia are branched triand tetra-saccharides of rhamnose, fucose, xylose, and glucose. The dTDP-L-rhamnose biosynthetic pathway is involved in the activation of this sugar to be used as a precursor in the synthesis of LPS. In S. maltophilia the enzymes catalyzing these conversions are encoded by rmlB, rmlA, rmlC and rmlD, respectively, and are arranged in the operon rmlBACD (Locus tag Smlt0647-0650). In this work, we have obtained a mutant defective in the biosynthesis of the rhamnose-containing O-antigen of S. maltophilia K279a to study the role of full length LPS in the intrinsic resistance to hydrophobic antibiotics such as polymyxins and in virulence-associated phenotypes. The I-Sce I homing endonuclease system was used to construct unmarked deletion of gene cluster rmlBACD in S. maltophilia K279a. SDS-PAGE analysis of whole-cell lysates of the S. maltophilia rmlBACD mutant revealed that the operon is needed for O-antigen biosynthesis. The resulting mutant strain does not produce the O-antigen and exhibits a non-mucoid colony morphology and cellular aggregation in liquid cultures. Significantly, the rmlBACD mutant displayed an increase in susceptibility to antimicrobial peptide agents, particularly at antibiotic concentrations that induce adaptive resistance in the K279a strain. With the rmlBACD mutant, we have also confirmed that incomplete LPS can alter the biofilm production of S. maltophilia, and it showed a modest decrease in virulence in an animal infection model. Complementation analyses (by introducing the wild-type rmlBACD full operon) indicated that rmlBACD full operon is required to restore full LPS biosynthesis in the mutant as well as its mucoid colony morphology. The complementation also confirmed the role of the O-antigen in resistance and biofilm formation in S. maltophilia. Overall, mutations in the rmlBACD operon leading to rough-type LPS in S. maltophilia, like K279a, highlight the contribution made by LPS to the antimicrobial resistance and virulence of this bacterium. This result provides further insights into the mechanisms of LPS biosynthesis and assembly in this bacterium. Acknowledgments This work has been supported by Spanish MICINN/FEDER (BIO2015-66674-R) and Catalan AGAUR (2009SGR-00108). Part of this work was financially supported by inhouse funding of the Research Center Borstel. Panel 122 Mobilization mechanism of pathogenicity islands by endogenous phages of Staphylococcus aureus isolates from cystic fibrosis patients Miglė Žiemytė, Katerina Guzman, Mercedes Cervera, Eva Gonzalez and Mª Ángeles Tormo-Mas Grupo Acreditado de Infección Grave del Instituto de Investigación Sanitaria la FE [email protected] Cystic fibrosis (CF) is a multisystem disease, but respiratory manifestations are the leading cause of morbidity and mortality, so that combating infectious exacerbations is one of the main strategies for the treatment of these patients Staphylococcus aureus is one of the most common pathogens of CF patients particularly in patients of young age. S. aureus is an important pathogenic bacteria associated with a huge variety of infections and multidrug-resistance. The enormous versatility of S. aureus as a pathogen is due to its ability to persist and multiply in different environments along with its ability to produce some virulence factors, which are encoded by mobile genetic elements (MGE), such as bacteriophages and pathogenicity Islands. The staphylococcal pathogenicity islands (SaPIs) are the prototypical members of a novel family of mobile genetic elements, the phage-inducible chromosomal islands (PICIs). These elements are intimately related to certain helper phages, whose life cycles they parasitize. Following infection by a helper phage or SOS induction of a helper prophage, the PICI genome excises, using the PICI-coded integrases (int) and excision functions (xis), replicates extensively using its replicon, and is efficiently packaged into infectious particles composed of phage virion proteins. These events conform the ERP (excisionreplication-packaging) cycle of the PICIs. In this study, we are interested in SaPIs mobilization mechanism of 199 S. aureus strains isolated from cystic fibrosis patients. This mechanism is important because it allows the mobilization and dissemination of virulence factors encoded in this SaPIs. In particular, we focused on SaPIs