
Transfusion Medicine - Moffitt Cancer Center
cancercontroljournal.org Vol. 22, No. 1, January 2015 H. LEE MOFFITT CANCER CENTER & RESEARCH INSTITUTE, AN NCI COMPREHENSIVE CANCER CENTER Safety of the Blood Supply German F. Leparc, MD Adverse Effects of Transfusion Radhika Dasararaju, MD, and Marisa B. Marques, MD Clinical Effects of Red Blood Cell Storage Lirong Qu, MD, PhD, and Darrell J. Triulzi, MD Transfusion Indications for Patients With Cancer Thomas Watkins, DO, PhD, Maria Katarzyna Surowiecka, MD, and Jeffrey McCullough, MD Platelet Transfusion for Patients With Cancer Craig H. Fletcher, MD, Melkon G. DomBourian, MD, and Peter A. Millward, MD Transfusion Support Issues in Hematopoietic Stem Cell Transplantation Claudia S. Cohn, MD, PhD Therapeutic Apheresis for Patients With Cancer Laura S. Connelly-Smith, MBBCh, DM, and Michael L. Linenberger, MD Using HLA Typing to Support Patients With Cancer Mark K. Fung, MD, PhD, and Kaaron Benson, MD Cancer Control is included in Index Medicus/MEDLINE H LEE MOFFITT CANCER CENTER & RESEARCH INSTITUTE INC 12902 Magnolia Drive Tampa FL 33612-9416 Jacksonville, FL Permit No. 4390 PAID Nonprofit Org. Save the Date THYROID CANCER CONFERENCE May 8–9, 2015 Sheraton Sand Key 1160 Gulf Boulevard Clearwater Beach, FL This conference is designed to provide the latest research and clinical updates to endocrinologists, thyroid surgeons, medical oncologists, and other health care professionals who are interested in the diagnosis, treatment, and management of thyroid cancer. COURSE DIRECTOR Bryan McIver, MD CONFERENCE CONTACTS Marsha Moyer: [email protected] Cindi Hughlett: [email protected] P ROV I D E D BY January 2015, Vol. 22, No. 1 Cancer Control 1 Editorial Board Members Editor: Lodovico Balducci, MD Senior Member Program Leader, Senior Adult Oncology Program Moffitt Cancer Center Deputy Editor: Julio M. Pow-Sang, MD Senior Member Chair, Department of Genitourinary Oncology Director of Moffitt Robotics Program Moffitt Cancer Center Editor Emeritus: Rami Komrokji, MD Associate Member Malignant Hematology Conor C. Lynch, PhD Assistant Member Tumor Biology Amit Mahipal, MD Assistant Member Clinical Research Unit Gastrointestinal Oncology Kristen J. Otto, MD Assistant Member Head & Neck Oncology John Horton, MB, ChB Professor Emeritus of Medicine & Oncology Michael A. Poch, MD Assistant Member Genitourinary Oncology Moffitt Cancer Center Journal Advisory Committee: Jeffery S. Russell, MD, PhD Assistant Member Endocrine Tumor Oncology Aliyah Baluch, MD Assistant Member Infectious Diseases Elizabeth M. Sagatys, MD Assistant Member Pathology - Clinical Dung-Tsa Chen, PhD Associate Member Biostatistics Jose E. Sarria, MD Assistant Member Anesthesiology Hey Sook Chon, MD Assistant Member Gynecological Oncology Saïd M. Sebti, PhD Senior Member Drug Discovery Jasreman Dhillon, MD Assistant Member Pathology - Anatomic Bijal D. Shah, MD Assistant Member Malignant Hematology Jennifer S. Drukteinis, MD Associate Member Diagnostic Radiology Lubomir Sokol, MD, PhD Senior Member Hematology/Oncology Timothy J. George, PharmD Pharmacy Residency Director Clinical Pharmacist - Malignant Hematology Hatem H. Soliman, MD Assistant Member Breast Oncology Clement K. Gwede, PhD Associate Member Health Outcomes & Behavior Jonathan R. Strosberg, MD Assistant Member Gastrointestinal Oncology Sarah E. Hoffe, MD Associate Member Radiation Oncology Sarah W. Thirlwell, RN Nurse Director Supportive Care Medicine Program John V. Kiluk, MD Associate Member Breast Oncology Eric M. Toloza, MD, PhD Assistant Member Thoracic Oncology Richard D. Kim, MD Associate Member Gastrointestinal Oncology Nam D. Tran, MD Assistant Member Neuro-Oncology Bela Kis, MD, PhD Assistant Member Diagnostic Radiology Jonathan S. Zager, MD Associate Member Sarcoma and Cutaneous Oncology Production Staff: Veronica Nemeth Editorial Coordinator Don Buchanan Graphic Designer Consultants: Sherri Damlo Medical Copy Editor and Art Coordinator Kelly Young Graphic Designer Associate Editor of Educational Projects John M. York, PharmD Akita Biomedical Consulting 1111 Bailey Drive Paso Robles, CA 93446 Phone: 805-238-2485 Fax: 949-203-6115 E-mail: [email protected] For Consumer and General Advertising Information: Veronica Nemeth Editorial Coordinator Cancer Control: Journal of the Moffitt Cancer Center 12902 Magnolia Drive – MBC-JRNL Tampa, FL 33612 Phone: 813-745-1348 Fax: 813-449-8680 E-mail: [email protected] Cancer Control is a member of the Medscape Publishers’ Circle®, an alliance of leading medical publishers whose content is featured on Medscape (www.medscape.com). Most issues and supplements of Cancer Control are available at cancercontroljournal.org CANCER CONTROL: JOURNAL OF THE MOFFITT CANCER CENTER (ISSN 1073-2748) is published by H. Lee Moffitt Cancer Center & Research Institute, 12902 Magnolia Drive, Tampa, FL 33612. Telephone: 813-745-1348. Fax: 813-449-8680. E-mail: [email protected]. Internet address: cancercontroljournal.org. Cancer Control is included in Index Medicus ®/MEDLINE® and EMBASE®/ Excerpta Medica, Thomson Reuters Science Citation Index Expanded (SciSearch®) and Journal Citation Reports/Science Edition. Copyright 2015 by H. Lee Moffitt Cancer Center & Research Institute. All rights reserved. Cancer Control: Journal of the Moffitt Cancer Center is a peer-reviewed journal that is published to enhance the knowledge needed by professionals in oncology to help them minimize the impact of human malignancy. Each issue emphasizes a specific theme relating to the detection or management of cancer. The objectives of Cancer Control are to define the current state of cancer care, to integrate recently generated information with historical practice patterns, and to enlighten readers through critical reviews, commentaries, and analyses of recent research studies. DISCLAIMER: All articles published in this journal, including editorials and letters, represent the opinions of the author(s) and do not necessarily reflect the opinions of the editorial board, the H. Lee Moffitt Cancer Center & Research Institute, Inc, or the institutions with which the authors are affiliated unless clearly specified. The reader is advised to independently verify the effectiveness of all methods of treatment and the accuracy of all drug names, dosages, and schedules. Dosages and methods of administration of pharmaceutical products may not be those listed in the package insert and solely reflect the experience of the author(s) and/or clinical investigator(s). January 2015, Vol. 22, No. 1 Cancer Control 1 Table of Contents Editorial Transfusion Medicine Issues Pertaining to Patients With Cancer 4 Kaaron Benson, MD Articles Safety of the Blood Supply 7 German F. Leparc, MD Adverse Effects of Transfusion 16 Radhika Dasararaju, MD, and Marisa B. Marques, MD Clinical Effects of Red Blood Cell Storage 26 Lirong Qu, MD, PhD, and Darrell J. Triulzi, MD Transfusion Indications for Patients With Cancer 38 Thomas Watkins, DO, PhD, Maria Katarzyna Surowiecka, MD, and Jeffrey McCullough, MD Platelet Transfusion for Patients With Cancer 47 Craig H. Fletcher, MD, Melkon G. DomBourian, MD, and Peter A. Millward, MD Transfusion Support Issues in Hematopoietic Stem Cell Transplantation 52 Claudia S. Cohn, MD, PhD Therapeutic Apheresis for Patients With Cancer 60 Laura S. Connelly-Smith, MBBCh, DM, and Michael L. Linenberger, MD 2 Cancer Control January 2015, Vol. 22, No. 1 Table of Contents Using HLA Typing to Support Patients With Cancer 79 Mark K. Fung, MD, PhD, and Kaaron Benson, MD Departments Special Report: Mobilization and Transplantation Patterns of Autologous Hematopoietic Stem Cells in Multiple Myeloma and Non-Hodgkin Lymphoma 87 Luciano J. Costa, MD, PhD, Shaji Kumar, MD, Stephanie A. Stowell, MPhil, and Shari J. Dermer, PhD Special Report: Functional Health Literacy, Chemotherapy Decisions, and Outcomes Among a Colorectal Cancer Cohort 95 Evan L. Busch, MPH, Christopher Martin, MSPH, Darren A. DeWalt, MD, and Robert S. Sandler, MD Pathology Report: Familial Gastrointestinal Stromal Tumor Syndrome: Report of 2 Cases With KIT Exon 11 Mutation 102 Derek H. Jones, MD, Jamie T. Caracciolo, MD, Pamela J. Hodul, MD, Jonathan R. Strosberg, MD, Domenico Coppola, MD, and Marilyn M. Bui, MD, PhD Tumor Biology: Current and Emerging Therapies for Bone Metastatic Castration-Resistant Prostate Cancer 109 Jeremy S. Frieling, David Basanta, PhD, and Conor C. Lynch, PhD Ten Best Readings Relating to Transfusion Medicine 121 About the art in this issue: Ray Paul began drawing and painting as a young boy, so art has always been part of his life. Animals and images from National Geographic magazine were his early subjects. Growing up in Ohio, dreams of Florida and all things tropical helped him survive the dreary winters and infused his palette with saturated hues. This and his fascination with science led him to Florida State University where he received a bachelor of science in biology in 1986. Returning to his first love, art, Paul earned a master of fine arts in painting from the University of Cincinnati in 1991. Paul currently resides in Tampa, Florida, where he maintains a studio. His work is a synthesis of life experiences and a desire to illuminate the subconscious. Elements of abstract expressionism, surrealism, pop art, biology, and psychedelic music all combine and intertwine, creating a unique style of abstract painting. Each work is an experiment as different paints mix and swirl, with meticulous layering providing the final touch. Paul was diagnosed with high-grade myxofibrosarcoma and is currently a patient at the Moffitt Cancer Center in Tampa and has undergone 9 surgeries, 2 courses of radiation therapy, chemotherapy, and participated in a clinical trial. This experience has led Paul to a greater understanding and appreciation of his work. He envisions his art to be a prescient, visual manifestation of the battle raging within and a powerful testament to the beauty of hope.To view more of his work or to contact the artist, please visit www.raypaulart.com. Cover: Pink Rorschach, triptych (detail: 3, right panel), 2010. Acrylic, latex, enamel on canvas, 20" × 16". Pages: 2-3Dendritic Swarm, 2013. Acrylic, latex, enamel on canvas, printed with an image of myxofibrosarcoma with metastases to the artist’s lung, 11" × 17". Petal Purple, 2010. Acrylic, latex, enamel on canvas, 12" × 12". Sweet Jane, 2007. Acrylic, latex, enamel on canvas, 37" × 3". Private collection of the Mathematical Oncology Department at Moffitt Cancer Center, Tampa, FL. Blue Marble, 2007. Acrylic, latex, enamel on wood, 49" × 64". Private collection. Flowers for Phoebe, 2010. Acrylic, latex, enamel on canvas, 48" × 48". Private collection. SP12-6796 × 40, 2013. Acrylic, latex, enamel on canvas, printed with an image of myxofibrosarcoma with metastases to the artist’s lung, 26" × 36". Dark Shadows, 2010. Acrylic, latex, enamel on canvas, 30" × 30". Currant, 2014. Acrylic, latex, enamel on canvas, 20" × 20". January 2015, Vol. 22, No. 1 Cancer Control 3 Editorial Transfusion Medicine Issues Pertaining to Patients With Cancer In the United States, blood transfusion was the most common procedure performed in hospitals in 2010 and occurred in 11% of hospital stays requiring at least 1 procedure.1 Red blood cells (RBCs) are the most likely blood component to be transfused due to their role in treating symptomatic anemia. RBC units are often provided to patients with cancer because anemia occurs in more than 40% of these patients.2 Most patients receiving chemotherapy can be expected to require RBCs, platelet transfusions, or both during the course of therapy.3 Currently, approximately 80% of all platelet transfusions are administered to patients with hypoproliferative thrombocytopenia, generally due to chemotherapy, hematopoietic stem cell transplantation, or underlying disease.4 Although blood transfusions can be life-saving measures and allow for more aggressive therapy, they are not without risk. This issue of Cancer Control addresses select issues related to transfusion medicine that pertain to patients with cancer. Measures to improve the safety of the blood supply have resulted in the lowest rates of transfusion-transmissible infection and disease since blood was first used. Although post-transfusion hepatitis occurred in about one-third of multiple-transfused patients in the 1960s, today transfusion-transmitted rates of hepatitis B and C viruses each result in about 1 case per 1 million units transfused.5,6 The risk of transfusion-transmitted HIV infection was around 1% in metropolitan areas like San Francisco in the early 1980s.7 Currently, the risk of HIV transmission via blood in the United States is less than 1 per 1 million units transfused.5 Mitigation strategies to lower the rates of nonviral transfusion adverse events have also allowed for reaction rates well below previous rates of about 5%; current averages are typically less than 1%. Measures such as leukoreduction, γ irradiation, and the preparation of male-only donor plasma have lowered risks of human leukocyte antigen (HLA) alloimmunization, transfusion-associated graft-vs-host disease, and transfusion-related acute lung injury, respectively. The annual transfusion reaction rates at the H. Lee Moffitt Cancer Center & Research Institute have consistently been 0.6% for the last 3 years, with many of these events being fever occurring during transfusion and likely due to neutropenia, not the blood infused. In this issue of Cancer Control, Dr Leparc reviews the blood donation process and safety measures currently employed to reduce the risks of transfusion transmitted disease. Drs Dasararaju and Marques cover 4 Cancer Control nonviral transfusion adverse events and their diagnosis, treatment, and prevention. Whether older units of blood result in significant harm to recipients has been questioned recently, and Drs Qu and Triulzi review the storage lesion and summarize key study results, including data from recent randomized controlled trials. Blood transfusion can be misused and unnecessary transfusion is a common problem. Drs Watkins, Surowiecka, and McCullough address the appropriate use of RBCs, plasma, and granulocytes, and Drs Fletcher, DomBourian, and Millward cover indications for platelet transfusion. Most blood components are given for therapeutic benefit due to anemia, bleeding, or both. Platelets are the one component commonly used prophylactically — generally in the oncology setting for patients with hypoproliferative thrombocytopenia. Whether selected populations of patients could be moved to receive therapeutic platelet transfusion alone is an area of current investigation and preliminary data suggest that this may be possible.8,9 Blood component support in patients receiving hematopoietic stem cell transplantation can present unique challenges and this topic is reviewed by Dr Cohn. She focuses on the pre-, peri-, and post-transplantation treatment periods, with special attention to ABO-incompatible recipient–donor pairings. The transfusion service must provide blood components to minimize hemolysis and other adverse events, such as transfusion-associated graft-vs-host disease and transfusion-transmitted cytomegalovirus infection. Apheresis technology can be applied in both the donor and patient settings. Apheresis platelets, plasma, and RBCs (the equivalent of 2 units of RBCs from 1 donor) are commonly collected from volunteer donors. Therapeutic aphereses are often performed for the treatment of several oncological diseases. As in the donor population, blood fractions such as plasma, leukocytes, RBCs, or platelets can be selectively removed. Unique to therapeutic apheresis is the option to modify the collected component as in extracorporeal photopheresis before reinfusion back to the patient. Drs Connelly-Smith and Linenberger review indications for use of therapeutic apheresis as well as a number of practical issues to consider when beginning these specialized procedures. The HLA system is crucial in our immune response; it may play a role in our response to certain medications, may increase our risk for certain diseases, and has a critical function in both solid organ and hematopoietic stem cell transplantation. Dr Fung and I address January 2015, Vol. 22, No. 1 HLA assays commonly performed today and their use in pharmacogenomics, disease association, and platelet transfusion and transplantation. The American Board of Internal Medicine Foundation has initiated a campaign known as Choosing Wisely to help health care professionals reduce the overuse of tests and procedures, and the AABB (formerly the American Association of Blood Banks) developed a list of 10 recommendations in support of this campaign as well as to promote better blood management among patients.10 The first recommendation is to avoid transfusing more blood than absolutely necessary, noting that a restrictive threshold of 7.0 to 8.0 g/dL should be used for the vast majority of stable, hospitalized patients without evidence of inadequate tissue oxygenation.10 In addition, single RBC unit transfusions — instead of the traditional minimum 2 units — should be the new standard for hospitalized patients without bleeding.10 Although blood transfusion has allowed for the aggressive treatment of patients with cancer, it is not without risk and must be judiciously used in this patient population. The next 3 AABB recommendations are also apropos to patients with cancer, cautioning health care professionals to avoid transfusing RBCs for iron deficiency in patients without hemodynamic instability.10 Iron deficiency anemia should be treated with oral and/or intravenous iron supplementation in such cases. Another recommendation relates to blood components, such as plasma, which the AABB notes should not be routinely used to reverse warfarin because vitamin K alone is often sufficient.10 Prothrombin complex concentrates offer advantages over plasma transfusion in more emergent situations such as in the setting of significant bleeding, nonelective surgery, or both. The AABB also recommends against performing serial blood counts on patients who are clinically stable.10 Hospitalized patients with new clinically significant conditions may require frequent blood counts to be performed; however, the AABB notes that serial counts in stable patients are unlikely to benefit patient care and are likely to result in iatrogenic anemia.10 In order to improve outcomes among our patients requiring blood transfusion, we must apply patient blood management (PBM), which is the use of efficacious and safe medical and surgical techniques to prevent anemia, treat anemia, or both, as well as decrease the risk of bleeding and optimize hemostasis.11 When providing blood transfusion therapy to our patients, we must provide the right blood component in the right amount at the right time to maximize the clinical benefit and minimize any potential adverse effects. In addition to the topics relating to transfusion medicine, 2 Special Reports are also included in this issue. In the first report, Dr Costa and coauthors discuss the mobilization and transplantation patterns of autologous hematopoietic stem cells in multiple myeloma and non-Hodgkin lymphoma. In the second, Dr Busch January 2015, Vol. 22, No. 1 and colleagues review the functional health literacy, chemotherapy decisions, and outcomes among a cohort of volunteers with colorectal cancer. Also included in the January issue of Cancer Control is a Pathology Report by Dr Jones and others who present 2 cases of familial gastrointestinal stromal tumor syndrome with KIT exon 11 mutations. Mr Frieling and colleagues have authored a Tumor Biology Report discussing the current and emerging therapies for bone metastatic castration-resistant prostate cancer. We hope you enjoy and benefit from reading this issue of Cancer Control. Kaaron Benson, MD Senior Member Director, Blood Bank, HLA, Chemistry, Specimen Processing, Implantable Tissues Department of Hematopathology and Laboratory Medicine H. Lee Moffitt Cancer Center & Research Institute Tampa, Florida [email protected] References 1. Pfuntner A, Wier LM, Stocks C. Most Frequent Procedures Performed in U.S. Hospitals, 2010. Rockville, MD: Agency for Healthcare Research and Quality; 2013. http://hcup-us.ahrq.gov/reports/statbriefs/sb149.pdf. Accessed November 25, 2014. 2. Shander A, Knight K, Thurer R, et al. Prevalence and outcomes of anemia in surgery: a systematic review of the literature. Am J Med. 2004; g116(suppl 7A):58S-69S. 3. Tas F, Eralp Y, Basaran M, et al. Anemia in oncology practice: relation to diseases and their therapies. Am J Clin Oncol. 2002;25(4):371-379. 4. Fung MK, Grossman BJ, Hillyer C, et al, eds. Technical Manual. 18 ed. Bethesda, MD: AABB; 2014. 5. Zou S, Stramer SL, Dodd RY. Donor testing and risk: current prevalence, incidence, and residual risk of transfusion-transmissible agents in US allogeneic donations. Transfus Med Rev. 2012;26(2):119-128. 6. Stramer SL. Tibor Greenwalt memorial award and lectureship: getting to 1 in a million and beyond. Paper presented at: AABB Annual Meeting; Philadelpha, PA; October 25–28, 2014 7. Busch MP, Young MJ, Samson SM, et al; Transfusion Safety Study Group. Risk of human immunodeficiency virus (HIV) transmission by blood transfusions before the implementation of HIV-1 antibody screening. Transfusion. 1991;31(1):4-11. 8. Wandt H, Schaefer-Eckart K, Wendelin K, et al. Therapeutic platelet transfusion versus routine prophylactic transfusion in patients with haematological malignancies: an open-label, multicentre, randomised study. Lancet. 2012;380(9850):1309-1316. 9. Stanworth SJ, Estcourt LJ, Llewelyn CA, et al. Impact of prophylactic platelet transfusions on bleeding events in patients with hematologic malignancies: a subgroup analysis of a randomized trial (CME). Transfusion. 2014;54(10):2385-2393. 10. Callum JL, Waters JH, Shaz BH, et al. The AABB recommendations for the Choosing Wisely campaign of the American Board of Internal Medicine. Transfusion. 2014;54(9):2344-2352. 11. Society for the Advancement of Blood Management. What is patient blood management? http://www.sabm.org/sites/www.sabm.org/files/sabmpbmawpresentation2012.pdf. Accessed November 25, 2014. Cancer Control 5 Department of Hematopathology and Laboratory Medicine H. Lee Moffitt Cancer Center & Research Institute The Department of Hematopathology and Laboratory Medicine at the H. Lee Moffitt Cancer Center & Research Institute performs a variety of routine and highly specialized diagnostic services and testing for the care of oncology and blood and marrow transplant patients with the mission of contributing to the prevention and cure of cancer. The department consists of the Hematology, Chemistry, Microbiology/Virology, Blood Bank, HLA, Histocompatibility, Flow Cytometry, Specimen Processing, Bone Marrow Service, and Molecular Diagnostics divisions. The laboratory is equipped with state-of-the-art instrumentation represented by more than 80 platforms. The Microbiology laboratory works with the Infection Control team to ensure low-infection rates for the immunocompromised patient population, and the HLA laboratory works closely with the Blood and Marrow Transplant Program to ensure the accurate and timely HLA typing of both patients and donors. Hematopathology/Laboratory Medicine Department Department Chair Lynn Moscinski, MD Blood Bank, HLA, Chemistry, Medical Director Kaaron Benson, MD Microbiology/Virology Medical Director Ramon Sandin, MD Moffitt International Plaza Medical Director Elizabeth Sagatys, MD Hematopathology Fellowship Director Ling Zhang, MD Hematopathologists Pedro Horna, MD Mohammad Hussaini, MD Haipeng Shao, MD, PhD Jinming Song, MD Jianguo Tao, MD, PhD Hailing Zhang, MD Xiaohui Zhang, MD, PhD Clinical Molecular Diagnostics Director Dahui Qin, MD, PhD Molecular/FISH Director Kenian Liu, PhD This academic department participates in the teaching of medical students, residents, and clinical hematopathology fellows in its accredited fellowship program. Research is also a significant component of the academic mission of the department and its pathologists are engaged in collaborating with other Moffitt Cancer Center investigators and researchers on clinical trials and the publication of study results. One of the main focuses of the department has been the identification and application of new prognostic markers in the myeloid malignancies of bone marrow, particularly myelodysplasia and acute myelogenous leukemia. The pathologists also work with other Moffitt programs in creating algorithmic pathways that provide a standardized tool to guide patient care and treatment plans as well as the translation of new discoveries into clinical diagnostics — new testing and methodologies are continuously being added. For more information about the Department of Hematopathology and Laboratory Medicine, please call 813-745-4162 (during normal business hours). www.MOFFITT.org 6 Cancer Control January 2015, Vol. 22, No. 1 Blood transfusion is an invasive medical procedure that carries inherent hazards, so a risk–benefit profile must be considered in patients with cancer. Ray Paul. Dendritic Swarm, 2013. Acrylic, latex, enamel on canvas printed with an image of myxofibrosarcoma with metastases to the artist’s lung, 11" × 17". Safety of the Blood Supply German F. Leparc, MD Background: The transfusion of blood components plays a significant role as supportive therapy in the treatment of patients with cancer. Although blood transfusions help manage complications arising from either the patient’s primary condition or associated with therapeutic intervention, their use introduces a new set of risks; therefore, health care professionals must be aware of the potential morbidity introduced by using blood components and endeavor to optimize outcomes by ordering transfusions only when the benefits outweigh the inherent risks. Methods: This article sought to review the published literature, including the epidemiology of diseases transmissible