induction mechanism. For it, we have classified the SaPIs presented in these isolates based on them integrase by PCR and southern-blot. Secondly, by sequence analysis of PCR products determined the regulation module, presented between int and xis genes. Moreover, by southern-blot analysis we have tested the SaPI induction by endogenous phages. Surprisingly, 9% of SaPIs were induced. Also, we characterized the mobilization mechanism used by endogenous phages to mobilize SaPIs. It leads to conclusion that by mutagenesis and transcriptional fusions the phage proteins are capable to bind to SaPIs Stl and in the same moment to be responsible for them induction. Panel 123 Sugar catabolism in Brucella suis biovar 5 occurs though the pentose phosphate and the Entner-Doudoroff pathways and is dispensable in vivo A. Zúñiga-Ripa1, L. Lázaro-Antón1, T. Barbier2, C.J. Bolten3, M.J. de Miguel4, C. Wittman3, M. Iriarte1, J.J. Letesson2 and I. Moriyón1. 1 Departamento de Microbiología e Instituto de Salud Tropical, Universidad de Navarra, Pamplona, Spain; 2Unité de Recherche en Biologie des Microorganismes, UNAmur, Namur, Belgium; 3Biochemical Engineering Institute, Saarland University, Saarbrücken, Germany; 4Unidad de Sanidad Animal, CITA, Zaragoza, Spain. [email protected] Brucella are facultative intracellular parasites of mammals causing brucellosis, a worldwide-distributed zoonosis. These bacteria behave as facultative intracellular parasites able to adjust their metabolism to extra and intracellular environments. Recently, we have shown that whereas the synthesis of phosphoenolpyruvate (PEP) from Krebs Cycle intermediates is required in vivo, upper gluconeogenic steps seem not to be necessary, suggesting that 6C-5C sugars can be obtained from the intracellular milieu (Zúñiga-Ripa et al. 2014). To catabolize these sugars, Brucella presents a complete pentose-phosphate pathway (PPP) but an incomplete glycolysis (EMP), since these bacteria lack the phosphofructokinase (Pfk). Moreover, these bacteria have the genes coding for the enzymes of the Entner-Doudoroff pathway (EDP), although no in vitro activity could be detected for the first enzyme in biochemical studies (Robertson and McCullough. 1968a, 1968b). Although this information is valuable, the catabolic pathways used by Brucella and their role during infection remain unclear. First, to determine the pathway(s) used by B. suis biovar 5 to catabolize glucose, we performed a 13C labelling experiment followed by a GC-MS analysis, and observed that glucose is assimilated via EDP and PPP. In contrast to the model usually considered in the literature describing the PPP as the major pathway, the EDP turned out to be the main glycolytic route. In agreement, the growth on glucose of an edd mutant (D-gluconate dehydratase, first enzyme of EDP) was strongly delayed, while the growth of a gnd mutant (6phosphogluconate dehydrogenase, first enzyme of the PPP) was altered to a lesser extent. Although no attenuation was observed for both mutants in macrophages, we were unable to obtain a mutant in both pathways. Thus, to confirm the importance of 6C-5C sugars in vivo, we constructed a mutant unable to grow on these sugars by blocking the synthesis of pyruvate (PYR). To this end, we studied the enzymes leading to this compound, including Edd, the pyruvate phosphate dikinase (PpdK) and the pyruvate kinase (Pyk). The simultaneous deletion of pyk, ppdK and edd suppresses Brucella growth on 6C-5C sugars. Finally, this triple mutant was not attenuated in the murine model, showing that these sugars do not play an essential role during the infectious process. References: - Zúñiga-Ripa A, Barbier T, Conde-Álvarez R, Martínez-Gómez E, Palacios-Chaves L, GilRamírez Y, Grilló MJ, Letesson JJ, Iriarte M, Moriyón I (2014). Brucella abortus depends on pyruvate phosphate dikinase and malic enzyme but not on Fbp and GlpX fructose-1,6bisphosphatases for full virulence in laboratory models. J Bact 196(16):3045-57. - Robertson, D. C and McCullough, W. G. (1968b). The glucose catabolism of the genus Brucella II. Cell-free studies with B. abortus (S-19). Arch Biochem Biophys 127:445-456. - Robertson, D. C. and McCullough, W. G. (1968a). The glucose catabolism of the genus Brucella I. Evaluation of pathways. Arch Biochem Biophys 127:263-273. Panel 124 Prevalence and antibiotic resistance pattern