via transfusion, performance characteristics for assays used for blood donor screening, surveillance activities to detect newly emergent pathogens, and biovigilance activities reported by public health authorities. Results: Effective measures have been implemented to significantly decrease the risk of transmissible diseases associated with transfusion. Reports of viral disease transmitted via transfusion have been nearly eliminated, particularly since the introduction of molecular-based detection technology. The transmission of bacteria and parasites still represents a threat to the use of cellular blood components. Transfusion-associated human prion disease has not been reported in the United States. Immune-mediated reactions due to donor–recipient incompatibility remain a challenge. Conclusions: Transmissible agents most commonly associated with risks due to transfusion are no longer a major threat; however, a significant challenge remains with regard to addressing the need for quick response mechanisms to manage emerging pathogens with the potential for rapid spread, either unintentionally (eg, globalization) or intentionally (eg, bioterrorism). The use of technology to reduce pathogens holds promise for further increasing the safety profile of blood transfusion. Background Although many of the risks associated with blood transfusions have been recognized ever since the be- From OneBlood and Creative Testing Solutions, St Petersberg, Florida. Submitted June 8, 2014; accepted September 11, 2014. Address correspondence to German F. Leparc, MD, OneBlood, 10100 Dr Martin Luther King Jr Steet North, St Petersburg, FL 33716. E-mail: [email protected] No significant relationship exists between the author and the companies/organizations whose products or services may be referenced in this article. January 2015, Vol. 22, No. 1 ginning of the use of transfusions, the emergence of HIV transmission brought the safety of the blood supply into the limelight of the public. Since then, significant resources have been committed to implementing strategies to reduce the risk of transfusion-transmitted disease. Although challenges remain, significant advances have been made. A tight, multilayered safety net has been woven into the system. Volunteer donors must meet strict criteria, aseptic techniques are used to collect blood in single-use disposable containers, testing is performed for various markers of present or past infection, and Cancer Control 7 passive and active biovigilance activities are in place before and after blood components are transfused. In addition to the risk of transmissible diseases, blood components carry inherent risks associated with the presence of immunoactive effectors that interact with the host (blood recipient). These agents vary and can include immunoglobulins (antibodies), antigenic substances (phenotypes on cellular elements and plasma proteins), and various biological response modifiers (eg, cytokines, chemokines). Unlike chemical drugs and biological agents manufactured in a controlled pharmacological setting, blood components exhibit significant biological variability that impacts the achievement of desired outcomes as well as the manifestation of undesired adverse events. When considering the safety of blood transfusion, one must discriminate between the safety of the product to be transfused (ie, the biological contents) and the safety of the transfusion process (ie, pretransfusion testing, product administration, dosage, presence of indications or contraindications, risk vs benefit of such an intervention). In this manuscript, the aspects surrounding the safety of the transfusion product alone have been considered. Transfusion-Transmitted Viral Agents Hepatitis B Virus Not long after the transfusion of whole blood or its components became available in routine medical and surgical treatments was its association with hepatitis virus infection recognized. The introduction of the test for hepatitis B surface antigen (HBsAg) in the early 1970s allowed the interdiction of units collected from individuals with either subclinical acute or chronic forms of the infection.1 However, the kinetics of the hepatitis B virus (HBV) infection leaves 2 different periods when screening for the presence of HBsAg fails to prevent transmission: (1) an early acute phase when the viral load is below the assay’s limit of detection, and (2) a late chronic phase when HBsAg levels gradually become undetectable although infectivity remains. The introduction of molecular techniques (nucleic acid testing [NAT]) decreases the serological period of infectivity by decreasing the limit of detection for the presence of the viral genome. Laboratories in numerous countries, including the United States, have also added a test to detect the presence of antibodies directed against a viral core protein (hepatitis B core antibody) to detect chronic carriers who may have levels of viremia below those detectable even by molecular techniques, a condition known as occult hepatitis B.2 The use of the 3 markers — HBsAg, hepatitis B core antibody, and HBV-NAT — has reduced the residual risk of transfusion-transmitted HBV infection to approximately 1 per 1 million donations, and the rate of clinical hepatitis continues to decrease as a 8 Cancer Control larger segment of the population becomes immunized through vaccination.3 Hepatitis C Virus Following the implementation of first- and second-generation assays for HBsAg and the development of serological tests for hepatitis A virus, a distinct viral entity other than the 2 associated with the majority of cases of post-transfusion hepatitis was detected. After decades of work, hepatitis C virus (HCV) was molecularly characterized in the late 1980s using cloning techniques.4 It was a significant achievement because the virus could not be sustained in cell cultures. Screening tests were developed to detect the presence of anti-HCV antibodies and helped identify asymptomatic HCV carriers in the blood donor population. The use of the screening assay represented a significant advance because approximately 80% of individuals who become infected with HCV remain viremic for the remainder of their lives (unless treated).5 Most of these individuals are also asymptomatic for decades; by contrast, about 20% of those infected with HCV have spontaneous viral clearance.5 However, because of the prolonged seroconversion period, which lasts for more than 50 days, approximately 1 out of 230,000 donations with no demonstrable antibodies contain viral RNA.6 The introduction of NAT to detect viremia soon after a 10-day, ramp-up viral replication period has reduced the residual risk of transfusion-transmitted HCV infection to 1 in 1.93 million donations.7 HIV As the worldwide HIV epidemic unfolded, blood transfusion was recognized early on as an efficient means of HIV transmission. The emergence of this retrovirus focused attention on the safety of the blood supply and the importance of the development of rapid-response mechanisms to detect the rise of new threats to protect blood recipients. A serological test to detect the presence of anti-HIV antibodies was introduced in 1985 that allowed the interdiction of the vast majority of HIV-infected units.8 Similar to other screening assays based on the detection of donor seroconversion, infection in the donor could not be detected for approximately 22 days; however, this period was reduced by 11 days with the introduction of NAT.9 As a result, the residual risk of HIV transmission associated with transfusion has been reduced to 1 in 2.135 million.10 West Nile Virus West Nile virus is an emergent pathogen first isolated in samples obtained from patients in Uganda in 1937. Its presence was unknown on the North American continent until 1999 when it was found in patients diagnosed with neurological disease.11 Several species of mosquitos function as vectors for the virus, which January 2015, Vol. 22, No. 1 finds its reservoir in migrating birds; humans and horses are accidental hosts. More than 90% of individuals infected with West Nile virus remain asymptomatic, and, of those affected, mild, flu-like symptoms without sequelae are the most common presentation; however, 0.6% of infected persons will progress to neuroinvasive disease that results in meningoencephalitis and possibly death.12 Individuals who remain asymptomatic but donate blood while viremic pose a risk to blood recipients, particularly among those who are immunosuppressed, the elderly, and infants. The risk of transmission is highest during the months when both the reservoir bird and mosquito populations peak, which occurs predominantly during summer, possibly extending to early fall in some regions of the United States. In 2002, the implementation of NAT for West Nile virus decreased the risk of transmission; however, residual risk rates are highly variable and depend on climactic and geographical factors that modify the location and duration of endemic areas.13 Annual reports indicate a paucity of cases linked to blood transfusion since 2010.14 confirmed transmissions of HHV-8 via the same route have not been linked to the presentation of diseases known to be associated with it (eg, Kaposi sarcoma, malignant lymphoproliferative disorders). Parvovirus B19 Infection with parvovirus B19, a nonenveloped erythrovirus, manifests differently in distinct patient populations. In utero, the infected fetus develops severe anemia that results in hydrops fetalis; in children, the infection results in the exanthematous fifth disease; and, in adults, it may result in mild disease, including fever, myalgia, rash, arthropathy, and, occasionally, red cell aplasia in vulnerable individuals with ongoing hemolytic processes (eg, autoimmune or drug-related anemias, sickle cell disease). Concerns exist regarding the hypothetical possibility of the compromise of hematopoietic tissue engraftment due to parvovirus infection during the early transplantation stages; however, no reports of such associations have been published. The transmission of parvovirus B19 via transfusion has been documented, but morbidity has been limited, even in immunocompromised patients.18 This is despite epidemiological evidence that the virus is a relatively common contaminant in the blood supply and the incidence of transfusion-transmitted parvovirus B19 infection is likely under-reported.19 Human T-Cell Lymphotropic Virus The potential for HIV transmissibility through blood transfusion has raised concern about other retroviruses that, although they are not as pathogenic, could spread to the general population through the use of blood components. Two strains of human T-cell lymphotropic virus (HTLV), HTLV-1 and HTLV-2, were targeted for detection. Although HTLV-1 has been linked to adult T-cell leukemia, HTLV-associated myelopathy, and tropical spastic paraparesis, no firm link to disease entities has been found for HTLV-2.15 The virus is transmitted through cellular components alone and infectivity of the product declines with storage time, particularly when stored beyond 10 days.15 A total of 1% of those infected will develop disease associated with the infection. An immunoassay approved by the US Food and Drug Administration (FDA) is used to detect the presence of anti-HTLV-1 and 2 antibodies, although its rate of specificity is not optimal.16 The residual risk of transmission is low (1 in 3 million).10 Hepatitis A and E Viruses Although hepatitis A and E viruses are both predominantly transmitted via the oral–fecal route, sporadic transmissions via blood transfusion have been reported. In most of the reported cases, mild, temporary liver inflammation has occurred. Because the incidence of transfusion-associated transmission is low in most developed countries (although some regions within developed countries may show significant endemicity rates), testing the blood supply is not warranted at this time.20 However, given that pathogen-reduction methods have not eliminated the risk of hepatitis A virus transmission, and the incidence of hepatitis E virus is increasing in Europe, implementing NAT screening methods is currently under investigation.21 Herpesvirus A significant segment of patients with cancer are particularly vulnerable to herpesviruses because of the immune compromise associated with cancer treatment. Cytomegalovirus (CMV) and human herpesvirus (HHV) 8 are cell-associated pathogens that can be transmitted through the transfusion of cellular blood components. Recommendations to mitigate the transmission include the use of CMV-seronegative, leukoreduced cellular blood components, or both.17 Although the clinical manifestations of transfusion-transmitted CMV have been reported, Dengue and Chikungunya Both members of the Arboviridae family are expanding their traditional geographical boundaries together with the range of their vector, the Aedes aegypti mosquito, and both are poised to extend their range further as another member of the Aedes group (A albopictus) is an even more efficient vector for the chikungunya virus when a specific mutation in the viral envelope is present. Dengue virus is a mosquito-borne, single, positive-stranded RNA flavivirus with a wide distribution across the tropical and subtropical regions of the January 2015, Vol. 22, No. 1 Cancer Control 9 world. Four serotypes of similar pathogenicity have been identified (a fifth serotype has been proposed but is pending further characterization). In specific regions, more than 1 serotype may coexist. Although the World Health Organization estimates that the disease burden is more than 100 million cases, this is likely an underestimation given the large population in the geographical span of its vectors.22 Typically, individuals infected for the first time develop fever, headache, muscle and joint pains, and a characteristic skin rash similar to measles. In a small proportion of cases the disease develops into life-threatening dengue hemorrhagic fever, resulting in thrombocytopenia, bleeding, and capillary leakage that may progress into dengue shock syndrome. The reason that some people experience more severe forms of dengue, such as dengue hemorrhagic fever, is multifactorial. Among the possible causes is a cross-serotypic immune response, which occurs when a person who was previously infected with dengue becomes infected for the second, third, or fourth time. Through a mechanism known as antibody-dependent enhancement, the previous antibodies to the old strain of dengue virus interfere with the immune response to the current strain, paradoxically leading to more viral entries and uptakes that correlate with the increased severity of the disease.23 Chikungunya is an alphavirus with a positive sense, single-stranded RNA genome. Following a short incubation period, fever, intense headache, maculopapular rash, and severe joint and muscle pain ensue. An outbreak in the Reunion Island in the Indian Ocean, a region at the center of the historical range for the disease that extends from East Africa to Southeast Asia, was reported in 2005 to 2006.24 Given the development of tourism in the region, outbreaks traced to tourists returning from Reunion Island were later reported in Europe.25 Concerns about the extension of the endemic areas beyond the African and Asian continents have proven valid. Epidemiological surveillance has now identified cases in the Caribbean and sporadic outbreaks are occurring in the southeastern United States.26 Reports of transfusion-transmitted dengue in endemic regions have been published,27 although no cases have been reported of chikungunya transmitted via blood transfusions. Due to the significant overlap between the regions where dengue and chikungunya and malarial parasites are endemic, travel overseas disqualifies most potential blood donors who return to the United States and are infected after being abroad. However, as the geographical range continues to extend for both viruses, the potential for blood-mediated transmission does exist. At the time of publication, no assays licensed by the FDA are available for either virus. Sporadic, local outbreaks of dengue have been reported in Hawaii, Florida, and 10 Cancer Control South Texas,28,29 as well as chikungunya transmission in Florida.26 Through cooperation with public health authorities, surveillance and suspension of blood collection from areas affected have been successful in avoiding the spread of blood-borne infections in the United States.26,28,29 Ebola Ebola is a filovirus that has recently caused disease outbreaks in several West African countries. In addition, imported cases in the United States and Western Europe have been reported and are associated with health care workers returning from epidemic areas.30 Ebola is transmitted when an infected patient is symptomatic following the incubation period. Currently, individuals returning from Ebola epidemic areas are deferred from donating blood for 1 year because malarial travel restrictions apply to the same regions.30-32 To address the potential transmission through local contact, blood centers are also asking individuals who have been identified by public health officials as possibly exposed to a patient infected with Ebola virus not to donate blood for 28 days following the last contact with the infected person.30-32 No FDA-licensed assays exist to detect Ebola infection in donors.30 No cases of transfusion-acquired Ebola infection have been reported.30-32 The use of convalescent plasma for treatment remains investigational.32 Bacterial Infections Contamination of blood components with bacteria poses a significant challenge, particularly for platelets, because they cannot be stored at sufficiently low temperatures that have a bacteriostatic effect. The source of bacteria may be endogenous (eg, subclinical bacterial endocarditis, osteomyelitis, syphilis, dental abscess) or, more commonly, tied to a skin contaminant. In addition, during storage, the number of bacteria present in the container may continue to significantly increase up to the time of transfusion.33 The result may be that a sample from a unit of platelets cultured earlier (typically 24 hours following blood collection) does not necessarily reflect the current bacterial load prior to transfusion. Although alternative “close to release” assays have become available, none has proven to be practical for use outside of the nonemergent clinical setting. Currently, culture methods are capable of interdicting approximately 50% of contaminated units; however, most of the contaminated units not removed from inventory are transfused in the initial storage period before the bacterial load reaches concentrations that could have clinical consequences for the recipient.34 Therefore, while the rate of bacterial detection in platelet units is approximately 1 in 5,000, the incidence rate of significant morbidity associated with the transfusion of bacterially contaminated plateJanuary 2015, Vol. 22, No. 1 lets ranges from 1 in 70,000 to 118,000.35 The severity of the reaction depends on the amount of bacteria as well as their pathogenicity, which is associated with their capacity to induce septic shock and disseminated intravascular coagulation. In general, endotoxin-producing, gram-negative bacteria have a high correlation with significant morbidity and mortality. The use of pathogen-reduction technologies applied to platelet components effectively eliminates the transfusion of units containing viable bacteria; however, no such technology has been licensed by the FDA for use in the United States. Serological testing for syphilis has been performed since the early beginnings of transfusion. Treponema pallidum has been transmitted via transfusion when blood was reinfused within 24 hours of collection, and introducing serological testing for hepatitis rendered the practice of immediate use unfeasible, thus eliminating the transmission of syphilis through transfusion. Blood donors are still screened for syphilis, although no cases associated with blood component transfusion have been reported since the late 1960s.36 Another member of the Spirochaeta family that has been the subject of studies is Borrelia burgdorferi, the causative agent of Lyme disease. Although the theoretical risk of transmission through transfusion has been posed, no case has been documented. Furthermore, in studies of recipients of components from DNA-positive donors, no evidence of infection was ever found.22 Rickettsial agents may be transmitted via blood transfusion.37 These obligate, intracellular bacterial organisms remain viable even after 2 or more weeks of storage. The species and disease entities reported to be associated with blood transfusion as the exposure event are Anaplasma phagocytophilum (human granulocytic anaplasmosis), Coxiella burnetii (Q fever), and Rickettsia rickettsii (Rocky Mountain spotted fever). Given that the number of cases reported is low, no specific preventive measures beyond proper biovigilance are recommended.22 1 in several million units.22 In the United States, about 5 cases of malaria associated with transfusion have been published since 2000.22 Most cases were traced to immigrants from endemic regions who then remained asymptomatic for several years after being considered successfully treated. Recipients of red blood cells from asymptomatic, infected donors develop symptoms 1 month or more following transfusion; because of the unusual transmission route and its protean clinical presentation, the diagnosis is typically made after ruling out several other potential causes. Parasite Diseases With Possible Transmission via Blood Transfusion The tick-borne intraerythrocytic parasite Babesia microti, as well as other closely related members of the Babesia species, such as B duncani and B divergens, have been transmitted by blood transfusion in almost 100 reported cases, making this species the most frequently transmitted parasite via transfusion in the United States.40 The parasite uses wild rodents and deer as mammalian hosts and Ixodes ticks as vectors. In endemic areas of New England and the upper Midwest, serology surveys have found seroprevalence rates of around 2%, mostly for B microti.41 In western states, B duncani is the predominant variant. The density of the deer population in suburban areas has increased in the last few decades, so the number of Malaria Because the number of autochthonous cases occurring in the United States in the last several decades has been limited to a handful, the risk of transmission is confined to the collection of blood from individuals who immigrate from or return from travel to endemic areas in which any of the 4 species of Plasmodium can be found.38 No assays have been licensed by the FDA to detect malarial parasites or antibodies in blood donors. Nevertheless, eliminating blood collection from at-risk individuals has reduced the risk of transfusion-transmitted malaria to approximately January 2015, Vol. 22, No. 1 American Trypanosomiasis (Chagas Disease) The transmission of Trypanosoma cruzi via blood transfusion was recognized in endemic countries (mostly countries in the Western Hemisphere, except the United States and Canada) early after transfusion therapy became available. Seroprevalence studies conducted in Latin America have shown that 12% to 25% of seronegative recipients of fresh whole blood were found to seroconvert after receiving cellular blood components from infected donors.39 Detectable clinical disease 20 to 40 days after transfusion is more common in patients who are immunosuppressed; however, among immunocompetent recipients, approximately 30% of those who carry the parasite will develop cardiac or gastrointestinal clinical features characteristic of Chagas disease at least 20 years following transfusion.39 Migrants from endemic areas were identified in 7 documented cases associated with transfusion in the United States and Canada, but more undetected, subclinical transmissions have likely occurred.39 As a result, blood establishments in both countries have implemented serological screening for all blood donors for the presence of anti–T cruzi antibodies to identify and interdict blood components with the potential for transmitting the parasite to recipients.39 Given this measure, the risk of transmission via transfusion is now considered negligible.39 Babesiosis Cancer Control 11 donors carrying the parasite in their blood has also risen and resulted in more cases linked to transfusions every year. Patients who are immunocompromised or asplenic are vulnerable to a severe form of babesiosis, which is characterized by fever, hemolytic anemia, thrombocytopenia, and, in the most severe of cases, disseminated intravascular coagulation and multiorgan failure. Immunocompetent individuals who acquire the parasite either by tick bite or transfusion may remain asymptomatic or they may present with mild, flu-like illness. Asymptomatic individuals may remain parasitemic for months or even years. The FDA has not licensed any assays to detect current or past infestation in blood donors, so donor screening is limited to questioning potential donors about a prior diagnosis of babesiosis. Although the detection of parasites through NAT assays is the most effective way to interdict parasitemic units, given its complexity and cost, the detection of antibodies to Babesia appears to be the most practical donor screening mechanism. At the time of publication, 2 different methodologies are under development. Leishmaniasis Leishmania donovani may be transmitted via transfusion and causes severe clinical disease in immunosuppressed and newborn recipients.42 All reported cases have occurred in hyperendemic areas of the world (eg, the Middle East). As a result, the temporary deferral of individuals returning from regions of the world in which Leishmania poses a threat to the population is used to eliminate potential transmission by blood transfusion. Human Prion Disease Disease entities associated with prion infection include classical and sporadic Creutzfeldt–Jakob disease (CJD), infectious CJD (kuru associated with cannibalism and its iatrogenic form), familial or heritable (Gerstmann–Sträussler–Scheinker syndrome), and variant CJD. Variant CJD prions alone have been associated with blood transfusion. Four cases were reported in recipients of nonleukoreduced red blood cells and in 1 patient with hemophilia who was treated with clotting-factor concentrates sourced from the United Kingdom.43 Three of the individuals transfused with red blood cells developed clinical variant CJD 6.3 to 8.5 years after transfusion. The patient with hemophilia and 1 red blood cell recipient had prions detected in tissue but no clinical disease. In all cases, the sources were asymptomatic donors at the time of donation but who later developed clinical variant CJD.43 Experimental transfusion models using sheep have showed that transmission occurs in 36% of exposed animal recipients.44 12 Cancer Control At the time of publication, no assays reliably detect presymptomatic or asymptomatic infection. For that reason, intervention to prevent variant CJD transmission via transfusion is limited to the exclusion of donors exposed to regions where variant CJD has the potential to enter the food supply as the agent of bovine spongiform encephalitis, which infected cattle in the United Kingdom. This exclusion extends to individuals who spent at least 3 months in the United Kingdom from 1980 through 1996, at least 5 years in Europe since 1980, or those who received transfusions in the United Kingdom or France since 1980. The residual risk of infection with variant CJD in the United States is estimated to be negligible. Immune-Mediated Reactions Hemolysis Due to Serological Incompatibility All transfusable blood components are labeled to indicate the blood type of the donor as well as the screening result for the detection of unexpected antibodies against red blood cell antibodies. Blood components containing plasma with unexpected (ie, other than anti-A and/or anti-B) isoagglutinins are not transfused. However, under certain circumstances, units of plasma or platelets incompatible with the red blood cells of the recipients may be transfused. To avoid hemolytic reactions under such circumstances (particularly for platelet components containing significant amounts of supernatant plasma), at least 1 of the following strategies should be used: •Limit the total volume of ABO-incompatible plasma by restricting the total plasma volume to be transfused, reducing the plasma volume, or platelet washing •Store in platelet additive solutions to reduce the residual plasma by two-thirds •Obtain isoagglutinin titers to eliminate donors with high levels of hemolysins Transfusion-Related Acute Lung Injury Transfusion-related acute lung injury is most commonly associated with the transfusion of blood components containing a plasma volume that exceeds 100 mL. In more than one-half of observed transfusion-related acute lung injury, antihuman leukocyte antigen (anti-HLA) antibodies (classes 1, 2, or both) or antihuman neutrophil (anti-HNA) antibodies can be detected in the transfused product. Although the pathophysiology of transfusion-related acute lung injury has not been elucidated, the antibodies in the donor that interact with the leukocytes of the recipient are considered to be a significant risk factor.45 As a result, mitigation strategies involving the selection of donors not likely to have developed those antibodies (eg, untransfused men, women who have never January 2015, Vol. 22, No. 1 been pregnant or never received a transfusion) and testing donors more likely to have developed antibodies (eg, women who have been pregnant) have been developed. Although HLA antibody screening assays are available, no assays can be practically applied to detect anti-HNA. Furthermore, antibodies are not detected in a large number of cases of transfusionrelated acute lung injury; thus, alternative pathways for neutrophil priming and activation involving lipid molecules, microaggregates expressing CD40 receptors, and microparticles formed during cellular component storage are underway.46 Graft-vs-Host Disease Graft-vs-host disease is a life-threatening complication of transfusion and is mediated by post-transfusion clonal amplification of the donor’s lymphocytes in the recipient. This action occurs as a result of a patient’s inability to suppress lymphocyte proliferation due to cellular immunodeficiency associated with his or her primary condition or immunosuppressive therapy. Vulnerable patient populations include premature infants, recipients of hematopoietic stem cell transplantation, patients treated with fludarabine, patients transfused with cellular components collected from direct blood relatives,47 and individuals with hereditary immunodeficiencies. Blood recipients included in any of the categories above should receive irradiated cellular blood components alone; the radiation dose must be sufficient to stop the clonal expansion of donor lymphocytes (estimated at 25 Gy). Pathogen-reduction procedures that use amotosalen followed by irradiation with ultraviolet light have been reported to be appropriate.48 The use of high-efficiency leukocyte depletion filters is not effective as a preventive measure. Additional Mitigation Strategies to Enhance Safety Although testing for markers of transmissible disease and applying special methods in the preparation of blood components provide a strong foundation and support the safety of the blood supply, blood establishments have implemented additional safety layers to enhance the therapeutic profile of transfusable blood components. Donor Recruitment and Selection The use of volunteer, nonremunerated blood donors is an effective means for obtaining safe source material for blood transfusion. To obtain donations from segments of the population with the lowest incidence levels for transmissible diseases, each donor must be subjected to an extensive medical questionnaire to assess his or her medical history, travel, and behavior associated with potential risks of exposure to pathogens that may be transmitted by transfusion. For example, January 2015, Vol. 22, No. 1 travel to endemic areas for tropical disease temporarily disqualifies individuals until appropriate incubation periods have lapsed, and a history positive for viral hepatitis, drug use, or male-to-male sexual contact will result in indefinite deferral from blood donation under current US government regulations. Eligibility and disqualification criteria are established through governmental regulations as well as a set of standards established by professional societies. In addition, a physical examination that includes vital signs is also performed. All donors are provided with instructions to allow them to report potential prodromic symptoms of infection within 72 hours following donation. Leukocyte Reduction Current routine methods of filtration remove leukocytes from cellular blood components. Affinity filters that use physical properties as well as electrical static charges to remove the target cells are efficient devices that eliminate more than 99.999% of the white blood cells in the original blood collection with minimal loss of red blood cells or platelets.49 Using differential centrifugation, high-efficiency apheresis instruments are also capable of harvesting large numbers of platelets with minimal loss of white blood cells. The depletion of leukocytes decreases the rates of immune sensitization and febrile nonhemolytic reactions. It also plays an important role in the prevention of CMV, HTLV-1, and HTLV-2 transmission and the removal of other intracellular pathogens. Pathogen Reduction and Inactivation The elimination of microorganism transmission through blood components is a goal of transfusion practice; however, the chemical or physical processes used to achieve that goal must maintain the viability and functionality of the treated product.50 Plasma intended for transfusion may be treated with either methylene blue or solvent or detergent solutions. The former inactivates most viruses, bacteria, and parasites after forming stable chemical bonds when exposed to visible light. The latter acts by disrupting the membranes of most microorganisms with the exception of nonenveloped viruses such as hepatitis A and parvovirus B19. Both types of methods have been implemented in Europe for several years and one was approved for use in the United States in 201351; however, neither method can be used on cellular components. Platelet components may be treated with a psoralen (amotosalen) or riboflavin and then subsequently subjected to ultraviolet irradiation to stabilize the disruptive bonds made by those chemicals with DNA/ RNA molecules. Pathogen-reduction systems for platelets are not available for use in the United States. An amotosalen-based system called Intercept (Cerus, ConCancer Control 13 cord, California) is in use in some countries in Europe and the Middle East, and Mirasol (Terumbo BCT, Lakewood, Colorado), which is a riboflavin-based system, is undergoing several clinical trials (NCT01740531, NCT01907906, and NCT00261924). Given the high hemoglobin content in red blood cell components, methods requiring ultraviolet irradiation are not feasible. Although some chemicals have been identified that achieve significant pathogen-reduction levels, the potential formation of red blood cell neoantigens resulting in the immune destruction of treated cells has hampered progress.52 These systems are still in experimental phases. Biovigilance Procedures to quickly detect the possible spread of transmissible diseases via blood transfusion provide yet another safety layer for protecting the blood supply. Collaborating with public health officials by sharing surveillance data (eg, serosurveys of sentinel chickens for flavivirus outbreaks), investigating recipients of units from donors who seroconvert on subsequent donations (a process called “donor lookback”), or retesting donors who had their blood transfused to a patient who experienced a post-transfusion transmissible disease are some of the methods that can be used. In the laboratory, using samples from serial bleedings in recent seroconverted individuals provides valuable insights into the biology of infection with a particular pathogen and the different serological markers needed to detect infection in asymptomatic but infectious individuals. Conclusions Transmissible agents most commonly associated with risks due to transfusion are no longer a major threat; however, addressing the need for quick response mechanisms to manage emerging pathogens that may unintentionally or intentionally spread remains a challenge. The use of technology to reduce pathogens holds promise for further increasing the safety profile of blood transfusion. In addition, blood transfusion plays an important role in supporting patients with cancer. A multilayered strategy has raised the safety profile of blood components to acceptable levels; however, treatment with blood transfusions must be considered within the broader context of risks and benefits that go beyond strict product safety. Many aspects related to the interactions between the allogeneic components transfused must be reviewed when assessing the risks and benefits of transfusion therapy for patients with cancer. References 1. Tobler LH, Busch MP. History of posttransfusion hepatitis. Clin Chem. 1997;43(8 pt 2):1487-1493. 14 Cancer Control 2. Kleinman SH, Kuhns MC, Todd DS, et al. Frequency of HBV DNA detection in US blood donors testing positive for the presence of anti-HBc: implication for transfusion transmission and donor screening. Transfusion. 2003;43(6):696-704. 3. Stramer SL, Notari EP, Krysztof DE, et al. Hepatitis B virus testing by minipool nucleic acid testing: does it improve blood safety? Transfusion. 2013;53(10 pt 2):2449-2458. 4. Alter HJ, Houghton M. Hepatitis C virus and eliminating post-transfusion hepatitis. Nat Med. 2000;6(10):1082-1086. 5. Micallef JM, Kaldor JM, Dore GJ. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J Viral Hepat. 2006;13(1):34-41. 6. Busch MP, Glynn SA, Stramer SA, et al; NHLBI-REDS Nat Study Group. A new strategy for estimating risks of transfusion-transmitted viral infections based on rates of detection of recently infected donors. Transfusion. 2005;45(2):254-264. 7. Bihl F, Castelli D, Marincola F, et al. Transfusion-transmitted infections. J Transl Med. 2007;5:25. 8. Galel SA, Lifson JD, Engleman EG. Prevention of AIDS transmission through screening of the blood supply. Annu Rev Immunol. 1995;13:201-227. 9. Stramer SL, Glynn SA, Kleinman SH, et al; National Heart, Lung, and Blood Institute Nucleic Acid Test Study Group. Detection of HIV-1 and HCV infections among antibody-negative blood donors by nucleic acid amplification testing. N Engl J Med. 2004;351(8):760-768. 10. Zou S, Stramer SL, Dodd RY. Donor testing and risk: current prevalence, incidence, and residual risk of transfusion-transmissible agents in US allogeneic donations. Transf Med Rev. 2012;26(2):119-128. 11. Centers for Disease Control and Prevention. Outbreak of West Nilelike viral encephalitis--New York, 1999. MMWR Morb Mortal Wkly Rep. 1999;48(38):845-849. 12. Sejvar JJ, Haddad MB, Tierney BC, et al. Neurologic manifestations and outcome of West Nile virus infection [Erratum appears in JAMA. 2003;290(10):1318]. JAMA. 2003;290(4):511-515. 13. Busch MP, Caglioti S, Robertson EF, et al. Screening the blood supply for West Nile virus RNA by nucleic acid amplification testing. N Engl J Med. 2005;353(5):460-467. 14. Centers for Disease Control and Prevention. Fatal West Nile virus transmission after probable transfusion-associated transmission--Colorado, 2012. MMWR Morb Mortal Wkly Rep. 2013;62(31):622-624. 15. Saedi M, Sasannejad P, Foroughipou M, et al. Prevalence of peripheral neuropathy in patients with HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP). Acta Neurol Belg. 2011;111(1):41-44. 16. Stramer SL, Notari EP IV, Zou S, et al. Human T-lymphotrophic virus antibody screening of blood donors: rates of false-positive results and evaluation of a potential donor reentry algorithm. Transfusion. 2011;51(4):692-701. 17. Vamvakas EC. Is white blood cell reduction equivalent to antibody screening in preventing transmission of cytomegalovirus by transfusion? A review of the literature and meta-analysis. Transfus Med Rev. 2005;19(3): 181-199. 18. Plentz A, Hahn J, Knöll A, et al. Exposure of hematologic patients to parvovirus B19 as a contaminant of blood cell preparation and blood products. Transfusion. 2005;45(11):1811-1815. 19. Schmidt M, Themann A, Drexler C, et al. Blood donor screening for parvovirus B19 in Germany and Austria. Transfusion. 2007;47(10):1775-1782. 20. Kuniholm MH, Purcell RH, McQuillan GM, et al. Epidemiology of hepatitis E virus in the United States: results from the Third National Health and Nutrition Examination Survey, 1988-994. J Infect Dis. 2009;200(1):48-56. 21. Cleland A, Smith L, Crossan C, et al. Hepatitis E virus in Scottish blood donors. Vox Sang. 2013;105(4):283-289. 22. Stramer SL, Hollinger FB, Katz LM, et al. Emerging infectious disease agents and their potential threat to transfusion safety. Transfusion. 2009;(49 suppl 2):1S-29S. 23. Leong AS, Wong KT, Leong TY, et al. The pathology of dengue hemorrhagic fever. Semin Diagn Pathol. 2007;24(4):227-236. 24. Josseran L, Paquet C, Zehgnoun A, et al. Chikungunya disease outbreak, Reunion Island. Emerg Infect Dis. 2006;12(12):1994-1995. 25. Parola P, de Lamballerie X, Jourdan J, et al. Novel chikungunya virus variant in travelers returning from Indian Ocean islands. Emerg Infect Dis. 2006;12(10):1493-1499. 26. Centers for Disease Control and Prevention. First chikungunya case acquired in the United States reported in Florida. http://www.cdc.gov/media/ releases/2014/p0717-chikungunya.html. Accessed October 10, 2014. 27. Tomashek KM, Margolis HS. Dengue: a potential transfusion-transmitted disease. Transfusion. 2011;51(8):1654-1660. 28. Adalja AA, Sell TK, Bouri N, et al. Lessons learned during dengue outbreaks in the United States, 2001-2011. Emerg Infect Dis. 2012;18(4): 608-614. 29. Centers for Disease Control and Prevention. Locally acquired dengue -Key West, Florida, 2009-2010. MMWR Morb Mortal Wkly Rep. 2010;59(19): 577-581. 30. American Association of Blood Banks; America’s Blood Centers; American Red Cross. Joint statement regarding Ebola and the safety of the blood supply. http://www.americasblood.org/media/50695/stmnt_ebola_joint_1410. pdf. Accessed November 4, 2014. January 2015, Vol. 22, No. 1 31. American Association of Blood Banks. Association bulletin #14-08. http://www.aabb.org/programs/publications/bulletins/Documents/ab14-08.pdf. Accessed November 4, 2014. 32. America’s Blood Centers. Q & A with ABC chief medical officer Louis Katz – Ebola virus: what you need to know. http://hospitals.unitedbloodservices. org/abcnews/ABC_10_24_14.pdf. Accessed November 4, 2014. 33. Brecher ME, Hay SN. Bacterial contamination of blood components. Clin Microbiol Rev. 2005;18(1):195-204. 34. Schezenmeier H, Walther-Wenke G, Müller TH, et al. Bacterial contamination of platelet concentrates: results of a prospective multicenter study comparing whole blood-derived platelets and apheresis platelets. Transfusion. 2007;47(4):644-652. 35. Eder AF, Kennedy JM, Dy BA, Notari EP, et al; American Red Cross Regional Blood Centers. Bacterial screening of apheresis platelets and residual risk of septic transfusion reactions: the American Red Cross experience (2004-2006). Transfusion. 2007;47(7):1134-1142. 36. Katz LM. A test that won’t die: the serologic test for syphilis. Transfusion. 2009;49(4)617-619. 37. Leiby DA, Gill JE. Transfusion-transmitted tick-borne infections: a cornucopia of threats. Transf Med Rev. 2004;18(4):293-306. 38. Kitchen AD, Chiodini PL. Malaria and blood transfusion. Vox Sang. 2006;90(2):77-84. 39. Benjamin RJ, Stramer SL, Leiby DA, et al. Trypanosoma cruzi infection in North America and Spain: evidence in support of transfusion transmission. Transfusion. 2012;52(9):1913-1921. 40. Leiby DA. Transfusion-transmitted Babesia spp.: bull’s-eye on Babesia microti. Clin Microbiol Rev. 2011;24(1):14-28. 41. Levin AE, Williamson PC, Erwin JL, et al. Determination of Babesia microti seroprevalence in blood donor populations using an investigational enzyme immunoassay. Transfusion. 2014;54(9):2237-2244. 42.Cardo LJ. Leishmania: risk to the blood supply. Transfusion. 2006;46(9):1641-1645. 43. Hewitt PE, Llewelyn CA, Mackenzie J, et al. Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang. 2006;91(3):221-230. 44. Dodd RY, Busch MP. Animal models of bovine spongiform encephalopathy and vCJD infectivity in blood: two swallows do not a summer make. Transfusion. 2002;42(5):509-512. 45. Toy P, Gajic G, Bacchetti P, et al; TRALI Study Group. Transfusion-related acute lung injury: incidence and risk factors. Blood. 2012;119(7):1757-1767. 46. Van Bruggen R, de Korte D. Prevention of non-immune mediated transfusion-related acute lung injury; from blood bank to patient. Curr Pharm Des. 2012;18(22):3249-3254. 47. Agbaht K, Altintas ND, Topeli A, et al. Transfusion-associated graftversus-host disease in immunocompetent patients: case series and review of the literature. Transfusion. 2007;47(8):1405-1411. 48. Mintz PD, Wehrli G. Irradiation eradication and pathogen reduction. Ceasing cesium irradiation of blood products. Bone Marrow Transplant. 2009;44(4):205-211. 49. Shapiro, MJ. To filter blood or universal leukoreduction: what is the answer? Crit Care. 2004;8(suppl 2):S27-S30. 50. Goodrich RP, Custer B, Kell S, et al. Defining “adequate” pathogen reduction performance for transfused blood components. Transfusion. 2010;50(8):1827-1837. 51. Octapharma USA. FDA approves Octaplas expanding Octapharma U.S. transfusion medicine therapies. http://www.octaplasus.com/uploads/octaplas_approval_01-18-13.pdf. Accessed October 10, 2014. 52. Wagner SJ. Developing pathogen reduction technologies for RBC suspensions. Vox Sang. 2011;100(1):112-121. January 2015, Vol. 22, No. 1 Cancer Control 15 Transfusions confer such risks as acute TRALI, alloimmunization, and iron overload, so they must be used only when benefits outweigh the risks. Ray Paul. Petal Purple, 2010. Acrylic, latex, enamel on canvas, 12" × 12". Adverse Effects of Transfusion Radhika Dasararaju, MD, and Marisa B. Marques, MD Background: Patients with malignancy comprise a unique group for whom transfusions play an important role. Because the need for transfusions may span a long period of time, these patients may be at risk for more adverse events due to transfusion than other patient groups. Methods: A literature search on PubMed that included original studies and reviews was performed. The results were summarized and complemented by our clinical experience. Long-term complications of transfusions, such as transfusion-associated graft-vs-host disease, alloimmunization, transfusion-related immunomodulation, and iron overload, are discussed. Results: Transfusion-related acute lung injury, transfusion-associated circulatory overload, and hemolytic transfusion reaction are deadly complications from transfusion. These adverse events have nonspecific presentations and may be missed or confused with a patient’s underlying condition. Thus, a high level of suspicion and close monitoring of the patient during and following the transfusion is imperative. Common reactions (eg, febrile nonhemolytic transfusion reaction, allergic reaction) are not life threatening, but they may cause discomfort and blood product wastage. Conclusions: Every transfusion carries risks of immediate and delayed adverse events. Therefore, oncologists should prescribe transfusion for patients with cancer only when absolutely necessary. Introduction Patients with malignancy comprise a unique group for whom transfusions play an important — and sometimes lifesaving — role. Typically, patients with cancer are pancytopenic, immunosuppressed, or both, and these conditions affect their transfusion needs as well as the interpretation of signs and sympFrom the Department of Pathology, University of Alabama at Birmingham, Birmingham, Alabama. Submitted June 16, 2014; accepted October 14, 2014. Address correspondence to Marisa B. Marques, MD, University of Alabama at Birmingham, Department of Pathology, WP P230G, 619 – 19th Street South, Birmingham, AL 35249. E-mail: mmarques @uab.edu No significant relationships exist between the authors and the companies/organizations whose products or services may be referenced in this article. 