of Salmonella species isolated from vended raw beef in Ghana Courage Kosi Setsoafia Saba1, Opoku Bright1, Jose Francisco Delgado Blas2, Bruno Gonzalez Zorn2 1 University for Development Studies, Tamale, Ghana 2 Departamento de Sanidad Animal, Facultad de Veterinaria, Universidad Complutense de Madrid, Spain [email protected] Even though there is evidence that Salmonellosis has reduced drastically in the developed, there is paucity of data as far the prevalence in developing countries are concerned, but there has been high reports of salmonellosis in many African countries. The sources of this infection are not well known. A cross-sectional study was carried out in June 2015 to September 2015 on beef samples collected from different sale points within the Tamale Metropolis of Ghana. The study was conducted with the aim of isolating and testing for susceptibility of Salmonella with common antibiotics. A total of 124 beef samples were collected from 124 beef venders, homogenised and subjected to isolation procedures which included pre-enrichment, enrichment and plating using XLD, MSRV and Nutrient agar and Simon citrate agar. Identification by MALDI-TOF mass spectrometry was performed to confirm all the salmonella isolates. All the isolates were serotyped. Thirtyseven out of 124 (29.84%) of the samples were confirmed to be Salmonella. Antibiotics used were ampicillin, gentamicin, tetracycline, cefotaxime, ciprofloxacin, trimethoprimsulfamethoxazole and amoxycillin clavulanic acid. Among the 37 Salmonella isolates that were tested against these antibiotics, all were susceptible to ciprofloxacin. In contrast, all the 37 (100%) isolates were resistant to tetracycline. However, 40.5%, 32.4%, 24.3%, 16.2% and 8.1% of the isolates were resistant to cefotaxime, trimethoprimsulfamethoxazole, ampicillin and gentamicin respectively. The overall prevalence (29,84%) of Salmonella in this study indicates that beef may be one of the potential sources of Salmonella infection through cross-contamination of other foodstuff. Panel 125 ÍNDICE DE PARTICIPANTES APELLIDOS Abascal Sáiz Aguilar Pérez Aguilar Pierlé Aguilera Herce Aínsa Claver Álvarez Escribano Amaral Piubeli Aragón Fernández Aragón Aranda Ares Arroyo Arévalo Díaz Argandoña Bertrán Baelo Álvarez Baquedano Mozos Barahona Martín Barroso García Bastet Berenguer Carlos Bernabé Balas Bernal Bayard Beuzón López Blanco Torres Bosch Reñé Botello Cambero Brenes Álvarez Broset Blasco Burgui Erice Burnat Clemente Bustamante García Caballero Sánchez Cámara Almirón Camargo Bernal Capel Malo Caro Astorga Carrilero Aguado Casadesús Pursals Catalán Moreno Cepeda Molero Cervera Alamar Claret Fernández Conchillo Solé Coronas Serna Correa Fiz Couso Liañez Crespo González de Dios Barranco Delgado Blas Díaz Romero NOMBRE Estefanía Clara Sebastián Julia José Antonio Isidro Francine Virgina Beatriz Manuel Sergio Montserrat Aida Ignacio Emma Rocío Lauréne José Cristina Joaquín Carmen Rosario Paula Sandra Emilia Manuel Esther Saioa Mireia CORREO ELECTRÓNICO [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] COMUNICACIONES 1 2 3 4 2 5 9, 89 36 6 7, 16, 39, 110 8 9, 66, 77, 79, 89, 116 10, 113 11 12, 95 13 14 11, 15, 19, 49 7, 16, 76, 110 17 [email protected] 61 [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected]. Ligia Zulema bo Carlos [email protected] Jesús [email protected] Sergio [email protected] Elena [email protected] Joaquín [email protected] Laura [email protected] Josep [email protected] Arancha [email protected] Massiel [email protected] Esther Mercedes [email protected] Laura [email protected] Oscar [email protected] Julia María [email protected] Florencia [email protected] Inmaculada [email protected] José Luis [email protected] Rubén [email protected] José [email protected] Francisco Alberto [email protected] 18 19 20 21 22, 52 23 8, 24 25 14, 26, 69 27 28 29 30 31, 67 48, 70, 98, 108, 114 26 32 33, 53, 123 101 34 35 36 37 37, 87 38 39, 54, 67, 76, 125 40 APELLIDOS Escobar Doncel Escudero GarcíaCalderón Espinosa Portero Fernández Calvet Fernández Herrero Fernández Morales Flores García Floriano Pardal NOMBRE Álvaro José Antonio Rocío Ariadna Luis Ángel Ana Enrique Belén María Gallegos Fernández Trinidad García García María Jesús María García Gutiérrez Teresa García Martínez Begoña García Pastor Lucía María de las García Quintáns Nieves García Romero Inmaculada