16 Cancer Control toms of possible reactions. Because their need for transfusions may span a long period of time, this patient population may be at risk of experiencing more adverse events due to transfusion than any other patient group. That being said, a 4-year study by Huh and Lichtiger1 revealed that reactions occurred less frequently in patients with cancer and that febrile nonhemolytic transfusion reactions (FNHTRs) and allergic reactions were the most common (51.3% and 36.7%, respectively). FNHTRs are particularly difficult to differentiate from the patient’s underlying illness, considering that many are already febrile before the transfusion. A thorough review of vital signs before and after the transfusion, associated signs and symptoms, and timing of the increased temperature are essential to make the correct diagnosis. To prevent FNHTRs, transfusion services strive to offer leukoreduced January 2015, Vol. 22, No. 1 products alone to patients with cancer. Leukoreduced red blood cells (RBCs) and platelets have the added advantage of mitigating the risk of cytomegalovirus (CMV) transmission because they are CMV safe.2 Table 1. — Signs and Symptoms of Acute Transfusion Reactions Sign/Symptom Possible Transfusion Reaction Fever FNHTRa AHTR TRALI Microbial contamination Itching Rash Urticaria Wheezing Facial edema Allergic reaction ecrease oxygen saturation D to < 90% on room air TACO TRALI Dyspnea Respiratory distress Cyanosis AHTR Allergic reaction Microbial contamination TACO TRALI Hypertension Tachycardia TACO Hypotension AHTR Allergic reaction Microbial contamination TRALI Pain at IV infusion site Abdominal/chest/flank pain AHTR Allergic reaction Premedication Prior to Transfusion In 2007, according to Geiger and Howard,3 physicians at a research hospital prescribed an antipyretic and an antihistamine (usually acetaminophen and diphenhydramine) prior to almost 70% of transfusions. This figure is higher than the rest of the United States at about 50%. Although the practice of premedication to prevent FNHTRs and allergic reactions is likely to continue, several published reports have questioned its validity. A prospective study of hematology/oncology patients suggested that premedication use can be decreased without increasing reaction rates and that prestorage leukoreduction, reduced plasma from platelet units, or both diminish but do not eliminate FNHTRs.4 Another study concluded that, although routine pretransfusion antipyretics reduce patient inconvenience and morbidity rates associated with FNHTRs, as well as decrease product wastage, the process is not cost effective.5 A randomized controlled trial of 315 patients with leukemia or post–stem cell transplantation without a history of transfusion reactions showed that premedication and bedside leukoreduction significantly decreased the risk of FNHTRs.6 And, more recently, a systematic review found no evidence to justify premedication to prevent FNHTRs and allergic reactions regardless of patient history.7 Acute Transfusion Reactions Although FNHTRs and allergic reactions are common and familiar to most health care professionals, these reactions are not as life threatening as acute hemolytic transfusion reactions (AHTRs), transfusion-associated circulatory overload (TACO), and transfusion-related acute lung injury (TRALI). According to the US Food and Drug Administration (FDA), 30 to 44 patients died due to transfusion reactions per year in the United States between 2009 and 2013.8 The top 3 causes of transfusion-related fatalities for the combined 5 years were TRALI at 38%, TACO at 24%, and AHTRs at 22%.8 The remaining deaths were caused by microbial contamination at 10%, anaphylaxis at 5%, and other causes, such as transfusion-associated graft-vshost disease (TA-GVHD) and hypotension, at 1%.8 The dilemma for the health care team caring for patients with cancer who develop a reaction is to determine: (1) If the signs and symptoms represent a true reaction or a coincidence (ie, fever), and (2) how serious a reaction is if it has occurred. The differentiation between the patient’s underlying status and a reaction to explain new signs and symptoms, as well as the type of reaction, is difficult to ascertain because of January 2015, Vol. 22, No. 1 AHTR = acute hemolytic transfusion reaction, FNHTR = febrile nonhemolytic transfusion reaction, IV = intravenous, TACO = transfusion-associated circulatory overload, TRALI = transfusion-related acute lung injury. a Fever is most often due to underlying infection among patients with cancer, especially if the blood product (red blood cells or platelets) is leukoreduced. From references 9 to 15. the nonspecific manifestations of transfusion-related adverse events (Table 1).9-15 Fever, chills, nausea, vomiting, pain, itching at the intravenous (IV) insertion site, variations in blood pressure, tachycardia, dyspnea, and restlessness are among the most common reasons a reaction is suspected. Although fever may indicate an FNHTR, it may also be a sign of a potentially fatal complication such as AHTR or sepsis. For this reason, transfusion administration guidelines must be strictly followed to avoid a reaction, such as an AHTR, caused by the infusion of the incorrect unit to the patient and to detect one as soon as it occurs.9,16 Because the severity of the reaction and its consequences are directly proportional to the volume of incompatible product transfused, early recognition and rapid intervention are essential to minimize harm. After stopping the transfusion at the earliest sign of reaction, the IV access line should be kept open with normal saline. The next critical step is to check that the blood product was intended for that recipient.9 Immediately thereafter, the remainder of the unit with the attached tubing and compatibility label or “bag tag” must be sent to the transfusion service (ie, blood bank) accompanied by a description of the clinical picture, vital signs before and during the transfusion, and a sample of the patient’s blood. Fresh Cancer Control 17 urine should also be sent if hemolysis is suspected. In the blood bank, a clerical check is repeated and pretransfusion data, such as ABO type, antibody screen, crossmatch result if packed RBCs were implicated, and any other pertinent history are reviewed. Because the laboratory workup is aimed at detecting or excluding hemolysis, such workup starts with the inspection of the plasma color followed by a direct antiglobulin test (DAT) and screening for free hemoglobin in plasma and urine.17 A newly positive post-transfusion DAT result compared with a negative DAT pretransfusion result suggests an AHTR.17 In such cases, the patient’s clinical team must be notified as soon as possible so aggressive hydration can be initiated to limit the deleterious effects of free plasma hemoglobin. A negative laboratory workup is expected for all other types of adverse effects of transfusions (Table 2).9-15,17-21 Acute Hemolytic Transfusion Reaction Most often, AHTRs are caused by immune incompatibility between the donor and the recipient (typically, antigen-positive RBCs are transfused to a patient with the corresponding antibodies).22 The most severe AHTR is due to immunoglobulin (Ig) M anti-A, usually from a processing error in which the wrong blood was sent to the transfusion service with the patient’s name, or from failing to perform a patient identification check at the bedside and transfusing a unit of RBCs intended for someone else.16 In the last 5 years, 13 patients died from an ABO-mediated AHTR.8 In addition, non-ABO antibodies caused more than twice as many fatal AHTRs in the same time-period (29 deaths).8 As seen in Table 2, a variety of symptoms may denote an AHTR.9-15,17-21 Because the volume of incompatible blood transfused correlates with the severity of the reaction, it is important for transfusionists to stay with the patient for the first few minutes of every transfusion and then advise the patient to immediately notify the nursing staff if any new symptoms occur. Other variables that affect the severity of an AHTR include the recipient antibody type and titer. Because AHTRs may also be delayed, patients should be instructed on how to report any symptoms that develop within 24 hours, especially if they were transfused as outpatients. Hemolysis due to anti-A and anti-B is mainly intravascular because IgM readily activates complement, inducing the formation of a membrane attack complex.22 In turn, complement activation leads to the release of vasoactive amines, histamine, and other inflammatory cytokines such as interleukins and tumor necrosis factor α, which activate coagulation and fibrinolysis. In addition, complement-activation products and cytokines cause hypotension. Free plasma hemoglobin is both damaging to the endothelium and a nitric oxide scavenger, causing vasoconstriction 18 Cancer Control and hypoxia.10 Hemolysis mediated by IgG antibodies (non-ABO) is mainly extravascular through phagocytosis of the transfused RBCs by splenic macrophages via their Fc receptors. However, in patients with high-titer IgG antibodies to RBC antigens, combined extravascular and intravascular hemolyses may occur. Patients with cancer are also at risk of an AHTR when receiving ABO-incompatible platelets with anti-A, anti-B, or both in the plasma.23 To minimize this risk, transfusion services are expected to avoid units with high-titer ABO antibodies, if known, because testing is not routine at all institutions. In the event that hemolysis is suspected following incompatible platelets, a post-transfusion DAT would provide useful information. Hemolysis can also occur from improper storage of RBCs, leading to thermal, mechanical, or osmolar injury and, rarely, bacterial contamination. The concomitant infusion of hypotonic solutions or medications with RBCs also results in hemolysis and is not recommended.24 Rh immunoglobulin (passively acquired IgG anti-D) or intravenous immunoglobulin (IVIG; which contains anti-A and anti-B) can also cause hemolysis, and this complication should be promptly recognized.25-27 A suspected AHTR is confirmed by a change in plasma color and a positive result on DAT for IgG, complement, or both.17 In such patients, an extended workup may include haptoglobin, lactate dehydrogenase, bilirubin, plasma-free hemoglobin, creatinine, and a disseminated intravascular coagulation profile. Management is mainly supportive with IV fluids, diuretics, vasopressors, and blood products if bleeding induced by disseminated intravascular coagulation ensues.9 Strict adherence to patient identification procedures, and proper specimen collection practices help prevent AHTRs and improve transfusion safety.16 Transfusion-Related Acute Lung Injury Twenty years ago, the American-European Consensus Conference published a definition of acute lung injury.28 Ten years later, TRALI was defined as new-onset acute lung injury within 6 hours of transfusion with a PaO2:FIO2 ratio of no more than 300 mm Hg or oxygen saturation of at least 90% on room air and bilateral infiltrates on chest radiography in the absence of left atrial hypertension.11,12 TRALI is most often caused by antibodies to human leukocyte antigens (HLAs) or human neutrophil antigens (HNAs) in the transfused blood product given to a patient whose leukocytes express the cognate antigen.29 It is believed that TRALI follows a 2-hit model: (1) Neutrophils are primed and sequestered in the lungs due to an underlying clinical condition, and (2) they become activated by the infusion of antibodies or biological response modifiers (ie, cytokines and lipids accumulated in the blood product).30,31 January 2015, Vol. 22, No. 1 Table 2. — Possible Diagnoses for Immediate Adverse Events of Transfusion Symptoms Diagnosis Symptoms AHTR Fever, chills, dyspnea Hypotension, shock, DIC Red or brown urine Chest/flank/abdominal pain Nausea and vomiting Pain at IV infusion site Renal ischemia and failure Oliguria/anuria Hypotension and dark urine may be the initial signs in anesthetized patients Microbial Contamination DAT positive (may be negative if all incompatible red cells destroyed) Hemolyzed plasma, hemoglobinuria Antibody screen positive; negative if due to ABO incompatibility Eluate with alloantibody or anti-A or anti-B Falling hematocrit level Haptoglobin decreased, LDH increased If DAT negative, consider thermal, osmotic, mechanical, or chemical cause Fever Chills Hypotension Shock DIC Vomiting, diarrhea TRALI Onset within 6 hours of transfusion Dyspnea Oxygen saturation < 90% Cyanosis Hypotension Fever Chills Hypoxia (PaO2/FIO2 ratio ≤ 300 mm Hg) Pulmonary artery pressure < 18 mm Hg TACO Urticaria Itching Rash Wheezing Negative for AHTR Mainly clinical diagnosis Anaphylactic Transfusion Reaction Negative for AHTR High brain natriuretic peptide Respiratory distress Dyspnea Bronchospasm Sweating Flushing Nausea, vomiting, abdominal cramps Substernal pain Hypotension Shock Localized angioedema Transfusion-Associated Dyspnea Dyspnea Cyanosis Onset within 24 hours of transfusion Negative for AHTR Gram stain and culture positive of implicated unit (usually platelets) Allergic Transfusion Reaction Negative for AHTR Transient leukopenia Chest radiography with bilateral pulmonary infiltrates Dyspnea Oxygen saturation < 90%, cyanosis Nonproductive cough, orthopnea Hypertension, tachycardia Left atrial hypertension, congestive heart failure New ST segment and T wave change on electrocardiography Diagnosis Negative for AHTR IgA deficiency with class-specific or subclass-specific anti-IgA (later determination) FNHTR Negative for AHTR, TACO, TRALI, and allergic reactions Temperature rise within 4 hours of transfusion, not caused by underlying condition, with or without chills or rigors Negative for AHTR AHTR = acute hemolytic transfusion reaction, DAT = direct antiglobulin test, DIC = disseminated intravascular coagulation, FNHTR = febrile nonhemolytic transfusion reaction, Ig = immunoglobulin, IV = intravenous, LDH = lactate dehydrogenase, TACO = transfusion-associated circulatory overload, TRALI = transfusion-related acute lung injury. From references 9 to 15 and 17 to 21. In addition to the lungs, neutrophils accumulate in other organs (eg, liver, central nervous system), likely contributing to the morbidity and mortality of TRALI.32 A case-nested study reported that patients with hematological malignancies undergoing induction chemotherapy were at increased risk for TRALI.33 In addition, TRALI may occur in patients with neutropenia, presumably by the infusion of vascular endothelial growth factor or antibodies to HLA class II that bind to pulmonary endothelium and cause pulmonary leak.34 Because plasma from females was implicated in January 2015, Vol. 22, No. 1 most initial cases of TRALI, almost all units of plasma currently manufactured in the United States are from male donors.35 Since this change, the risk of TRALI from plasma is comparable with that from RBC and platelet products.8 TRALI is a diagnosis of exclusion because it is clinically indistinguishable from other causes of respiratory distress (see Table 2). Thus, when patients develop sudden dyspnea, hypoxia, and hypotension during or within 6 hours of transfusion, the possibility of TRALI must be considered. Although fever is also Cancer Control 19 common, it may not initially occur. In addition to the laboratory workup to exclude hemolysis, a complete blood count may show acute neutropenia, which is a useful marker of TRALI.18,19 Chest radiography supports the diagnosis of TRALI with newly developed bilateral pulmonary infiltrates, but the infiltrates can also be seen in cases of TACO and other causes of acute lung injury. Treatment for TRALI consists of respiratory support and pressors. Although some patients receive corticosteroids, steroids have not been proven to be beneficial and diuretics are not indicated.31 Mortality rates range from 5% to 25%, and, with vigorous respiratory support, 80% of patients recover within 48 to 96 hours.36 Confirmation of TRALI occurs when anti-HLA or anti-HNA in the serum of the donor matches the phenotype of the patient.36 Any donor implicated in a case of TRALI should be indefinitely deferred from donating blood in the future. Transfusion-Associated Circulatory Overload The true morbidity and mortality rates of TACO are unknown due to the uncertain prevalence of TACO. Because TACO is now the second leading cause of transfusion-associated fatality in the United States, it is likely that awareness of its life-threatening potential has increased.8 By contrast to TRALI, which is difficult to prevent except by minimizing transfusions and avoiding donors with HLA and HNA antibodies, TACO is conceivably preventable.13,37,38 Health care professionals should identify transfusion recipients unable to effectively process the volume challenge and either avoid transfusions altogether, prescribe the smallest possible number of units, and/or ensure a slow infusion rate. The risk of TACO increases with age and the number of units transfused, especially in patients with congestive heart failure, chronic pulmonary disease, anemia, or those receiving plasma products.37,38 TACO should be suspected when the patient develops new or exacerbated respiratory distress, pulmonary edema, or evidence exists of left or right heart failure or elevated central venous pressure (see Table 2). These signs and symptoms usually present within 2 hours of the transfusion onset but may take up to 6 hours to manifest.13 It is often difficult to distinguish TACO from TRALI, although hypertension (not hypotension) is expected. If available, a high brain natriuretic peptide level or pro–brain natriuretic peptide may help diagnose TACO.20 In addition to slow infusion rates and close monitoring for the development of symptoms, concurrent infusion of other fluids should be avoided. Furthermore, peritransfusion diuretics can considerably decrease the risk of TACO.38 Transfusion-Associated Dyspnea Transfusion-associated dyspnea is defined as acute respiratory distress occurring within 24 hours of trans20 Cancer Control fusion that is not explained by the patient’s underlying medical condition and does not meet the criteria for TRALI, TACO, or an allergic reaction.21,39 Microbial Contamination Although bacterial contamination of RBCs is extremely rare, bacterial overgrowth in platelet units continues to be possible despite the implementation of various detection methods in the last 10 years.40 Bacterially contaminated platelets are the most common transfusion-transmitted disease and present a particular risk to patients with cancer due to their considerable exposure to platelets and their frequent immunocompromised state. Introduction of skin flora into the collected unit during phlebotomy, storage of the unit at room temperature or, rarely, asymptomatic donor bacteremia, all contribute to the risk. Although the presence of bacteria is often unsuspected, Fig 1 shows a unit in which the growth of methicillin-resistant Staphylococcus aureus caused fibrin clots and helped to prevent the unit from being issued from our transfusion service. Subsequent culture confirmed the clinical suspicion of bacterial contamination. In the last 5 years, S aureus infections have accounted for the majority of deaths due to infected platelet units, although other gram-positive and gram-negative organisms have also been implicated.8 Parasites that infect RBCs, such as Babesia microti or various malarial species, are the most likely etiology of infection from RBCs.41 Awareness of these transfusion-transmitted infections is of particular importance for oncologists. Babesiosis or malaria would not be suspected as the cause of unexplained fever in patients who lack the usual risk factors (eg, travel to an endemic area). Furthermore, the diagnosis requires a high level of suspicion and expert review of the patient’s peripheral blood (Fig 2). Thus, it is imperative that transfusion-transmitted infections be included in the differential diagnosis of fever in patients with cancer and should be followed by the specific diagnostic laboratory evaluation as soon as symptoms develop. Splenectomized patients are at significantly increased risk of developing severe babesiosis, which carries a grave prognosis. In such circumstances, RBC exchange may be indicated to decrease the parasite burden in critically ill patients.42 Because donor testing does not include assays for babesiosis and malaria, prevention is based on history of exposure, which can be ineffective. Polymerase chain reaction and indirect immunofluorescence are being investigated to screen donors but are not yet in use.41,43 Due to their immunocompromised state, patients with cancer are also at risk for other infections, including those due to CMV, parvovirus B19, and West Nile virus. Because leukoreduction nearly eliminates the risk of CMV infection and polymerase chain reaction January 2015, Vol. 22, No. 1 Fig 1. — Unit of apheresis platelets contaminated with methicillin-resistant Staphylococcus aureus discovered on day 5. Fig 2. — Peripheral blood smear of a patient with babesiosis (hematoxylin and eosin, × 1000). Courtesy of James Kelley, MD, PhD. for West Nile virus infection is routinely performed in donors, these infections are no longer significant concerns.2,44 However, parvovirus B19 remains a threat.45 Transfusion through indwelling central venous catheters with subclinical microbial colonization may lead to a septic reaction.46 Allergic Transfusion Reactions Minor allergic reactions manifested as pruritus and rash are common transfusion reactions, but they are benign and usually easily treated. However, allergic reactions can also represent life-threatening systemic anaphylaxis with hypotension and respiratory distress.14 Typically, they are IgE-mediated type 1 hypersensitivity reactions, leading to mast cell activation and the release of inflammatory mediators. Complement fixation and macrophage-derived cytokines may also contribute to allergic symptoms. Although the exact offending agent is typically unknown, these reactions occur when the patient has been presensitized to an immunologically active compound in the plasma of the donor. Examples of allergens include foods, medications, and polymorphic forms of serum January 2015, Vol. 22, No. 1 proteins other than IgA, like haptoglobin, C3, C4, transferrin, and albumin. The passive transfer of IgE antibodies to common environmental allergens and anaphylatoxins or platelet biological response mediators (eg, cytokines, chemokines) generated during storage also plays a role.14 For patients with mild symptoms such as pruritus or rash, transfusion may be restarted under close supervision and at a slower rate following treatment with an antihistaminic and if symptomatic improvement is seen; however, this practice is controversial. Severe allergic reactions are caused by antibodies to plasma proteins (eg, IgA, haptoglobin). IgA-related anaphylactic reactions occur in IgA-deficient patients with serum IgA levels below 0.05 mg/dL who have developed class-specific IgA antibodies, even without any previous pregnancy or transfusion (“naturally occurring”).14,15 Anaphylaxis causes bronchoconstriction that results in respiratory distress, wheezing, stridor, angioedema, and hypotension (see Tables 1 and 2). Prompt action should be taken to maintain oxygenation and improve blood pressure.9,14,15 Epinephrine may be intravenously or intramuscularly administered in addition to corticosteroids and antihistaminics. If bronchospasm is present, then respiratory symptoms may not respond to epinephrine; adding a β2 agonist or aminophylline may be required.9,14,15 Severe reactions should be further investigated to determine their etiology and to prevent their occurrence in future transfusions. Patients with an IgA deficiency and anti-IgA should be transfused products from IgA-deficient donors alone or given RBC washed units.14 For platelets, plasma reduction decreases the incidence of allergic reactions.14 In emergent situations, regular products may be given after premedication with antihistamines and steroids if the risk of withdrawing the transfusion is higher than the risk of anaphylaxis. The newly approved platelet additive solution, PAS C, replaces most of the plasma in the unit, decreasing the risk of allergic reactions and FNHTRs.47 Febrile Nonhemolytic Transfusion Reaction FNHTRs are the most common immediate adverse event of transfusion in patients with cancer.1 They are characterized by a temperature of 100.4°F (38°C) or an increase of 1.8°F or 1°C from the pretransfusion value, with or without chills, during or within 4 hours following the completion of the transfusion, occurring more often with platelets than RBCs (see Tables 1 and 2).9 FNHTRs are a consequence of the passive transfer of stored cytokines or due to recipient antibodies against HLAs, HNAs, or platelet antigens that stimulate the release of cytokines. When receiving leukoreduced products, FNHTR is a diagnosis of exclusion and other possibilities like Cancer Control 21 AHTR, microbial contamination, TRALI, medication adverse events, or an underlying infection should be considered first, because prestorage leukoreduction makes FNHTRs unlikely.48 For patients experiencing recurrent FNHTRs despite leukoreduction, washed RBCs in 2 L saline and premedication with an antipyretic may be useful.9 In addition, these patients could be given a narcotic analgesic for chills, rigor, or both. Delayed Adverse Events of Transfusions Delayed Hemolytic Transfusion Reaction Delayed hemolytic transfusion reactions (DHTRs) can be expected between 3 and 10 days following a transfusion of apparently compatible RBCs in patients with RBC antibodies with a low titer and which went undetectable during pretransfusion testing. Following the transfusion of RBCs containing the antigen the patient had been presensitized against, an anamnestic response occurs with a rapid increase in the antibody titer between 1 and 2 weeks. Because these antibodies are IgG and recognize antigens of the Kidd, Duffy, Kell, Rh, and MNS systems, extravascular hemolysis is expected.49 Patients may complain of weakness and jaundice, and the laboratory workup will show a drop in hematocrit level, circulating microspherocytes, increased levels of lactate dehydrogenase and bilirubin, and a positive result on DAT.17 Using a type and screen procedure, a new RBC alloantibody can be identified unless the antibody has bound to the transfused RBCs. In those cases, an elution is essential to determine the antibody specificity. A positive DAT result following the transfusion due to a new alloantibody but without signs of hemolysis occurs more often than a DHTR and is termed a delayed serological transfusion reaction.50 Transfusion-Associated Graft-vs-Host Disease Recipients of transfusion who are immunocompromised are at risk for developing TA-GVHD, a potentially fatal complication.51,52 The transfusion of viable T lymphocytes and the patient’s inability to mount an immune response, either due to immunosuppression or due to similarity in HLA (such as when a donor is a first-degree relative), allows the lymphocytes to survive and proliferate in the recipient. Patients with lymphoid malignancies (particularly Hodgkin lymphoma), those undergoing chemotherapy with purine analogs or fludarabine, or those with cellular immunodeficiency, as well as neonates, are at risk for developing TA-GVHD.52 Clinically, TA-GVHD is similar to GVHD post–stem cell transplantation, but it occurs earlier (≤ 2 weeks of the transfusion) and suppresses bone marrow.52-54 TA-GVHD presents as a rash with fever, diarrhea, cholestasis, nausea, vomiting, and pancytopenia. Diagnosis is usually clinical, supported by 22 Cancer Control biopsies from the skin, liver, or gastrointestinal tract, and sometimes with molecular techniques to determine genetic chimerism. The mortality rate is high because no effective treatment has been ascertained and the neutropenia caused by TA-GVHD is profound. The best strategy for health care professionals is to prevent the occurrence of TA-GVHD by irradiating the cellular blood components.55 Post-Transfusion Purpura Post-transfusion purpura is a rare immunological phenomenon characterized by sudden thrombocytopenia that takes place 2 to 14 days following a blood transfusion.56 It is caused by platelet alloantibodies (mostly anti-HPA-1a) in a patient previously sensitized from pregnancy or transfusion. Because the thrombocytopenia is typically severe (< 10 × 109/L), patients complain of petechial, purpura, or mucosal bleeds. The diagnosis of post-transfusion purpura is confirmed by the detection of platelet-specific alloantibodies in the serum.57 Most cases are self-limited and the platelet count recovers within 3 weeks. IVIG alone or in combination with corticosteroids is the mainstay of treatment.56 Patients with severe bleeding may benefit from platelet transfusions, preferably with units lacking the offending antigen. Red Blood Cell Alloimmunization The transfusion of RBCs may induce alloantibodies, potentially causing major problems in chronically transfused patients such as those with myelodysplastic syndromes.58 Chronically transfused patients who are also minorities may be at greater risk when receiving RBCs from a primarily Caucasian donor population, as is typically seen in patients with sickle cell disease. Although clinical factors that affect the rate of alloimmunization have been suggested, predicting which patients will form 1 or more alloantibodies after each RBC transfusion is not possible.59 Sanz et al60 reported that alloimmunization occurred in 15% of transfusion-dependent patients with myelodysplastic syndromes or chronic myelomonocytic leukemia and that the incidence of alloimmunization increased with the number of donor units. Platelet Alloimmunization Because platelets express HLA- and platelet-specific antigens, they may also induce alloantibodies.61 Sensitization may occur from pregnancy, transfusion, or transplantation and lead to platelet refractoriness (lack of appropriate response from transfusion). Although clinical factors, such as fever, sepsis, disseminated intravascular coagulation, splenomegaly, and active bleeding, as well as drug use, are more likely to cause decreased response from platelet transfusions than alloantibodies, the latter may be difficult January 2015, Vol. 22, No. 1 to overcome. Because ABO incompatibility may compromise post-transfusion platelet count increments, patients may benefit from a trial of ABO-compatible platelets before the initiation of HLA-matched platelet transfusions.23,61 The best strategy to prevent platelet refractoriness is to avoid alloimmunization by using exclusively leukoreduced RBCs and platelets. Alloimmunization to the D antigen (Rh) may be another concern if Rh-negative patients receive Rh-positive platelet transfusions. Rh antigens are not expressed on platelets, but they are present in the few RBCs in each unit of platelets. Although one study has concluded that the risk of developing anti-D is negligible and does not warrant the use of Rh immunoglobulin to prevent it,62 health care professionals should make a decision on a case-by-case basis when treating a patient who may become pregnant in the future. Transfusion-Related Immunomodulation Several lines of evidence, both in vitro and in vivo, have suggested that allogeneic transfusions alter the recipient’s immune system and his or her ability to respond to infections and tumor antigens.58,63 However, transfusion-related immunomodulation (TRIM) continues to be a debatable complication of transfusion.64 TRIM may be multifactorial and possibly mediated by allogeneic mononuclear cells, leukocyte-derived soluble mediators, or soluble HLA peptides, among others. A review by Refaai and Blumberg65 of TRIM summarizes the effects of transfusion in the immune system as the following: • Decreased Th1 and increased Th2 cytokine production in vitro • Reduced responses in mixed lymphocyte culture • Decreased proliferative response to mitogens or soluble antigens in vitro, thus causing impaired delayed-type hypersensitivity skin responses • Increased CD8 T cells or suppressor function in vitro • Decreased natural killer cells and activity in vitro • Decreased CD4 helper T cells • Decreased monocyte/macrophage function in vitro and in vivo • Enhanced production of anti-idiotypic antibodies suppressive of mixed lymphocyte response in vitro • Decreased cell-mediated cytotoxicity against target cells in vitro • Humoral alloimmunization to cell-associated and soluble antigens • Increased T-regulatory cells and function Iron Overload In addition to patients with hemoglobinopathies (eg, thalassemia, sickle cell disease), those with January 2015, Vol. 22, No. 1 myelodysplastic syndromes and aplastic anemia often require chronic transfusion support. Transfusion dependency in myelodysplastic syndromes has been associated with worse outcomes, including decreased rates of survival.66 Chronic transfusions cause significant iron overload because iron absorption is tightly regulated and the body has limited ability to excrete excess iron.67 Considering that 1 unit of RBCs has 200 to 250 mg of iron, most patients will develop iron overload after transfusion of 10 to 20 units. Deposition of iron in the parenchymal tissues and reticuloendothelial cells causes progressive end-organ damage such as hepatomegaly and liver dysfunction, heart failure, hypogonadism, diabetes mellitus, skin pigmentation, and arthropathy. For this reason, the debate regarding iron chelation therapy in myelodysplastic syndromes is currently ongoing despite the lack of data from randomized controlled trials.68 Conclusions Transfusion safety encompasses the continuum from donor qualification and screening to the appropriate choice of blood components and the monitoring of patients for adverse events.69 Patients with malignancy constitute a unique group, especially when diseaseor treatment-induced bone marrow failure causes severe pancytopenia and demands transfusions. Furthermore, their clinical condition may contribute to transfusion reactions while making their recognition more challenging. Although extensive and strong evidence supports a restrictive transfusion approach, the data are limited to patients without malignancies; therefore, extrapolation is not possible.70 Nonetheless, a judicious approach to transfusion, as well as the administration of single units followed by patient assessment, will help to decrease the likelihood of adverse events in patients with cancer undergoing transfusion. References 1. Huh YO, Lichtiger B. Transfusion reactions in patients with cancer. Am J Clin Path. 1987;87(2):253-257. 2. Vamvakas EC. Is white blood cell reduction equivalent to antibody screening in preventing transmission of cytomegalovirus by transfusion? A review of the literature and meta-analysis. Transf Med Rev. 2005;19(3):181-199. 3. Geiger TL, Howard SC. Acetaminophen and diphenhydramine premedication for allergic and febrile nonhemolytic transfusion reactions: good prophylaxis or bad practice? Transf Med Rev. 2007;21(1):1-12. 4. Patterson BJ, Freedman J, Blanchette V, et al. Effect of premedication guidelines and leukoreduction on the rate of febrile nonhaemolytic platelet transfusion reactions. Transf Med (Oxford, England). 2000;10(3):199-206. 5. Ezidiegwu CN, Lauenstein KJ, Rosales LG, et al. Febrile nonhemolytic transfusion reactions. Management by premedication and cost implications in adult patients. Arch Path Lab Med. 2004;128(9):991-995. 6. Kennedy LD, Case LD, Hurd DD, et al. A prospective, randomized, double-blind controlled trial of acetaminophen and diphenhydramine pretransfusion medication versus placebo for the prevention of transfusion reactions. Transfusion. 2008;48(11):2285-2291. 7. Marti-Carvajal AJ, Solá I, González LE, et al. Pharmacological interventions for the prevention of allergic and febrile non-haemolytic transfusion reactions. Cochrane Database Syst Rev. 2010(6):CD007539. 8. US Food and Drug Administration. Transfusion/donation fatalities. http:// www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/ReportaProblem/ Cancer Control 23 TransfusionDonationFatalities. Accessed November 5, 2014. 9. Tinegate H, Birchall J, Gray A, et al; BCSH Blood Transfusion Task Force. Guideline on the investigation and management of acute transfusion reactions. Br J Haematol. 2012;159(2):143-153. 10. Liu C, Zhao W, Christ GJ, et al. Nitric oxide scavenging by red cell microparticles. Free Radic Biol Med. 2013;65:1164-1173. 11. Kleinman S, Caulfield T, Chan P, et al. Toward an understanding of transfusion-related acute lung injury: statement of a consensus panel. Transfusion. 2004;44(12):1774-1789. 12. Toy P, Popovsky MA, Abraham E, et al. Transfusion-related acute lung injury: definition and review. Crit Care Med. 2005;33(4):721-726. 13. Andrzejewski C Jr, Casey MA, Popovsky MA. How we view and approach transfusion-associated circulatory overload: pathogenesis, diagnosis, management, mitigation, and prevention. Transfusion. 2013;53(12):3037-3047. 14. Hirayama F. Current understanding of allergic transfusion reactions: incidence, pathogenesis, laboratory tests, prevention and treatment. Br J Haematol. 2013;160(4):434-444. 15. Zilberstein J, McCurdy MT, Winters ME. Anaphylaxis. J Emerg Med. 2014;47(2):182-187. 16. Maskens C, Downie H, Wendt A, et al. Hospital-based transfusion error tracking from 2005 to 2010: identifying the key errors threatening patient transfusion safety. Transfusion. 2014;54(1):66-73. 17. Zantek ND, Koepsell SA, Tharp DR Jr, et al. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87(7):707-709. 18. Nakagawa M, Toy P. Acute and transient decrease in neutrophil count in transfusion-related acute lung injury: cases at one hospital. Transfusion. 2004;44(12):1689-1694. 19. Marques MB, Tuncer HH, Divers SG, et al. Acute transient leukopenia as a sign of TRALI. Am J Hematol. 2005;80(1):90-91. 20. Zhou L, Giacherio D, Cooling L, et al. Use of B-natriuretic peptide as a diagnostic marker in the differential diagnosis of transfusion-associated circulatory overload. Transfusion. 2005;45(7):1056-1063. 21. Bux J, Sachs UJ. Pulmonary transfusion reactions. Transfus Med Hemother. 2008;35(5):337-345. 22. Stowell SR, Winkler AM, Maier CL, et al. Initiation and regulation of complement during hemolytic transfusion reactions. Clin Dev Immunol. 2012;2012:307093. 23. Cid J, Harm SK, Yazer MH. Platelet transfusion - the art and science of compromise. Transfus Med Hemother. 2013;40(3):160-171. 24. US Food and Drug Administration. Guidance for industry: circular of information for the use of human blood and blood components. http://www. fda.gov/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/ guidances/blood/ucm364565.htm. Accessed November 5, 2014. 25. Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following Rh(0)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood. 2005;106(5):1532-1537. 26. Pintova S, Bhardwaj AS, Aledort LM. IVIG--a hemolytic culprit. N Engl J Med. 2012;367(10):974-976. 27. Michelis FV, Branch DR, Scovell I, et al. Acute hemolysis after intravenous immunoglobulin amid host factors of ABO-mismatched bone marrow transplantation, inflammation, and activated mononuclear phagocytes. Transfusion. 2014;54(3):681-690. 28. Bernard GR, Artigas A, Brigham KL, et al; Consensus Committee. Report of the American-European Consensus conference on acute respiratory distress syndrome: definitions, mechanisms, relevant outcomes, and clinical trial coordination. J Crit Care. 1994;9(1):72-81. 29. Popovsky MA, Moore SB. Diagnostic and pathogenetic considerations in transfusion-related acute lung injury. Transfusion. 1985;25(6):573-577. 30. Silliman CC, Bjornsen AJ, Wyman TH, et al. Plasma and lipids from stored platelets cause acute lung injury in an animal model. Transfusion. 2003;43(5):633-640. 31. West FB, Silliman CC. Transfusion-related acute lung injury: advances in understanding the role of proinflammatory mediators in its genesis. Expert Rev Hematol. 2013;6(3):265-276. 32. Cherry T, Steciuk M, Reddy VV, et al. Transfusion-related acute lung injury: past, present, and future. Am J Clin Path. 2008;129(2):287-297 33. Silliman CC, Boshkov LK, Mehdizadehkashi Z, et al. Transfusion-related acute lung injury: epidemiology and a prospective analysis of etiologic factors. Blood. 2003;101(2):454-462. 34. Finlayson J, Grey D, Kavanagh L, et al. Transfusion-related acute lung injury in a neutropenic patient. Intern Med J. 2011;41(8):638-641. 35. Eder AF, Dy BA, Perez JM, et al. The residual risk of transfusion-related acute lung injury at the American Red Cross (2008-2011): limitations of a predominantly male-donor plasma mitigation strategy. Transfusion. 2013;53(7):1442-1449. 36. Vlaar AP, Juffermans NP. Transfusion-related acute lung injury: a clinical review. Lancet. 2013;382(9896):984-994. 37. Menis M, Anderson SA, Forshee RA, et al. Transfusion-associated circulatory overload (TACO) and potential risk factors among the inpatient US elderly as recorded in Medicare administrative databases during 2011. Vox Sang. 2014;106(2):144-152. 38. Alam A, Lin Y, Lima A, et al. The prevention of transfusion-associated circulatory overload. Transfus Med. 2013;27(2):105-112. 24 Cancer Control 39. Centers for Disease Control and Prevention. National Healthcare Safety Network Biovigilance Component Hemovigilance Module Surveillance Protocol. Version 2.1.3. Atlanta, GA: National Center for Emergency and Zoonotic Infections Diseases; 2014. http://www.cdc.gov/nhsn/PDFs/hemovigModuleProtocol_current.pdf. Accessed November 5, 2014. 40. Brecher ME, Blajchman MA, Yomtovian R, et al. Addressing the risk of bacterial contamination of platelets within the United States: a history to help illuminate the future. Transfusion. 2013;53(1):221-231. 41. Young C, Chawla A, Berardi V, et al; Babesia Testing Investigational Containment Study Group. Preventing transfusion-transmitted babesiosis: preliminary experience of the first laboratory-based blood donor screening program. Transfusion. 2012;52(7):1523-1529. 42. Schwartz J, Winters JL, Padmanabhan A, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145-284. 43. Johnson ST, Van Tassell ER, Tonnetti L, et al. Babesia microti real-time polymerase chain reaction testing of Connecticut blood donors: potential implications for screening algorithms. Transfusion. 2013;53(11):2644-2649. 44. American Red Cross. Infectious disease testing for donated blood. http:// www.redcrossblood.org/learn-about-blood/blood-testing. Accessed November 5, 2014. 45. Yu MY, Alter HJ, Virata-Theimer ML, et al. Parvovirus B19 infection transmitted by transfusion of red blood cells confirmed by molecular analysis of linked donor and recipient samples. Transfusion. 2010;50(8):1712-1721. 46. Ricci KS, Martinez F, Lichtiger B, et al. Septic transfusion reactions during blood transfusion via indwelling central venous catheters. Transfusion. 2014;54(10):2412-2418. 47. Cohn CS, Stubbs J, Schwartz J, et al. A comparison of adverse reaction rates for PAS C versus plasma platelet units. Transfusion. 2014;54(8):19271934. 48. Wang RR, Triulzi DJ, Qu L. Effects of prestorage vs poststorage leukoreduction on the rate of febrile nonhemolytic transfusion reactions to platelets. Am J Clin Path. 2012;138(2):255-259. 49. Tormey CA, Stack G. Limiting the extent of a delayed hemolytic transfusion reaction with automated red blood cell exchange. Arch Pathol Lab Med. 2013;137(6):861-864. 50. Ness PM, Shirey RS, Thoman SK, et al. The differentiation of delayed serologic and delayed hemolytic transfusion reactions: incidence, long-term serologic findings, and clinical significance. Transfusion. 1990;30(8):688-693. 51. Alter HJ, Klein HG. The hazards of blood transfusion in historical perspective. Blood. 2008;112(7):2617-2626. 52. Higgins MJ, Blackall DP. Transfusion-associated graft-versus-host disease: a serious residual risk of blood transfusion. Curr Hematol Rep. 2005;4(6):470-476. 53. Agbaht K, Altintas ND, Topeli A, et al. Transfusion-associated graftversus-host disease in immunocompetent patients: case series and review of the literature. Transfusion. 2007;47(8):1405-1411. 54. Sunul H, Erguven N. Transfusion-associated graft-versus-host disease. Transfus Apher Sci. 2013;49(2):331-333. 55. Uchida S, Tadokoro K, Takahashi M, et al. Analysis of 66 patients with definitive transfusion-associated graft-versus-host disease and the effect of universal irradiation of blood. Transfus Med. 2013;23(6):416-422. 56. Shtalrid M, Shvidel L, Vorst E, et al. Post-transfusion purpura: a challenging diagnosis. Isr Med Assoc J. 2006;8(10):672-674. 57. Heikal NM, Smock KJ. Laboratory testing for platelet antibodies. Am J Hematol. 2013;88(9):818-821. 58. Heal JM, Phipps RP, Blumberg N. One big unhappy family: transfusion alloimmunization, thrombosis, and immune modulation/inflammation. Transfusion. 2009;49(6):1032-1036. 59. Bauer MP, Wiersum-Osselton J, Schipperus M, et al. Clinical predictors of alloimmunization after red blood cell transfusion. Transfusion. 2007;47(11):2066-2071. 60. Sanz C, Nomdedeu M, Belkaid M, et al. Red blood cell alloimmunization in transfused patients with myelodysplastic syndrome or chronic myelomonocytic leukemia. Transfusion. 2013;53(4):710-715. 61. Pavenski K, Freedman J, Semple JW. HLA alloimmunization against platelet transfusions: pathophysiology, significance, prevention and management. Tissue Antigens. 2012;79(4):237-245. 62. O’Brien KL, Haspel RL, Uhl L. Anti-D alloimmunization after D-incompatible platelet transfusions: a 14-year single-institution retrospective review. Transfusion. 2014;54(3):650-654. 63. Blumberg N, Heal JM. Effects of transfusion on immune function. Cancer recurrence and infection. Arch Pathol Lab Med. 1994;118(4):371-379. 64. Vamvakas EC, Blajchman MA. Transfusion-related immunomodulation (TRIM): an update. Blood Rev. 2007;21(6):327-348. 65. Refaai MA, Blumberg N. Transfusion immunomodulation from a clinical perspective: an update. Expert Rev Hematol. 2013;6(6):653-663. 66. Goldberg SL, Chen E, Corral M, et al. Incidence and clinical complications of myelodysplastic syndromes among United States Medicare beneficiaries. J Clin Oncol. 2010;28(17):2847-2852. 67. Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol. 2007;69:69-85. 68. Mitchell M, Gore SD, Zeidan AM. Iron chelation therapy in myelodysplastic January 2015, Vol. 22, No. 1 syndromes: where do we stand? Expert Rev Hematol. 2013;6(4):397-410. 69. Dzik WH. Emily Cooley lecture 2002: transfusion safety in the hospital. Transfusion. 2003;43(9):1190-1198. 70. Carson JL, Hebert PC. Should we universally adopt a restrictive approach to blood transfusion? It’s all about the number. Am J Med. 2014;127(2):103-104. January 2015, Vol. 22, No. 1 Cancer Control 25 The clinical significance of storing RBCs is controversial, although new data suggest the RBC storage lesion does not affect clinical outcomes in select patients receiving transfusions. Ray Paul. Sweet Jane, 2007. Acrylic, latex, enamel on canvas, 37" × 37". Private collection of the Mathematical Oncology Department at Moffitt Cancer Center, Tampa, FL. Clinical Effects of Red Blood Cell Storage Lirong Qu, MD, PhD, and Darrell J. Triulzi, MD Background: Well-characterized biochemical, structural, and physiological changes occur when red blood cells (RBCs) are stored for a period of time and are collectively called the storage lesion. Methods: Key study results are summarized and contrasted and new data from recently completed randomized controlled trials will be discussed. Results: It is unclear whether in vitro changes to RBCs that occur during storage are clinically relevant. The clinical effects of RBC storage have been the focus of observational studies in recent years. However, these studies lack any consensus, possibly because of methodological limitations. Conclusions: The clinical significance of storing RBCs is controversial, although new data from randomized controlled trials of neonates and patients undergoing cardiac surgery suggest that the duration of RBC storage is not associated with adverse clinical outcomes in these patient populations. Introduction According to a survey conducted in part by the US Department of Health and Human Services, more than 13 million units of red blood cells (RBCs) were transfused in the United States in 2011.1 The mean time of storage duration for a unit of RBCs at transfusion was 17.9 days.1 The maximum allowable storage duration for RBC is defined by the US Food and Drug Administration (FDA) and depends on the storage media. In the United States, AS-1, AS-3, and AS-5 are frequently used as additive solutions and can be stored for up to 42 days at a temperature of 33.8 to 42.8°F (1–6°C). During the storage of RBCs, From the Department of Pathology, University of Pittsburgh, and the Institute for Transfusion Medicine, Pittsburgh, Pennsylvania. Submitted July 17, 2014; accepted November 12, 2014. Address correspondence to Darrell J. Triulzi, MD, Department of Pathology, Institute for Transfusion Medicine, 3636 Boulevard of Allies, Pittsburgh, PA 15213. E-mail: [email protected] No significant relationships exist between the authors and the companies/organizations whose products or services may be referenced in this article. 26 Cancer Control well-characterized biochemical, metabolic, structural, inflammatory, and physiological changes occur that are collectively known as the “storage lesion.” Although the storage lesion has been well documented and demonstrated in vitro, the clinical relevance of these changes on patient outcomes remains unclear. Ongoing interest exists in the relationship between the duration of RBC storage and clinical outcomes among recipients of transfusions, beginning with the publication of a small, randomized, single-center trial in 1989 that compared the effects of fresh whole blood (< 12 hours old) with stored blood (2–5 days old) in patients who underwent cardiac surgery.2 Interest in the subject was further galvanized by the publication of a retrospective study by Koch et al3 who reported that patients who underwent cardiac surgery and received “older” blood (> 14 days old) had worse outcomes than those who received fresher blood (≤ 14 days old). This heightened awareness of the controversy resulted in an increase of observational studies, small studies, randomized controlled trials (RCTs), and phase 3 RCTs to address this issue. January 2015, Vol. 22, No. 1 One systematic review provided a detailed summary of relevant publications in adult patients during the last 3 decades (1983–2012).4 The authors of the review identified 55 studies for detailed qualitative synthesis, most of which were retrospectively performed at a single institution; 8 (14.5%) were small randomized studies, 3 of which were conducted using healthy volunteers. Twenty-six of the studies (47%) suggested that stored RBCs were adversely affected in at least 1 clinical end point, whereas the remaining 29 studies (53%) revealed no difference in effect. The authors concluded that the evidence did not definitively indicate that fresher RBCs were clinically superior to older RBCs.4 It is worth noting that they did not perform a quantitative meta-analysis due to the considerable heterogeneity among studies and a concern of numerous biased studies they identified in their systematic review.4 Two other meta-analyses of studies from mostly observational data have resulted in conflicting results.5,6 Wang et al5 performed a meta-analysis on 21 studies published between 2001 and 2011, including 6 studies of cardiac surgery and 6 studies reviewing trauma, that totaled 409,966 patients. They showed that RBC storage was associated with an increased risk of mortality (pooled odds ratio [OR], 1.16; 95% confidence interval [CI]: 1.07–1.24; P = .0001), pneumonia (pooled OR, 1.17; 95% CI: 1.08–1.27; P = .0001), and multiple organ dysfunction syndrome (pooled OR, 2.26; 95% CI: 1.56–3.25; P < .0001).5 A meta-analysis by Vamvakas et al6 of studies that included adjusted results for mortality found that the storage duration was not associated with an increased risk of mortality. There are several possible explanations for the conflicting conclusions from these mostly observational studies. A retrospective study design does not control for known or unrecognized factors that may be clinically important, including baseline patient characteristics, underlying disease, volume transfused, transfusion of other blood components, and follow-up period. Sicker patients receive more blood transfusions than their counterparts, and, thus, have a greater likelihood of receiving at least 1 older unit. An observational study cannot determine whether worse outcomes are due to the need for transfusion or the transfusion itself (confounded by indication). Varied rates of mortality (eg, 7 vs 28 days) and morbidity end points (outcomes) were reported among studies, making comparisons difficult. In addition, various definitions of length of RBC storage were used to define “fresher” versus “older.” This issue is particularly problematic when multiple units of various durations of storage are transfused. For example, some used less than 7 days, less than 10 days, or less than 14 days as “fresher,” and more than 14 days, more than 21 days, or longer as “older” RBCs. Other January 2015, Vol. 22, No. 1 studies used the mean age, oldest unit, or oldest of multiple units. It is worth emphasizing that no clinical evidence supports these “fresh” and “old” definitions. Investigators have inferred that the kinetics of in vitro changes during storage correlate with the in vivo effectiveness of RBCs when defining the age of RBC storage. This approach does not account for the fact that the kinetics of the in vitro changes are variable depending on the parameter, and none has been shown to be clinically relevant. Variable preparations of RBCs, including differing storage media or modifications (eg, leukoreduction), have been used in studies over the years. Leukocytes in the blood products have been reported to affect clinical outcomes via immunomodulation.7 Rather than replicate a recent systematic review on the subject,4 in this article we will summarize and contrast key results from previous studies, describe the results of recent publications, and discuss the available data from recently completed RCTs. In Vitro Changes In the United States, the FDA has indicated that the maximum “shelf-life” for a unit of RBCs is 42 days when stored in additive solution (eg, AS-1, AS-3, AS-5). The limit on RBC storage duration is primarily based on degree of hemolysis (< 1%) at the end of storage and the percent (minimum, 75%) of the RBCs remaining in the circulation 24 hours following transfusion.8,9 The many changes encompassed by the storage lesion occur in a time-dependent manner with kinetics that vary depending on the parameter.8,9 There is a progressive decrease in intracellular 2,3-diphosphoglycerate (DPG) and adenosine triphosphate with a concomitant accumulation of extracellular free hemoglobin and free iron. A decrease in 2,3-DPG reduces oxygen delivery to tissue, although this change is reversible after transfusion. Irreversible changes to the RBC membrane, including the release of microvesicles, reduce deformability and may increase the likelihood of occluding