Garmendia García Juncal Gibert González Isidre Giraldo Suárez Rafael Godoy Olivares Liliana González Zorn Bruno Gonzalo Asensio Govantes Romero Guerrero Moreno Guzmán Hoefer Hüttener Queiroz Jiménez Gutiérrez Jiménez Zurdo Juárez Giménez Leal Morales Linares Clemente López Igual López Maury López Pagán López Torrejón Madrid Xufré Mañas Torres Mariscal Romero Marqués Martín Martín Basanta Martín Montañés Martínez Gómez Martínez Martínez Jesús Fernando Ricardo Katerina Andreas Mário Elena José Ignacio Antonio Antonio Pedro Rocío Luis Nieves Gema Cristina Carmen Vicente Silvia Marta Carlos Estrella Lourdes Francisco Juan Cristina Encarnación Mario Pilar Martínez Mojica Martínez Ovejero Mellado Durán Mencía Caballero Menéndez Gil CORREO ELECTRÓNICO [email protected] COMUNICACIONES 101 [email protected] 41 [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] 42 43, 72, 96 32, 63 44 8, 24, 28, 80, 92 50 [email protected] 45 [email protected] 46 [email protected] 105 [email protected] [email protected] 26, 47, 94, 102 48 [email protected] 11, 49 [email protected] [email protected] [email protected] [email protected] [email protected] 50 43, 72, 96 34, 122 [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] 51 7, 16, 31, 39, 54, 67, 76, 110, 125 22, 52, 88 42, 58, 78, 101 33, 53, 123 31, 54 55 56 57 55, 62 58 59 60 61 62, 83 63 92 82 64, 95 22, 52, 88 6, 65 9, 66 [email protected] Inaugural [email protected] [email protected] [email protected] [email protected] 31, 39, 54, 67 68, 105 11, 49 69 APELLIDOS Mérida Floriano Mesa Marín Moleres Apilluelo Molina Martín Moreno Amador Moreno Cabezuelo Moreno Sánchez Moyano Ortega Muro Pastor Naranjo Fernández Navarrete Ruiz de Clavijo NOMBRE Ángela Jennifer Javier María María de Lourdes José Ángel Ismael Gabriel Alicia María Emilia Blanca Joaquín José Nieves Morión Mercedes Olmedo Verd Elvira Pacheco Sánchez Daniel Paytubi Casabona Sònia Pedraz López Lucas Pérez Etayo Lara Pérez Mendoza Daniel Pérez Pérez María Esther Pérez Rodríguez Alejandro Pérez Sánchez Irene Prieto Mariscal Juana María Prieto Márquez Ana Isabel Ramos León Félix Ramos Morales Francisco Rapún Araiz Beatriz Reyes Ramírez Francisca Rivilla Palma Rafael Rodríguez Arce Irene Rodríguez Galán Olga Rodríguez Olivenza David Rodríguez Osorio Carlos Rodríguez Rosado Ana Isabel José Rufián Plaza Sebastián Ruiz Albert Javier Ruiz Romero Gabriel Saba Courage Sáenz Lahoya Sonia Salvador Bescós Miriam Sánchez Alonso Patricia G. Sánchez Leyre Barrionuevo Sánchez Hevia Dione Lara Sánchez-Porro Cristina Álvarez María Sánchez Romero Antonia Santamaría Gómez Javier Santero Santurino Eduardo Nieto Gutiérrez CORREO ELECTRÓNICO [email protected] [email protected] [email protected] [email protected] COMUNICACIONES 70 71 43, 72 35, 56 [email protected] 73 [email protected] [email protected] [email protected] [email protected] [email protected] 74 75 31, 39, 76 5, 21, 81 9, 77 [email protected] 58, 78 [email protected] 9, 66, 77, 79, 89, 116 [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] 80 21, 81 82 62, 83 84 85 86 37, 87 [email protected] 61 [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] 61 101 125 102 6, 103 104, 117 [email protected] 68, 105 [email protected] 106 [email protected] 107, 119, 120 [email protected] 108 [email protected] [email protected] 109 13, 38, 50 88 90 91 8, 92 4, 93 94 38 64, 95 43, 96 97 98 99 100 APELLIDOS Santos López Suárez Rodríguez Thomas López Toledo Arana NOMBRE Alfonso Mónica Daniel Alejandro María Tormo Mas Ángeles Torrado Maya Alejandro Torrents Serra Eduard Urdaneta Páez Verónica Valero Blanco Eva María Vargas Macías Carmen Vázquez Cruz Candelario Vázquez Fernández Roberto Ventosa Ucero Antonio Vera Gargallo Blanca Villanueva San Maite Martín Vioque Peña Agustín Yero Corona Daniel Žiemytė Miglė Zúñiga Ripa Amaia CORREO ELECTRÓNICO [email protected] [email protected] [email protected] [email protected] COMUNICACIONES 7, 16, 31, 54, 76, 110 111 31, 54, 67, 76 14, 26, 33, 69, 102 [email protected] 33, 53, 123 [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] [email protected] 112 10, 84, 113 114 115 9, 66, 77, 79, 89, 116 104, 117 118 107, 119, 120 120 [email protected] 14, 102, 121 [email protected] [email protected] [email protected] [email protected] 5, 21, 81 34, 122 33, 53, 123 65, 85, 124




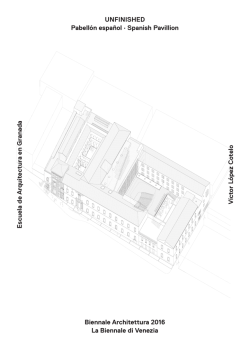

© Copyright 2026