microvasculature. Extracellular hemoglobin that is free or contained in microvesicles may scavenge nitric oxide, and iron may increase circulating, nontransferrin-bound iron and, thus, could promote inflammation.8-10 There is a progressive accumulation of lactic acid and potassium and a steady decrease in pH during storage. In addition, the accumulation of other biological by-products, including cytokines, lipids, histamines, and enzymes, may induce febrile transfusion reactions, increase oxidative membrane damage, and activate or suppress the immune system.10 Although the in vitro changes are clear and demonstrable (eg, loss of 2,3-DPG by day 14 of storage), no data exist on the in vivo clinical significance of these changes nor a cutoff point of storage duration Cancer Control 27 to define “older” RBCs. Therefore, defining “older” RBCs (or the age of multiple transfused units) for clinical study is arbitrary, and a fact that, in part, may explain the varied storage duration cutoffs used in many published studies. Cardiac Surgery Clinical studies of the duration of RBC storage in patients who underwent cardiac surgery are shown in Table 1.2,3,11-22 The most well-known of these studies is that of Koch et al,3 which is a single center, retrospective study of 6,002 patients who underwent cardiac surgery and received transfusions with RBCs between 1998 and 2006. A total of 2,872 patients received 8,802 units of blood stored for 14 days or less (“fresher”), and 3,130 patients received 10,782 units of blood stored for more than 14 days (“older”). Re- cipients of older RBCs (median, 20 days) had higher rates of hospital mortality compared with those who received fresher RBCs (median, 11 days; 2.8% vs 1.7%; P = .004). Compared with those who received fresher RBCs, recipients of older RBCs also had a higher rate of 1-year mortality (7.4% vs 11.0%; P < .001), were more likely to require prolonged support on mechanical ventilation (MV) beyond 72 hours (9.7% vs 5.6%; P < .001), and more likely to have renal failure (2.7% vs 1.6%; P = .003), sepsis or septicemia (4.0% vs 2.8%; P = .01), or multisystem organ failure (0.7% vs 0.2%; P = .007).3 Of note, the conclusions of this study have been debated and challenged primarily because of the observational nature of the study and the presentation of unadjusted analyses.23 Several studies of patients who underwent cardiac surgery were unable to find similar associations. Table 1. — Effects of the Duration of RBC Storage Among Patients Undergoing Cardiac Surgery Study Design Definition of Storage Duration Clinical Setting No. of Patients Increased Risk for Adverse Events With Longer Storage? Andreasen19 Retrospective multicenter < 14 vs ≥ 14 days Cardiac surgery 1,748 Yes for postoperative wound infection and septicemia Baltsvias22 Retrospective single center ≤ 7 vs > 14 days Cardiac surgery (pediatric) 570 No for mortality No for other end points Koch3 Retrospective single center ≤ 14 vs > 14 days Cardiac surgery 6,002 Yes for mortality and composite of 17 clinical outcomes Leal-Noval17 Retrospective single center Mean age Cardiac surgery 585 Yes for pneumonia; No for ICU LOS and other end points McKenny14 Retrospective single center ≤ 14 vs > 14 days Cardiac surgery 1,153 No for mortality No for ICU LOS Redlin21 Retrospective single center ≤ 3 vs 4–14 days Cardiac surgery (pediatric) 139 Yes for transfusion requirements Sanders20 Retrospective single center < 14 vs > 14 days Cardiac surgery 444 Yes for LOS and renal failure Vamvakas16 Retrospective single center Mean age Cardiac surgery 256 Yes for postoperative infections Vamvakas18 Retrospective single center Mean age, oldest, mean of 2 oldest units Cardiac surgery 268 No for postoperative ICU and hospital LOS van de Watering11 Retrospective single center Mean age, youngest, oldest, < 18 vs > 18 days CABG 2,732 No for mortality and ICU LOS in multivariate analysis van Straten13 Retrospective single center < 14 vs > 14 days CABG 3,597 No for early or late postoperative mortality Voorhuis15 Retrospective single center ≤ 14 vs > 14 days Cardiac surgery 821 No for mortality No for other end points Wasser2 Randomized single center < 12 hours vs 2–5 days Cardiac surgery 237 No for coagulation rests, postoperative bleeding, or transfusion requirements Yap12 Retrospective single center Median age, oldest, < 30 vs ≥ 30 days Cardiac surgery 670 No for postoperative mortality, renal failure, pneumonia, duration of MV, or ICU LOS CABG = coronary artery bypass graft, ICU = intensive care unit, LOS = length of stay, MV = mechanical ventilation, RBC = red blood cell. 28 Cancer Control January 2015, Vol. 22, No. 1 In a multivariate analysis of 2,732 patients who received coronary artery bypass graft (CABG), van de Watering et al11 showed that patients who exclusively received older RBCs (mean storage, 24.3 ± 3.5 days) had similar rates of 30-day survival or length of stay (LOS) in the intensive care unit (ICU) as patients who received fresher RBCs (mean storage, 12.7 ± 2.8 days). Yap et al12 found no association between the duration of storage of RBCs and any of the study end points, such as early postoperative mortality, renal failure, pneumonia, LOS in the ICU, and hours of MV support, in 670 consecutive patients who had nonemergency CABG, aortic valve replacement, or both. In a retrospective analysis of 3,597 patients who underwent CABG, van Straten et al13 did not find length of RBC storage (cutoff, 14 days) to be a risk factor for early or late mortality. McKenny et al14 analyzed 1,153 patients who underwent cardiac surgery and found no association between duration of storage (cutoff, 14 days) and postoperative mortality rates in multivariate analyses. Voorhuis et al15 analyzed the data of 821 patients who underwent cardiac surgery and found that the transfusion of RBCs stored for more than 14 days was not associated with adverse outcomes. The incidences of the primary outcome (composite end point of death, myocardial infarction, and stroke) were 8.6% and 4.5% in the “any older” group (RBC age, 21 ± 5 days) and the “fresher” group (RBC age, 13 ± 2 days), respectively (adjusted OR, 1.68; 95% CI: 0.65–4.34).15 Rates of prolonged ICU stays were 12.3% and 6.3% in the “any older” and “fresher” groups, respectively (adjusted OR, 1.58; 95% CI: 0.69–3.66).15 In addition to the reports on mortality outcomes, several investigators assessed the possible association between length of RBC storage and other clinical parameters, such as occurrence of infections, ICU and hospital LOS, as well as organ failure in transfused patients. Vamvakas et al16 reported on an independent relationship between the storage duration of RBCs and postoperative pneumonia or wound infections among 256 patients who received transfusions and underwent CABG surgery, discovering that the risk of pneumonia increases by 1% for every additional day of mean storage length of RBCs (P < .005). Leal-Noval et al17 found that the oldest unit transfused (> 28 days), but not the mean age of all transfused units, correlated with the development of postoperative pneumonia (OR, 1.06; 95% CI: 1.01–1.11). However, they did not find a correlation between the mean duration of storage with prolonged ICU LOS (> 4 days), prolonged length of MV support (> 24 hours), or perioperative infarction, mediastinitis, or sepsis.17 A subsequent study by Vamvakas et al18 reported no association between duration of RBC storage and postoperative ICU and hospital LOS. Andreasen et al19 investigated 1,748 patients receiving CABG and found that, compared with patients not receiving transfusions, the January 2015, Vol. 22, No. 1 adjusted ORs for severe infection among all transfusion recipients and recipients of fresher (< 14 days; n = 953) or older (≥ 14 days; n = 548) RBCs were 1.6 (95% CI: 0.9–2.8), 1.1 (95% CI: 0.6–2.1), and 2.3 (95% CI: 1.2–4.2), respectively. In a retrospective study of 444 patients undergoing cardiac surgery, Sanders et al20 found that patients who exclusively received older RBCs (> 14 days) or any older blood (older blood, fresher blood, or a mixture of both) had a longer postoperative LOS and a higher incidence of new renal complications than patients who received transfusions with fresher RBCs. Two recent retrospective studies reviewed data from pediatric patients who underwent cardiac surgery.21,22 Redlin et al21 evaluated 139 pediatric patients undergoing cardiac surgery, and, of those patients, 26 received RBC units stored for no more than 3 days, and 113 received RBCs stored for between 4 and 14 days. The latter group required additional transfusions of RBCs and fresh frozen plasma than the former group (19 vs 25 mL/kg [P = .003] and 73% vs 35% [P = .0006], respectively). In another retrospective review of 570 patients receiving transfusion with 1 or 2 units of blood, Baltsavias et al22 found no difference in mortality, length of ICU stay, MV duration, postoperative infection, and major organ dysfunction between the fresher group (median [interquartile range] storage duration, 6 days [range, 5–7 days]) and the older group (storage duration, 14 days [range, 11–19 days]). Trauma Studies on the effects of RBC storage among patients with trauma have generated conflicting results (Table 2).24-37 Zallen et al24 performed a retrospective review of a prospectively collected database and found that the mean age of blood or number of units for more than 14 or 21 days was an independent risk for multiorgan failure among the 63 patients studied. Keller et al25 reported that the number of units older than 14 days was associated with an increased hospital LOS, but not the duration of MV support in 86 patients with trauma. Offner et al26 found the number of transfused units that had been stored for 14 or 21 days was associated with an increased risk for infection. Weinberg et al27 reported that transfusion of large (but not small) volumes of older blood was associated with increased rates of mortality among 1,813 patients with severe trauma (mean injury severity score [ISS], 26]. In a report of patients with less severe traumatic injury (mean ISS, 14.4), Weinberg et al28 found that the transfusion of older blood (> 14 days old) was associated with a slightly increased rate of mortality (OR, 1.12; 95% CI: 1.02–1.23). The same authors later reported that a higher mortality rate occurred in patients receiving transfusions with at least 3 units of older blood (≥ 14 days) compared Cancer Control 29 with those who received at least 3 units of fresher RBCs (adjusted RR, 1.57; 95% CI: 1.14–2.15).29 In a retrospective analysis of 202 patients with trauma who received at least 5 units of RBCs, Spinella et al30 found higher rates of mortality among patients transfused with older RBCs (maximum storage age, ≥ 28 days) than with fresher RBCs (maximum storage age, ≤ 27 days; 26.7% vs 13.9%; P = .02). They also found an association between the maximum age of transfused RBCs (> 21 or 28 days) and deep venous thrombosis.30 Vandromme et al31 retrospectively analyzed the data of 1,183 patients with trauma who received transfusions and found that transfusions of exclusively older RBCs (≥ 14 days) significantly increased the risk of pneumonia (adjusted RR, 1.42; CI: 1.01–2.02), whereas transfusions of exclusively fresher RBCs (adjusted RR, 1.02; CI: 0.62–1.67) or mixed RBCs (adjusted RR, 1.35; 95% CI: 0.98–1.87) did not. In a prospective, observational study, Leal-Noval et al32 assessed the effects of RBC transfusion on cerebral tissue oxygenation (PtiO2) in 66 patients with traumatic brain injury. The duration of RBC storage was divided into 4 groups: less than 10 days (n = 18), 10 to 14 days (n = 15), 15 to 19 days (n = 17), and more than 19 days (n = 16). They observed a significant increase in PtiO2 after the transfusion of RBCs stored for less than 19 days, but they saw no significant changes in PtiO2 after the transfusion of RBCs stored for more than 19 days. Kiraly et al33 performed a prospective, nonrandomized study in 32 trauma patients in the ICU who were hemodynamically stable and nonseptic. Seventeen of those patients transfused with older blood (≥ 21 days) demonstrated a significant decline in the area under the curve for tissue oxygen saturation (StO2) as measured by near-infrared spectroscopy.33 Patients transfused with fresher blood (< 21 days old) and a control group not receiving transfusions had no similar rate of decline in the area Table 2. — Effects of the Duration of RBC Storage Among Patients Undergoing Trauma Study Design Definition of Storage Duration No. of Patients Hassan36 Retrospective single center < 14 vs ≥ 14 days Juffermans37 Retrospective single center > 14 days 196 Yes for occurrence of new infection Keller25 Retrospective single center Mean age, oldest unit, 2 oldest units > 7, 14, 21, 28 days 86 Yes for hospital LOS for RBCs > 14 days No for MV Prospective single center < 21 vs ≥ 21 days 32 Yes for decrease in tissue oxygenation Leal-Noval Prospective, observational single center < 10 days 10–14 days 15–19 days > 19 days 66 Yes for failure to increase cerebral tissue oxygenation (>19 days in the RBC group) Murrell35 Retrospective single center Weighted mean age 275 Yes for ICU LOS No for hospital mortality Offner26 Retrospective single center > 14 days > 21 days 61 Yes for infection Spinella30 Retrospective single center Max age ≥ 14, 21, 28 days 202 Yes for hospital mortality and deep venous thrombosis Vandromme31 Retrospective single center < 14 vs ≥ 14 days 1,183 Yes for pneumonia 27 Weinberg Retrospective single center < 14 vs ≥ 14 days 1,813 Yes for mortality rate (for a large volume of older RBCs) Weinberg28 Retrospective single center < 14 vs ≥ 14 days 430 Yes for mortality Weinberg Retrospective single center < 14 vs ≥ 14 days 1,647 Yes for mortality (> 3 units transfused) Weinberg34 Prospective single center Range: 7–42 days 93 Yes for decline in tissue oxygenation with RBC age Zallen24 Retrospective single center Mean age 63 Yes for multiorgan failure Kiraly33 32 29 820 Increased Risk for Adverse Events With Longer Storage? Yes for complicated sepsis No for mortality ICU, intensive care unit, LOS = length of stay, MV = mechanical ventilation, RBC = red blood cell. 30 Cancer Control January 2015, Vol. 22, No. 1 under the curve for StO2. They found a moderate correlation between increasing the duration of RBC storage and decreasing the oxygenation (r = 0.5).33 Weinberg et al34 evaluated microvascular perfusion in 93 stable patients with trauma (mean ISS, 26.4) during RBC transfusion and found that the transfusion of relatively older RBCs was associated with a decline in both StO2 and perfused capillary density. Other studies have produced mixed results (see Table 2). Murrell et al35 studied 275 patients and found that older blood was associated with longer stays in the ICU (RR, 1.15; 95% CI: 1.11–1.20) but not higher rates of hospital mortality (OR, 1.21; 95% CI: 0.87–1.69). In a cohort of 820 patients with trauma, Hassan et al36 found that the total number of RBC units but not the number of older (> 14 days old) RBC units transfused was associated with an increased rate of mortality. However, the number of older units was a significant risk factor for severe sepsis or septic shock, particularly when more than 7 units were transfused (OR, 1.9; 95% CI: 1.1–3.4; P = .03).36 In a retrospective study of 196 patients with trauma who received transfusions, Juffermans et al37 found a modest correlation between transfused RBCs stored for longer than 14 days and the occurrence of new infections (OR, 1.04; 95% CI: 1.01–1.07). Critical Care Regarding the adverse effects of prolonged RBC storage on clinical outcomes, conflicting analyses have also been reported among patients who are critically ill (Table 3).38-53 Purdy et al38 reported that, of the 31 patients they studied who were admitted to the ICU with severe sepsis, the median age of RBC units Table 3. — Effects of the Duration of RBC Storage Among Patients in Critical Care Study Design Aubron43 Definition of Storage Duration Clinical Setting No. of Patients Increased Risk for Adverse Events With Longer Storage? Retrospective 2 centers Mean, maximum, minimum age ICU 8,416 Prospective single center Median ICU 44 No for tissue oxygen saturation Dessertaine Retrospective single center Median of max age ICU 534 No for mortality No for infections Fernandes49 Prospectively randomized, single center Continuous variable ICU (sepsis) 15 No for gastric mucosal pH Gajic44 Retrospective single center < 15 days 15–20 days > 20 days ICU 181 No for acute lung injury Janz46 Retrospective single center Median ICU (sepsis) 97 Yes for acute lung injury/ARDS Juffermans41 Retrospective single center ≤ 14 vs > 14 days ICU (sepsis) 67 Storage time as a confounder Katsios Prospective single center > 7, > 14, and > 21 days as cutoff ICU 261 No for occurrence of deep venous thrombosis Kopterides53 Prospective single center < 14 vs > 14 days ICU (sepsis) 37 No for change in lactate:pyruvate ratio Kor45 Randomized single center ≤ 5 days vs standard issue RBCs (median, 21 days) ICU 100 No difference in pulmonary function Marik48 Prospective single center < 15 vs ≥ 15 days ICU (sepsis) 23 Yes for gastric mucosal pH Pettila Prospective multicenter Quartile of max age ICU 757 Higher hospital mortality with older RBCs Purdy38 Retrospective single center Median age ICU (sepsis) 31 Yes for mortality Sakr51 Prospective single center Continuous variable ICU (sepsis) 35 No for microvascular flow Taylor Prospective single center Maximum age ICU 449 No for nosocomial infections Walsh50 Prospectively randomized, single center ≤ 5 vs ≥ 20 days ICU 22 No for gastric mucosal pH Creteur52 39 47 42 40 No for mortality No for ICU or hospital LOS ARDS = acute respiratory distress syndrome, ICU = intensive care unit, LOS = length of state, RBC = red blood cell. January 2015, Vol. 22, No. 1 Cancer Control 31 transfused to survivors was 17 days (range, 5–35 days) compared with 25 days (range, 9–36 days) for nonsurvivors (P < .0001). In another study of 534 patients in the ICU, Dessertaine et al39 found no association between the storage duration of transfused RBCs (defined as the maximum age of 23 days [median]) and rate of mortality or nosocomial infection. Furthermore, in a prospective study of 449 patients in the ICU, Taylor et al40 found that the maximum age of transfused RBCs was not associated with an increased risk for nosocomial infection. Juffermans et al41 retrospectively reviewed 67 patients in the ICU with sepsis and found that the total amount of transfused RBCs was associated with secondary infection (OR, 1.18; 95% CI: 1.01–1.37), and the duration of RBC storage was identified as a confounder of the effects of the amount of RBCs on infection. Pettila et al42 performed a prospective, multicenter, observational study of 757 patients who were critically ill (mixture of medical/surgical patients) in 47 ICUs in Australia and New Zealand and found that patients who received older blood (average age, 17.6 days) had higher rates of hospital mortality compared with those who received fresher blood (average age, 7.5 days; OR, 2.01; 95% CI: 1.07–3.77) after adjusting for disease severity and number of RBCs transfused. Aubron et al43 evaluated 8,416 patients who received a median of 4 RBC units (interquartile range, 2–7) in the ICU at 2 hospitals. After a multivariate analysis, the duration of RBC storage was not independently associated with rate of mortality. Furthermore, no clinically relevant relationship was seen between the mean age of transfused RBCs and length of ICU stay.43 Several investigators have assessed the relationship between duration of RBC storage and pulmonary function or the risk of acute lung injury (ALI) in patients in the ICU. In a retrospective analysis of 181 patients in the ICU, Gajic et al44 found no association between mean age or age of the oldest unit transfused and occurrence of ALI. Kor et al45 performed a small, double-blind, randomized, single-center trial of 100 patients in the ICU on MV support to compare the effects of fresher RBCs (median age, 4 days) with standard RBCs (median age, 26.5 days). They found no significant difference in the primary outcome of pulmonary function assessed by the partial pressure of arterial oxygen to the fraction of inspired oxygen concentration ratio as well as the immunological and coagulation status between the 2 groups. A similar rate of mortality was seen among the fresher and standard-issue RBC groups, but the study was not powered for this outcome.45 Janz et al46 assessed whether the duration of RBC storage was associated with the risk of developing ALI in a cohort of 96 patients in the ICU who were septic and had 32 Cancer Control received transfusions. They found that the median storage duration of transfused RBCs in patients with ALI/acute respiratory distress syndrome (ARDS) was longer (24.5 days; interquartile range, 20–31 days) compared with patients without ALI/ARDS (21 days; interquartile range, 15–27 days; P = .018). The same association was not seen in the 176 trauma patients in the ICU who received transfusions or in 125 patients in the ICU who were not septic, had no trauma, and had received transfusions.46 In a prospective study of 261 patients in the ICU, Katsios et al47 reported no association between the storage duration of transfused RBCs and the development of deep venous thrombosis. A previous study of patients with trauma found an association between the maximum age of transfused RBCs (> 21 or 28 days) and the occurrence of deep venous thrombisis, but no multivariate analysis was performed.30 Several studies have addressed whether older RBCs adversely affect microcirculation or tissue oxygenation (see Table 3).38-53 Marik et al48 found an inverse relationship between the duration of RBC storage and the maximal changes in gastric mucosal pH (pHi) in 23 patients in the ICU who were septic (r = –0.71; P < .001). However, later studies did not confirm this finding. In a small, randomized study of 15 septic patients, Fernandes et al49 compared the transfusion of either 1 RBC unit (mean age, 12.8 ± 8.1 days) or 500 mL of albumin solution and found no correlation between the storage duration of transfused RBCs and pHi. In another double-blind, randomized study of 22 patients in the ICU, Walsh et al50 compared the effects of transfusing 2 units of leukoreduced RBCs, which were stored for either no more than 5 days (median, 2 days) or for at least 20 days (median, 28 days) and found no difference in pHi or gastric-to-arterial PCO2 gap. Sakr et al51 assessed transfusion-induced changes in the sublingual microcirculation of 35 patients in the ICU with sepsis following the administration of either 1 or 2 units of leukoreduced RBCs (mean storage, 24 days; interquartile range, 12–28 days) and found no effect of the duration of RBC storage on the changes of microvascular flow. Creteur et al52 evaluated 44 patients in the ICU for oxygenation and microvascular reactivity using near-infrared spectroscopy and found no association between the duration of RBC storage and oxygen variables. Kopterides et al53 reviewed the data from 37 patients in the ICU with sepsis and found no relationship between the duration of RBC storage and change in the lactate:pyruvate ratio (microdialysis). Cancer and Other Patient Populations Several studies have addressed the effects of RBC storage among patients with cancer, those undergoing liver transplantation, those receiving transfusions, and January 2015, Vol. 22, No. 1 mixed patient populations, among others (Table 4).54-69 Several earlier studies reported on the effects of RBC storage duration on postoperative infections in patients undergoing surgery for colorectal cancer.54-56 In a retrospective study of 466 consecutive patients who underwent resection for colorectal cancer be- tween the years 1980 and 1992 in Norway, Edna et al54 analyzed 290 patients who received a transfusion of nonfiltered blood and found that infections were more likely to occur in the patients who received transfusions (31%) than in patients who did not receive transfusions (13%; P < 0.001). However, Table 4. — Effects of the Duration of RBC Storage Among Oncology and Other Patient Populations Study Design Definition of Storage Duration Basora67 Retrospective single center Median, oldest unit Cata57 Retrospective single center < 13 days 13–18 days > 18 days Cywinski62 Retrospective single center Dunn61 Retrospective single center Edgren63 Retrospective multicenter Edna54 Clinical Setting No. of Patients Increased Risk for Adverse Events With Longer Storage? Knee arthroplasty 335 No for postoperative wound infection Prostate cancer 316 No for 5-year cancer recurrence ≤ 15 vs > 15 days Liver transplantation 637 Yes for mortality and graft failure Median Liver transplantation 509 No for 2-year mortality No for postoperative infection or organ rejection 0–9 days 10–19 days 20–29 days 30–42 days Mix 364,037 Yes for 5% increased mortality for RBCs stored > 30 days (2-year follow-up) Retrospective single center Median age Colorectal cancer 240 No for postoperative infections Eikelboom64 Retrospective single center Continuous variable; quartile; 10-day interval Cardiovascular disease 4,933 Yes for hospital mortality for highest age quartile of RBCs Kekre58 Retrospective single center < 14 vs ≥ 15 days Cancer 1,929 No for overall survival Kekre59 Retrospective single center Mean age, ≥ 15 days HSCT 555 No for nonrelapse mortality No for LOS or ICU admission Lee68 Retrospective single center ≤ 14 vs > 14 days Breast reconstruction 74 Yes for postoperative complications Middelberg69 Retrospective single center ≤ 17 vs > 17 days Mix (inpatients and outpatients) 8,971 No for increased mortality with fresh (compared with old) RBCs Mynster55 Retrospective multicenter Max age Colorectal cancer 225 Yes for infection (for RBCs > 20 days) Mynster56 Retrospective multicenter < 21 vs ≥ 21 days Colorectal cancer 740 No for mortality No for cancer recurrence (recipients of ≥ 21 days had less recurrence) Robinson65 Retrospective single center Mean age Percutaneous coronary intervention 909 Yes for 30-day morality Saager66 Retrospective single center ≤ 14 days 14–28 days > 28 days Noncardiac surgery 6,994 No for mortality Yuruk60 Prospectively randomized, single center < 1 vs 3–4 weeks Hematology patients 20 Same increase in perfused vessel density in both groups HSCT = hematopoietic stem cell transplantation, ICU = intensive care unit, LOS = length of stay, RBC = red blood cell. January 2015, Vol. 22, No. 1 Cancer Control 33 the median storage time of blood transfused to patients with infectious complications (18 days; range, 2–35 days) was no different from those without infections (20 days; range, 1–37 days; P = .10).54 Mynster et al55 retrospectively analyzed transfusion data from a prospectively acquired database of 303 patients who underwent resection of colorectal carcinoma in Denmark; 225 of these patients received transfusions. The overall infection rate was higher in patients who received a transfusion (40%) compared with those who had not received a transfusion (24%; P = .011). A multivariate analysis showed that the transfusion of blood stored for at least 21 days correlated with the rate of postoperative infection (intra-abdominal abscess, anastomotic leakage, wound infection, pneumonia, and septicemia). The OR was 2.35 (95% CI: 1.27–4.37; P = .007).55 The same investigators published another analysis of 740 patients from the same prospectively acquired database and discovered a higher rate of cancer recurrence in patients who received blood exclusively stored for less than 21 days (hazard ratio [HR]: 1.5; 95% CI: 1.1–2.2) than patients who received blood stored for at least 21 days.56 One decade later, Cata et al57 studied 316 patients who underwent surgery for prostate cancer and did not find an association between the duration of RBC storage and the 5-year biomedical (defined by prostate-specific antigen level) recurrence of cancer following radical prostatectomy. At 5 years, the recurrence-free survival rates were 74%, 71%, and 76% for patients who received younger (≤ 13 days), middle (13–18 days), and older (≥ 18 days) aged RBCs, respectively (P = .82; Wald test).57 A retrospective study by Kekre et al58 analyzed the data from 1,929 patients with cancer who received a transfusion within 1 year of a diagnosis of cancer and found that the overall survival rates were not associated with the storage duration of transfused RBCs. Median survival rates were 1.2, 1.7, and 1.1 years for the patients who received exclusively fresher (< 14 days), intermediate (14–28 days), or older (> 28 days) units of RBCs, respectively (P = .36).58 The same authors had also studied the data of 555 patients who received transfusions during hematopoietic stem cell transplantation and found that the proportion of older units (≥ 15 days) and the mean age of the transfused RBCs did not correlate with rates of 100-day nonrelapse mortality, LOS, or ICU admission (P > .05).59 Yuruk et al60 performed a small randomized trial of 20 hematology patients and found that changes in microcirculatory density and hemorheologic properties were similar between the fresher RBC group (median, 7 days [interquartile range, 5–7 days]) and older RBC groups (median, 23 days [interquartile range, 22–28 days]). Two studies of patients receiving liver transplantation also generated conflicting conclusions. Dunn 34 Cancer Control et al61 retrospectively analyzed 509 patients receiving liver transplantations and reported that no independent association existed between the duration of RBC storage and postoperative infections, organ rejection, or death. Patients who received more blood had an increased risk for death.61 However, Cywinski et al62 analyzed 637 patients receiving liver transplantations and found that the risk for graft failure and mortality was significantly higher in recipients of older RBC units (> 15 days old) compared with recipients of fresher RBC units (HR, 1.65; 95% CI: 1.18–2.31). The authors concluded that patients who intraoperatively received older RBCs had an increased risk for adverse outcomes.62 Several large studies have reported outcomes on mixed patient populations. Edgren et al63 reported on a large data set of 404,959 transfusion episodes in more than 300,000 patients (mostly trauma and surgical) derived from the Swedish and Denmark Scandinavian Donations and Transfusions’ database from 1995 to 2002.63 During a 2-year follow-up, an increased mortality rate of 5% was seen among patients transfused with RBCs that were stored for 30 to 42 days compared with patients who received RBCs that were stored for 10 to 19 days (HR, 1.05; 95% CI: 0.97–1.12).63 No dose effect, no pattern for cause of mortality, and no change in mortality over time were seen, leading the authors to state that the small excess risk was most consistent with weak confounding.63 Two studies examined patients with cardiovascular disease.64,65 Eikelboom et al64 studied 4,933 patients with cardiovascular disease and found a higher risk of mortality (RR, 1.48; 95% CI: 1.07–2.05) in recipients of the highest quartile of transfused storage duration (31–42 days). Robinson et al65 analyzed 909 patients receiving transfusions following a percutaneous coronary intervention and found that the duration of RBC storage was associated with a slightly higher rate of 30-day mortality (HR, 1.02; 95% CI: 1.01–1.04; P = .002), and recipients of only older RBCs (> 28 days old) had an even a greater risk of mortality (HR, 2.49; 95% CI: 1.45–4.25; P = .001). In a retrospective analysis of nearly 7,000 patients who received transfusions for general (noncardiac) surgery, Saager et al66 found no association between the median duration of storage and risk of postoperative mortality (HR, 0.99; 95% CI: 0.94–1.04; P = .64). In a retrospective analysis of 335 patients who underwent knee arthroplasty, Basora et al67 found no independent association between the age of transfused RBCs and postoperative wound infection. However, Lee et al68 analyzed 261 patients who underwent surgery for breast reconstruction and found that postoperative complications (vascular thrombosis, hematoma, and flap congestion) were higher in patients who received older blood (44.1%; RBC age > 14 days) compared with those who received fresher blood (20.0%, RBC age January 2015, Vol. 22, No. 1 ≤ 14 days) or no transfusion (12.8%; P < .05). In a retrospective analysis of 8,971 patients who received transfusions, including both inpatients and outpatients, Middelburg et al69 found an almost 2-fold increase in mortality rate for recipients of fresher RBCs compared with older RBCs (HR, 0.56; 95% CI: 0.32–0.97 for RBC stored > 24 days compared with < 10 days). A similar report of the adverse events of fresher blood was previously published by Mynster et al.56 They found a higher rate of cancer recurrence in patients who received RBCs stored for less than 21 days (HR, 1.5; 95% CI: 1.04–2.18).56 These 2 studies suggest that fresher RBCs may have potentially detrimental effects in some patient populations. The results from observational studies and small RCTs, both of which represent the body of literature on the subject, are mixed, and these studies have substantial methodological limitations. The strong correlation between the decision to transfuse and the severity of the underlying illness limits the interpretations from observational studies. Randomized Controlled Trials More than 25 years ago, the first prospective RCT of RBC storage was conducted and involved 237 patients who were randomized to receive 2 units of either fresh whole blood (< 12 hours) or stored blood (2–5 days) at the end of the extracorporeal circulation in a primary coronary bypass operation.2 No differences were seen in postoperative bleeding, coagulation tests, or transfusion requirements between the 2 groups.2 Since then, several small, prospective RCTs have been conducted; however, these trials are limited by their low numbers of participants (Table 5).2,45,49,50,60,70-72 Given the inconclusive findings from retrospective studies and the small number of study participants from the historical RCTs, 2 large, properly powered, multicenter RCTs have been reported. One study evaluated 377 infants born premature with very-low birth weight (< 1250 g) in a neonatal ICU who required at least 1 RBC transfusion.70 A total of 188 patients received fresh RBCs (median storage, 5.1 days; standard deviation [SD], 2.0), and 189 patients received RBCs stored according to standard of care (median storage, 14.6 days; SD, 8.3).70 The RR was 1.00 (95% CI: 0.82–1.21) for the primary outcome of the study, a composite measure of necrotizing enterocolitis, retinopathy of prematurity, intraventricular hemorrhage, bronchopulmonary dysplasia, and death. The rates of clinically suspected infection were 77.7% and 77.2% for the fresher RBC and standard-care RBC groups, respectively (RR, 1.01; 95% CI: 0.90–1.12).70 Rates of positive cultures were 67.5% and 64.0% for Table 5. — Randomized Controlled Trials of RBC Storage Duration Study Design Definition of Storage Duration Clinical Setting No. of Patients Increased Risk for Adverse Events With Longer Storage? Fergusson70 Prospectively randomized multicenter < 7 days vs standard of care Premature infants 377 No for mortality No for rate of complications Fernandes49 Prospectively randomized, single-center Continuous variable ICU (sepsis) 15 (10 transfused, 5 received albumin) No for gastric mucosal pH Kor45 Randomized single center ≤ 5 days vs standard-issue RBCs (median, 21 days) ICU 100 No difference in pulmonary function Steiner72 Prospectively randomized, multicenter ≤ 10 vs ≥ 21 days Cardiac surgery 1,098 No for changes in MODS, adverse events, or 28-day mortality rate Yuruk60 Prospectively randomized, single center < 1 week vs 3–4 weeks Hematology patients 20 Same increase in perfused vessel density in both group Walsh50 Prospectively randomized, single center ≤ 5 days vs ≥ 20 days ICU (on MV) 22 No for gastric mucosal pH Wasser2 Randomized single center < 12 hours vs 2–5 days Cardiac surgery 237 No for coagulation rests, postoperative bleeding, or transfusion requirements Weiskopf71 Randomized single center < 2 hours vs 3–4 weeks Healthy volunteers 35 No in pulmonary gas exchange variables ICU = intensive care unit, MODS = multiorgan dysfunction score, MV = mechanical ventilation, RBC = red blood cell. January 2015, Vol. 22, No. 1 Cancer Control 35 the fresher RBC and standard RBC groups, respectively (RR: 1.06; 95%: 0.91–1.22).70 Hence, this RCT demonstrated that the use of fresher RBCs compared with standard care did not decrease or increase the rate of complications or death in this population of premature, very-low birth weight neonates.70 A study conducted by Steiner et al72 enrolled 1,613 patients undergoing cardiac surgery, 1,418 of whom were randomized to receive leukoreduced RBCs stored for either no more than 10 days (fresher) or 21 days or longer (older). A total of 1,096 patients receiving transfusions within 96 hours following surgery were evaluable and analyzed for changes in multiorgan dysfunction score from prior to surgery to the highest composite change in multiorgan dysfunction score through day 7 (or death or discharge, if earlier), adverse events, and 28-day mortality. A total of 538 patients received a median of 4 units of fresh RBCs (median storage age, 7 days), and 560 patients received a median of 3 units of older RBCs (median storage age, 28 days). No difference was seen in the median composite change in multiorgan dysfunction score at day 7 (8.48 in the fresh RBC group vs 8.66 in the older RBC group; P = .42).72 A total of 53% and 51% of the patients in the fresh and old groups, respectively, developed a serious adverse event. All-cause mortality rates at day 28 were 4% in patients who received fresh RBCs and 5% in patients who received old RBCs.72 The study concluded that RBC storage duration was not significantly associated with 7-day changes in multiorgan dysfunction score, serious adverse events, or 28-day mortality rates among patients undergoing cardiac surgery.72 Another recent trial, the Age of Blood Evaluation (ABLE) study, is a multicenter RCT of critically ill Canadian patients and was funded by the Canadian Institute of Health Research (MCT-90648). This study involved 2,420 patients in the ICU randomized to receive RBCs less than 7 days old or standard-issue RBCs. The primary end point was 90-day mortality. The preliminary results of this study were presented at a recent critical care meeting in Toronto, Canada.73 The average age of the RBCs in the fresh group was 6.1 ± 4.9 days compared with 22.0 ± 8.5 days in the standard-issue group. The 90-day mortality rate in the intent-to-treat patients was not different between the groups (absolute risk reduction, 1.57% [95% CI: –2.25–5.40]). Thus, the preliminary results from this study are consistent with the other large randomized trials showing no difference in clinical outcomes associated with longer compared with shorter stored RBCs. Conclusions Observational studies of the clinical effects of the storage duration of red blood cells are conflicting and have methodological limitations; in addition, they are 36 Cancer Control confounded by indication. These limitations are best addressed by randomized controlled trials. A number of published and reported randomized controlled trials — 1 in adults, 1 in pediatrics, and 1 recently reported — provide strong evidence that the storage duration of red blood cells does not have measurable adverse effects on the clinical outcomes in select transfused patient populations. Additional randomized controlled trials are underway in the critically ill and medical/surgical patient populations. Together, these randomized controlled trials will define whether the storage duration of red blood cells has clinical relevance. References 1. Whitaker BI, Hinkins S; US Department of Health and Human Services. The 2011 National Blood Collection and Utilization Survey Report. Bethesda, MD: US Department of Health and Human Services; 2011. http://www.hhs.gov/ ash/bloodsafety/2011-nbcus.pdf. Accessed September 11, 2014. 2. Wasser MN, Houbiers JG, D’Amaro J, et al. The effect of fresh versus stored blood on post-operative bleeding after coronary bypass surgery: a prospective randomized study. Br J Haematol. 1989;72(1):81-84. 3. Koch CG, Li L, Sessler DI, et al. Duration of red-cell storage and complications after cardiac surgery. N Engl J Med. 2008;358(12):1229-1239. 4. Lelubre C, Vincent JL. Relationship between red cell storage duration and outcomes in adults receiving red cell transfusions: a systematic review. Crit Care. 2013;17(2):R66. 5. Wang D, Sun J, Solomon SB, et al. Transfusion of older stored blood and risk of death: a meta-analysis. Transfusion. 2012;52(6):1184-1195. 6. Vamvakas EC. Purported deleterious effects of “old” versus “fresh” red blood cells: an updated meta-analysis. Transfusion. 2011;51(5):1122-1123. 7. Lannan KL, Sahler J, Spinelli SL, et al. Transfusion immunomodulation--the case for leukoreduced and (perhaps) washed transfusions. Blood Cells Mol Dis. 2013;50(1):61-68. 8. Hess JR. Red cell changes during storage. Transfus Apher Sci. 2010;43(1):51-59. 9. Koch CG, Figueroa PI, Li L, et al. Red blood cell storage: how long is too long? Ann Thorac Surg. 2013;96(5):1894-1899. 10. Spinella PC, Sparrow RL, Hess JR, et al. Properties of stored red blood cells: understanding immune and vascular reactivity. Transfusion. 2011;51(4):894-900. 11. van de Watering L, Lorinser J, Versteegh M, et al. Effects of storage time of red blood cell transfusions on the prognosis of coronary artery bypass graft patients. Transfusion. 2006;46(10):1712-1718. 12. Yap CH, Lau L, Krishnaswamy M, et al. Age of transfused red cells and early outcomes after cardiac surgery. Ann Thorac Surg. 2008;86(2):554-559. 13. van Straten AH, Soliman Hamad MA, van Zundert AA, et al. Effect of duration of red blood cell storage on early and late mortality after coronary artery bypass grafting. J Thorac Cardiovasc Surg. 2011;141(1):231-237. 14. McKenny M, Ryan T, Tate H, et al. Age of transfused blood is not associated with increased postoperative adverse outcome after cardiac surgery. Br J Anaesth. 2011;106(5):643-649. 15. Voorhuis FT, Dieleman JM, de Vooght KM, et al. Storage time of red blood cell concentrates and adverse outcomes after cardiac surgery: a cohort study. Ann Hematol. 2013;92(12):1701-1706. 16. Vamvakas EC, Carven JH. Transfusion and postoperative pneumonia in coronary artery bypass graft surgery: effect of the length of storage of transfused red cells. Transfusion. 1999;39(7):701-710. 17. Leal-Noval SR, Jara-López I, Garcia-Garmendia JL, et al. Influence of erythrocyte concentrate storage time on postsurgical morbidity in cardiac surgery patients. Anesthesiology. 2003;98(4):815-822. 18. Vamvakas EC, Carven JH. Length of storage of transfused red cells and postoperative morbidity in patients undergoing coronary artery bypass graft surgery. Transfusion. 2000;40(1):101-109. 19. Andreasen JJ, Dethlefsen C, Modrau IS, et al.; Denmark Transfusion Study Group. Storage time of allogeneic red blood cells is associated with risk of severe postoperative infection after coronary artery bypass grafting. Eur J Cardiothorac Surg. 2011;39(3):329-334. 20. Sanders J, Patel S, Cooper J, et al. Red blood cell storage is associated with length of stay and renal complications after cardiac surgery. Transfusion. 2011;51(11):2286-2294. 21. Redlin M, Habazettl H, Schoenfeld H, et al. Red blood cell storage duration is associated with various clinical outcomes in pediatric cardiac surgery. Transfus Med Hemother. 2014;41(2):146-151. 22. Baltsavias I, Faraoni D, Willems A, et al. Blood storage duration and morbidity and mortality in children undergoing cardiac surgery. A retrospective analysis. Eur J Anaesthesiol. 2014;31(6):310-316. January 2015, Vol. 22, No. 1 23. Triulzi DJ, Yazer MH. Clinical studies of the effect of blood storage on patient outcomes. Transfus Apher Sci. 2010;43(1):95-106. 24. Zallen G, Offner PJ, Moore EE, et al. Age of transfused blood is an independent risk factor for postinjury multiple organ failure. Am J Surg. 1999;178(6):570-572. 25. Keller ME, Jean R, LaMorte WW, et al. Effects of age of transfused blood on length of stay in trauma patients: a preliminary report. J Trauma. 2002;53(5):1023-1025. 26. Offner PJ, Moore EE, Biffl WL, et al. Increased rate of infection associated with transfusion of old blood after severe injury. Arch Surg. 2002;137(6): 711-717. 27. Weinberg JA, McGwin G Jr, Griffin RL, et al. Age of transfused blood: an independent predictor of mortality despite universal leukoreduction. J Trauma. 2008;65(2):279-284. 28. Weinberg JA, McGwin G Jr, Marques MB, et al. Transfusions in the less severely injured: does age of transfused blood affect outcomes? J Trauma. 2008;65(4):794-798. 29. Weinberg JA, McGwin G Jr, Vandromme MJ, et al. Duration of red cell storage influences mortality after trauma. J Trauma. 2010;69(6):1427-1432. 30. Spinella PC, Carroll CL, Staff I, et al. Duration of red blood cell storage is associated with increased incidence of deep vein thrombosis and in hospital mortality in patients with traumatic injuries. Crit Care. 2009;13(5):R151. 31. Vandromme MJ, McGwin G Jr, Marques MB, et al. Transfusion and pneumonia in the trauma intensive care unit: an examination of the temporal relationship. J Trauma. 2009;67(1):97-101. 32. Leal-Noval SR, Muñoz-Gómez M, Arellano-Orden V, et al. Impact of age of transfused blood on cerebral oxygenation in male patients with severe traumatic brain injury. Crit Care Med. 2008;36(4):1290-1296. 33. Kiraly LN, Underwood S, Differding JA, et al. Transfusion of aged packed red blood cells results in decreased tissue oxygenation in critically injured trauma patients. J Trauma. 2009;67(1):29-32. 34. Weinberg JA, MacLennan PA, Vandromme-Cusick MJ, et al. The deleterious effect of red blood cell storage on microvascular response to transfusion. J Trauma Acute Care Surg. 2013;75(5):807-812. 35. Murrell Z, Haukoos JS, Putnam B, et al. The effect of older blood on mortality, need for ICU care, and the length of ICU stay after major trauma. Am Surg. 2005;71(9):781-785. 36. Hassan M, Pham TN, Cuschieri J, et al. The association between the transfusion of older blood and outcomes after trauma. Shock. 2011;35(1):3-8. 37. Juffermans NP, Vlaar AP, Prins DJ, et al. The age of red blood cells is associated with bacterial infections in critically ill trauma patients. Blood Transfus. 2012;10(3):290-295. 38. Purdy FR, Tweeddale MG, Merrick PM. Association of mortality with age of blood transfused in septic ICU patients. Can J Anaesth. 1997;44(12): 1256-1261. 39. Dessertaine G, Hammer L, Chenais F, et al. Does red blood cell storage time still influence ICU survival? [In French]. Transfus Clin Biol. 2008;15(4): 154-159. 40. Taylor RW, O’Brien J, Trottier SJ, et al. Red blood cell transfusions and nosocomial infections in critically ill patients. Crit Care Med. 2006;34(9): 2302-2308. 41. Juffermans NP, Prins DJ, Vlaar AP, et al. Transfusion-related risk of secondary bacterial infections in sepsis patients: a retrospective cohort study. Shock. 2011;35(4):355-359. 42. Pettila V, Westbrook AJ, Nichol AD, et al. Age of red blood cells and mortality in the critically ill. Crit Care. 2011;15(2):R116. 43. Aubron C, Bailey M, McQuilten Z, et al. Duration of red blood cells storage and outcome in critically ill patients. J Crit Care. 2014;29(3):476 e471-478. 44. Gajic O, Rana R, Mendez JL, et al. Acute lung injury after blood transfusion in mechanically ventilated patients. Transfusion. 2004;44(10):1468-1474. 45. Kor DJ, Kashyap R, Weiskopf RB, et al. Fresh red blood cell transfusion and short-term pulmonary, immunologic, and coagulation status: a randomized clinical trial. Am J Respir Crit Care Med. 2012;185(8):842-850. 46. Janz DR, Zhao Z, Koyama T, et al. Longer storage duration of red blood cells is associated with an increased risk of acute lung injury in patients with sepsis. Ann Intensive Care. 2013;3(1):33. 47. Katsios C, Griffith L, Spinella P, et al. Red blood cell transfusion and increased length of storage are not associated with deep vein thrombosis in medical and surgical critically ill patients: a prospective observational cohort study. Crit Care. 2011;15(6):R263. 48. Marik PE, Sibbald WJ. Effect of stored-blood transfusion on oxygen delivery in patients with sepsis. JAMA. 1993;269(23):3024-3029. 49. Fernandes CJ Jr, Akamine N, De Marco FV, et al. Red blood cell transfusion does not increase oxygen consumption in critically ill septic patients. Crit Care. 2001;5(6):362-367. 50. Walsh TS, McArdle F, McLellan SA, et al. Does the storage time of transfused red blood cells influence regional or global indexes of tissue oxygenation in anemic critically ill patients? Crit Care Med. 2004;32(2):364-371. 51. Sakr Y, Chierego M, Piagnerelli M, et al. Microvascular response to red blood cell transfusion in patients with severe sepsis. Crit Care Med. 2007;35(7):1639-1644. 52. Creteur J, Neves AP, Vincent JL. Near-infrared spectroscopy technique to evaluate the effects of red blood cell transfusion on tissue oxygenation. Crit January 2015, Vol. 22, No. 1 Care. 2009;(13 suppl 5):S11. 53. Kopterides P, Theodorakopoulou M, Nikitas N, et al. Red blood cell transfusion affects microdialysis-assessed interstitial lactate/pyruvate ratio in critically ill patients with late sepsis. Intensive Care Med. 2012;38(11):1843-1850. 54. Edna TH, Bjerkeset T. Association between transfusion of stored blood and infective bacterial complications after resection for colorectal cancer. Eur J Surg. 1998;164(6):449-456. 55. Mynster T, Nielsen HJ; Danish RANX05 Colorectal Cancer Study Group. The impact of storage time of transfused blood on postoperative infectious complications in rectal cancer surgery. Scand J Gastroenterol. 2000;35(2):212-217. 56. Mynster T, Nielsen HJ. Storage time of transfused blood and disease recurrence after colorectal cancer surgery. Dis Colon Rectum. 2001;44(7): 955-964. 57. Cata JP, Klein EA, Hoeltge GA, et al. Blood storage duration and biochemical recurrence of cancer after radical prostatectomy. Mayo Clin Proc. 2011;86(2):120-127. 58. Kekre N, Mallick R, Allan D, et al. The impact of prolonged storage of red blood cells on cancer survival. PLoS One. 2013;8(7):e68820. 59. Kekre N, Chou A, Tokessey M, et al. Storage time of transfused red blood cells and impact on clinical outcomes in hematopoietic stem cell transplantation. Transfusion. 2011;51(11):2488-2494. 60. Yuruk K, Milstein DM, Bezemer R, et al. Transfusion of banked red blood cells and the effects on hemorrheology and microvascular hemodynamics in anemic hematology outpatients. Transfusion. 2013;53(6):1346-1352. 61. Dunn LK, Thiele RH, Ma JZ, et al. Duration of red blood cell storage and outcomes following orthotopic liver transplantation. Liver Transpl. 2012;18(4):475-481. 62. Cywinski JB, You J, Argalious M, et al. Transfusion of older red blood cells is associated with decreased graft survival after orthotopic liver transplantation. Liver Transpl. 2013;19(11):1181-1188. 63. Edgren G, Kamper-Jorgensen M, Eloranta S, et al. Duration of red blood cell storage and survival of transfused patients (CME). Transfusion. 2010;50(6):1185-1195. 64. Eikelboom JW, Cook RJ, Liu Y, et al. Duration of red cell storage before transfusion and in-hospital mortality. Am Heart J. 2010;159(5):737-743e731. 65. Robinson SD, Janssen C, Fretz EB, et al. Red blood cell storage duration and mortality in patients undergoing percutaneous coronary intervention. Am Heart J. 2010;159(5):876-881. 66. Saager L, Turan A, Dalton JE, et al. Erythrocyte storage duration is not associated with increased mortality in noncardiac surgical patients: a retrospective analysis of 6,994 patients. Anesthesiology. 2013;118(1):51-58. 67. Basora M, Pereira A, Soriano A, et al. Allogeneic blood transfusion does not increase the risk of wound infection in total knee arthroplasty. Vox Sang. 2010;98(2):124-129. 68. Lee HK, Kim DH, Jin US, et al. Effect of perioperative transfusion of old red blood cells on postoperative complications after free muscle sparing transverse rectus abdominis myocutaneous flap surgery for breast reconstruction. Microsurgery. 2014;34(6):434-438. 69. Middelburg RA, van de Watering LM, Briët E, et al. Storage time of red blood cells and mortality of transfusion recipients. Transfus Med Rev. 2013;27(1):36-43. 70. Fergusson DA, Hébert P, Hogan DL, et al. Effect of fresh red blood cell transfusions on clinical outcomes in premature, very low-birth-weight infants: the ARIPI randomized trial. JAMA. 2012;308(14):1443-1451. 71. Weiskopf RB, Feiner J, Toy P, et al. Fresh and stored red blood cell transfusion equivalently induce subclinical pulmonary gas exchange deficit in normal humans. Anesth Analg. 2012;114(3):511-519. 72. Steiner ME, Triulzi DJ, Assmann SF et al. Randomized trial results: red cell storage age is not associated with a significant difference in multiple-organ dysfunction score or mortality in transfused cardiac surgery patients [Abstract]. Transfusion. 2014;54(suppl):15A. 73. Hébert P. Age of BLood Evaluation (ABLE) trial in the resuscitation of critically ill patients. Paper presented at: Critical Care Canada Forum 2014; Toronto, Ontario, Canada; October 29–November 1, 2014. http://www.criticalcarecanada. com/presentations/2014/age_of_blood_evaluation_able_trial_in_the_resuscitation_of_critically_ill_patients.pdf. Accessed November 13, 2014. Cancer Control 37 Indications for RBC transfusions have been revised because patients can now be maintained at lower hemoglobin levels. Ray Paul. Blue Marble, 2007. Acrylic, latex, enamel on wood, 49" × 64". Private collection. Transfusion Indications for Patients With Cancer Thomas Watkins, DO, PhD, Maria Katarzyna Surowiecka, MD, and Jeffrey McCullough, MD Background: During the last few years, considerable focus has been given to the management of anemia and coagulopathies. This article provides current concepts of red blood cell (RBC) and plasma coagulation factor replacements. Methods: The literature was reviewed for clinical studies relevant to RBC transfusion indications and outcomes as well as for the uses of coagulation factor replacement products for coagulopathies most likely encountered in patients with cancer. Results: Most patients without complications can be treated with a hemoglobin level of 7 g/dL as an indication for RBC transfusion. However, the effects of disease among patients with cancer may cause fatigue, so transfusions at higher hemoglobin levels may be clinically helpful. Leukoreduced RBCs are recommended as standard therapy for all patients with cancer, most of whom do not develop coagulopathy. Transfusions to correct mild abnormalities are not indicated in this patient population. Data are inconclusive regarding the value of coagulation factor replacement for invasive procedures when the international normalized ratio is below 2. Conclusions: Indications for RBC transfusion have become more conservative as data and experience have shown that patients can be safely and effectively maintained at lower hemoglobin levels. Coagulation factor replacement is unnecessary for most modest coagulopathies. Introduction Transfusion is an important part of cancer therapy. Red blood cells (RBCs) may be needed because of myelosuppression for chemotherapy or anemia in the setting of chronic disease. Because of myelosuppression, platelets are often part of the continuum of care for patients with cancer. Typically, plasma is From the Department of Laboratory Medicine and Pathology, University of Minnesota Medical School, Minneapolis, Minnesota. Address correspondence to Jeffrey McCullough, MD, Department of Laboratory Medicine and Pathology, University of Minnesota Medical School, Mayo Mail Code 609, 420 Delaware Street South East, Minneapolis, MN 55455. E-mail: [email protected] Submitted June 26, 2014; accepted October 31, 2014. No significant relationships exist between the authors and the companies/organizations whose products or services may be referenced in this article. 38 Cancer Control not needed because coagulopathy is not a major aspect of cancer or its therapy. However, in infrequent situations in which coagulation factor replacement is needed, plasma can be vital to the treatment of patients with cancer. Most infections can be managed with antimicrobials; however, granulocyte transfusions may sometimes be considered for recalcitrant infections in patients with neutropenia. The uses and indications for all of these blood components have undergone changes over the last few years. This report is a summary of those changes and the current clinical indications and uses of RBCs, plasma, and granulocytes. Transfusions RBC transfusion is common in the treatment of patients with cancer.1-4 Overall, patients with oncological and January 2015, Vol. 22, No. 1 hematological malignancies use around 34% of the RBC supply.1 In patients with cancer, as is similar with any other patient population, the indication for RBC transfusion is to alleviate symptomatic anemia. The decision to transfuse should not be driven by the hemoglobin concentration, and no single criterion can be used as an indication for RBC transfusion. Thus, the patient’s clinical status should be of utmost consideration.5 Anemia may occur in 90% of patients during chemotherapy and, furthermore, cancer treatments often cause the loss, destruction, and decreased production of RBCs — all of which lead to anemia.6 In particular, lung and gynecological cancers are associated with anemia because the treatments for such cancers include platinum-based therapies.7 Anemia in cancer may decrease quality of life and increase cancer-induced fatigue. Cancer-associated anemia may also be an indicator of poor clinical outcomes.8 If urgent correction of anemia is unnecessary, then erythropoietin treatment may be a valid alternative to RBC transfusion and can decrease RBC transfusion rates.9 In general, RBC transfusions are used to treat (1) tissue hypoxia due to inadequate RBC mass, (2) acute anemia due to trauma or surgical blood loss, (3) anemia in patients receiving chemotherapy, and (4) cardiovascular decompensation of chronic anemia. They are also used to ensure the optimal tissue oxygenation in patients with anemia undergoing radiation therapy. RBC transfusion is not indicated for the correction of anemia due to iron deficiency, as a source of nutritional supplementation, or in volume expansion. Dynamic physiological changes in patients with anemia help allow the decreased RBC mass to continue to oxygenate tissue. In brief, these changes include increased blood flow (as blood viscosity decreases) and increased oxygen offloading in hypoxic tissues (as the concentration of 2,3-diphosphoglycerate increases in the RBCs). In the setting of anemia, the overall blood volume is maintained with increased plasma volume. Compensatory cardiac output changes maintain adequate perfusion. As a result of these dynamic physiological changes, symptoms of anemia rarely manifest until hemoglobin values significantly dip. Animal studies indicate that extreme hemodilution can be tolerated in healthy animals.10 One study showed that 6 of 7 baboons survived hematocrit levels down to 4% and that they maintained adequate cardiac compensation at hematocrit levels as low as 10%.11 In addition, humans can tolerate very low levels of hemoglobin.12,13 RBC transfusions have been used for decades; recently, the effectiveness of RBC transfusions has been evaluated in randomized trials so that the best evidence can be ascertained to guide transfusion decisions. In some patient populations, nontransfused patients have better outcomes than transfused patients and patients who receive fewer units do better than January 2015, Vol. 22, No. 1 those who receive more RBC units.14-16 The following clinical trials described below illustrate this concept. Results from a trial by Hébert et al17 changed the practice of transfusion. This study was a randomized controlled trial that compared a “liberal” transfusion strategy (defined as a post-transfusion goal of a hemoglobin concentration between 10 and 12 g/dL with a transfusion indication of a hemoglobin concentration of 10 g/dL) with a “restrictive” transfusion strategy (defined as a post-transfusion hemoglobin concentration between 7 and 9 g/dL with a transfusion indication of a hemoglobin concentration of 7 g/dL). The trial included 838 volunteers and demonstrated an overall in-hospital mortality rate significantly lower in the participants who received the restrictive transfusion. The 30-day mortality rates were not significantly different between 2 groups.17 However, clinically “less ill” volunteers (Acute Physiology and Chronic Health Evaluation score < 20) or those who were younger (< 55 years of age) had significantly lower 30-day mortality rates than those who were “more severely ill” or older following the restrictive transfusion strategy.17 Thus, the study results demonstrated that a restrictive transfusion strategy is at least equivalent to a liberal transfusion strategy in all groups, except among those with severe ischemic heart disease, and the restrictive transfusion strategy was potentially better in “less ill” and younger people.17 This was the first well-structured clinical trial of RBC transfusion suggesting that maintaining patients at lower hemoglobin levels might be beneficial.17 Vincent et al18 evaluated the 28-day mortality rate of 3,534 patients from 146 western European intensive care units (ICUs). The mortality rates among study volunteers were 22.7% and 17.1% among those receiving transfusions and those not receiving transfusions, respectively.18 The study controlled for patients with a similar degree of organ dysfunction. The receipt of an RBC transfusion in the ICU increased a patient’s odds of dying by a factor of 1.37.18 Corwin et al19 analyzed anemia and blood transfusions among 4,892 study volunteers who were critically ill in US ICUs. The study results showed that the number of RBC transfusions was an independent predictor of longer ICU stay, longer length of hospital stay, and increased mortality rates. In another trial, Carson et al20 compared the effect of a transfusion threshold of 10 g/dL with 8 g/dL in cardiovascular patients undergoing surgical hip fracture repair. The trial involved 2,016 patients older than 50 years of age, and the primary outcome was death or the inability to walk across a room without human assistance on 60-day follow-up. Rates of death or an inability to independently walk after 60 days, in-hospital morbidity rates, and in-hospital complications were similar in the 2 groups. Thus, the Cancer Control 39 liberal transfusion strategy did provide clinical benefit over the restrictive (9 g/dL) strategy. Evidence does not support a benefit to a post-transfusion hemoglobin concentration above 10 g/dL.17,21 However, this level may be helpful in pediatric patients with cancer who have acute blood loss or cyanotic heart disease due to the additional challenges of this patient population.22 Despite well-performed clinical trials, no universal RBC transfusion criterion exists. A restrictive transfusion strategy is at least equivalent to a liberal transfusion strategy in the majority of clinical scenarios. However, in clinical practice, the underlying condition of the patient and his or her transfusion goals and desired outcomes should be considered. RBC transfusion may be indicated in a patient with symptoms of anemia and a hemoglobin level below 7 g/dL. Transfusion with hemoglobin concentrations between 7 and 10 g/dL may be indicated when significant underlying comorbidities exist, such as cardiac disease, respiratory disease, bone marrow failure, or other hematological diseases; this is because anemia may not be well tolerated in these patients.23,24 Traditionally, single unit transfusions were not recommended; however, as the hemoglobin indication has decreased and transfusion has become more conservative, it has become clear that the transfusion of 1 unit can be effective and sufficient. Single unit vs 2-unit transfusions can reduce blood use as much as 25% with no adverse clinical consequences.25 Specifically, the AABB (formerly American Association of Blood Banks) recommends transfusion at a hemoglobin concentration of 7 to 8 g/dL for hospitalized patients who are stable, 8 g/dL for those with cardiovascular disease, and higher hemoglobin (unspecified) concentrations for patients with acute coronary syndromes.26 Reactions or adverse events due to RBC transfusion are uncommon and may occur in 1% to 3% of transfusions.27 The most common adverse event is febrile nonhemolytic transfusion reaction, which typically is due to human leukocyte antigen (HLA) antibodies in the recipient or an allergic reaction to plasma proteins. The most severe yet rare reaction is acute hemolysis, usually due to ABO incompatibility due to administration error. Because the changes that occur during RBC storage have become better understood, concern has developed as to whether RBCs nearing the end of the routine 42-day storage might have undergone changes, thus making them risky for certain patients. The focus of research has been on patients with cardiovascular disease or those undergoing surgery; presently, however, no data suggest this is a concern for patients with malignancy.28 For more information, please refer to the article by Drs Qu and Triulzi in this issue. RBC transfusion has an immune-modulating ef40 Cancer Control fect.29 RBC transfusion may be associated with increased risks of postoperative infections, longer durations of hospital stay, and longer stays in the ICU.28,30,31 RBC transfusion has also been linked to longer durations of mechanical ventilation, increased incidences of multiple organ failures, and an overall increase in health care costs.28,30-33 However, these issues have not been resolved. In the previous few years, concerns have been raised that RBC transfusions might exacerbate cancer; however, no consensus has yet to be made on this issue. For more information about this topic, please read the article by Drs Dasararaju and Marques in this issue. The rationale for the transfusion of RBCs is to increase the delivery of oxygen to the tissues, but physiological changes with RBC storage may limit this goal. In addition, the ability of transfused RBCs to deliver oxygen to areas most in need of oxygenation may be decreased.33 The physiological changes that occur in stored RBCs (collectively called the RBC storage lesion) may limit, to some degree, the ability of the transfused RBCs to enter the microcirculation and may decrease vasodilation by altering the bioavailability of nitric oxide. During storage, RBCs undergo changes that result in their removal from the circulation within 24 hours of transfusion. However, some RBCs recover biochemical normalcy and survive normally.34 Other changes to stored RBCs include microparticle formation, changes in shape, decreased concentration of RBC 2,3-diphosphoglycerate, decreased pH, and the decreased availability of adenosine triphosphate and glucose. In combination, the physiological changes resulting from the RBC storage lesion may limit the delivery of oxygen by the transfused RBCs. However, no consensus exists on whether RBCs stored for long periods of time are deleterious to any patient group; thus, RBCs of any storage age can be used for patients with cancer. Leukoreduced RBCs have decreased rates of transfusion reactions, HLA alloimmunization, and have the potential benefit of modifying the transfusion-related immune modulation (TRIM) effect (if it exists). Thus, leukoreduced RBCs are recommended as the standard blood product for routine use in patients with cancer. Frequent transfusions for cancer and chemotherapy treatments over an extended period of time may result in iron overload. Treatment regimens for many solid organ cancers avoid this complication because the transfusion-dependent period is shorter in duration due to chemotherapy and irradiation regimens.34,35 As transfusion dependence increases during treatment, the risk of transfusion-transmitted infection, allergic response, and severe transfusion reactions increase with each unit transfused. Health care professionals must weigh any benefit from RBC transfusions against these risks. January 2015, Vol. 22, No. 1 Special Red Blood Cell Products Patients with cancer may require specially prepared RBC products due to frequent comorbidities. Leukoreduced Blood Components The leukocyte content of different blood products widely varies (as high as 1 × 109 in whole blood to < 0.6 × 106 in fresh frozen plasma [FFP]). Leukoreduced blood products are blood products produced by filtration or apheresis to decrease the number of leukocytes remaining in the product to below 5 × 106 leukocytes/component.36 Leukoreduced blood components are beneficial in 3 ways: (1) decreased frequency of febrile nonhemolytic transfusion reactions, (2) decreased HLA sensitization of recipients, and (3) decreased likelihood of cytomegalovirus (CMV) transmission via transfusion.37 Leukoreduction may significantly reduce febrile nonhemolytic transfusion reactions and may decrease cardiopulmonary transfusion reactions (transfusion-related acute lung injury and transfusion-associated circulatory overload).38,39 Presumably, this occurs through reduced levels of bioactive lipids and soluble CD40L in leukoreduced RBCs, which would have been produced by leukocytes had they remained in the blood product.40 As the RBCs age in storage media, they develop well-established changes that include decreased deformability and decreased levels of adenosine triphosphate and 2,3 diphosphoglycerate. Donor leukocytes release cytokines and lipid mediators capable of affecting neutrophils in a time-dependent course during RBC storage.41 Prestorage leukoreduction decreases the release of metabolites and cellular components into the RBC product. Leukoreduction may also be effective in decreasing alloimmunization and platelet transfusion refractoriness. This is especially relevant to patients with cancer as they may receive numerous RBC and platelet transfusions during their treatment cycle. A study published in 1997 examined 1,047 patients with acute myeloid leukemia.42 Those who received leukoreduced platelets had decreased levels of lymphocytotoxic antibodies and lower rates of refractoriness to platelet transfusion when compared with the study controls who received unmodified pooled platelet concentrates.42 Leukoreduction may decrease the TRIM effect of blood transfusion that may lead to possible increased cancer recurrence. Evidence suggesting that blood transfusion may decrease immune function was established more than 30 years ago, showing that survival rates were increased following renal transplantation.29 Other, more controversial data exist regarding RBC transfusion and tumor recurrence perioperatively. Vamvakas and Carven31 showed that patients with colorectal cancer who received RBC transfusion perioperatively had longer lengths of hospital stays when January 2015, Vol. 22, No. 1 adjusted for multiple confounding factors related to the severity of their illness, difficulty of operation, and risks for postoperative infections. In addition, Blajchman32 reported adverse effects on tumor recurrence in 50% of nonrandomized trials. Further data suggest that an immunomodulatory role in transfusion is related to a dose-dependent association (ie, increased RBC transfusion) with postoperative bacterial infections and RBC transfusion.30 Cytomegalovirus Infection and Safe Blood Components Transfusion-transmitted CMV is a possible risk for severe infectious complications in severely immunosuppressed patients with cancer who have not been previously infected with CMV. Donor screening questionnaires cannot exclude CMV seropositive volunteers, and CMV has a high seroprevalence; 40% to 50% of adults have CMV antibodies. Furthermore, regional blood centers may have difficulty obtaining CMV negative products locally because the majority of adults in these areas may be CMV seropositive. An additional issue with CMV seronegative donors is that some may still carry CMV in their leukocytes or plasma. For transfusion recipients who are immunologically competent, CMV infection is not life threatening. However, CMV infection in immunocompromised patients with cancer can result in potentially fatal sequelae, including delayed hematopoietic stem cell engraftment, pneumonia, and severe gastrointestinal inflammation. However, even in these patients the risk is low. By using CMV-safe leukocyte blood cells, one study found that CMV infection was reduced from 2.4% to 1.3% and CMV disease was reduced from 2.4% to 0%.43 CMV is leukotropic and is not present in the RBCs, platelets, or plasma of healthy donors. Leukoreduction decreases the likelihood of CMV transmission, and leukoreduced products are generally regarded as being safe from CMV infection and equivalent to CMV antibody negative blood.43-46 Irradiated Blood Components A serious and typically fatal complication of blood transfusion is transfusion-associated graft-vs-host disease (TA-GVHD). The transfusion of viable allogeneic T-lymphocytes in blood products to an immunosuppressed individual has the potential for TA-GVHD, a complication that can be prevented by irradiating the blood components. The actual incidence of TA-GVHD is low in most patients with cancer, and the irradiation of blood products is indicated in few, small, but well-defined populations of patients at risk. The types of patients who require irradiated cellular blood products include neonates, patients with congenital immune deficiencies Cancer Control 41 (eg, severe combined immunodeficiency syndrome, Wiskott–Aldrich syndrome), those with a hematological malignancy who are undergoing chemotherapy, and recipients of allogeneic and autologous bone marrow transplantations, partial HLA-matched products (often directed donations from genetic relatives), and all granulocyte products. Patients with HIV/AIDS and patients with solid organ tumors do not require irradiated RBCs.47 Leukoreduction does not prevent TA-GVHD. Although patients with suppressed immune systems are at the most risk for TA-GVHD than any other patient group, rare instances exist in which transfusion between similar HLA-type individuals has resulted in TA-GVHD in immune-competent individuals. These include situations in which the donor and recipient share an HLA haplotype such that the patient does not recognize the donor cells as foreign and, thus, does not eliminate them, creating the potential for TA-GVHD.48,49 Washed Red Blood Cells The indications for washed RBC products in patients with cancer are largely in line with the requirements for washed products in general medical settings. Overall, the goal for washing RBC products is to decrease plasma elements, including antibodies, plasma proteins, and electrolytes, that may have adverse effects on the recipient. Patients with severe immunoglobulin A deficiency have the potential to have anaphylactic transfusion reactions. Washing RBCs removes immunoglobulin A from the unit. Rarely, patients who experience recurrent febrile nonhemolytic and urticarial transfusion reactions may also benefit from washed RBCs. Additional indications for washed RBCs may include rapid or large volume (> 25 mL/kg) transfusions in small volume or in patients with small stature. Washed products may be indicated for transfused products following irradiation, because some patients with cancer and poor renal function may have difficulty with the increased extracellular potassium in RBCs after the irradiation. Some patients with cancer may also require volume-reduced RBC products. If a patient with cancer has a compromised renal or circulatory system that cannot accommodate the increased volume of the transfused RBC unit, then volume reduction may be indicated. Fresh Frozen Plasma FFP is plasma that has been separated from whole blood or obtained by plasmapheresis and frozen at –0.4°F (–18°C) or below within 8 hours of collection. At this temperature, FFP can be stored for up to 12 months after donation. A unit of FFP has a volume of about 200 to 250 mL and contains all of the coagu42 Cancer Control lation proteins present in whole blood. FFP does not contain RBCs and, thus, can be administered without regard for the Rhesus (Rh) type of the patient. However, because plasma contains antibodies, it should be ABO matched to avoid possible hemolysis. Additional plasma preparations used clinically include plasma frozen within 24 hours of collection (FP24), which contains reduced levels of labile coagulation factors V and VIII. FP24 is frequently used by blood blanks interchangeably with FFP when a clinical need exists for fibrinogen replacement. FFP transfusions are typically undertaken in the setting of bleeding or in preparation for an invasive procedure when laboratory coagulation screening test results are abnormal.50,51 These are typically defined as prothrombin and partial thromboplastin times greater than 1.5 times the normal limit. The usual dose of FFP is 10 to 15 mL/kg body weight, but the dose may be higher in the setting of massive blood loss. This dose would be 3 to 4 units of FFP; however, in practice most health care professionals use 2 units and, thus, patients are often underdosed. A dose should be given at least every 6 hours until hemostasis is achieved or coagulation parameters are stabilized.51 The need for additional FFP is based on the repletion of factor VII, which has the shortest half-life of all the coagulation factors. If FFP is given for bleeding, then its effectiveness can be best assessed by monitoring the clinical response of the patient. If it is given to correct abnormal coagulation parameters, then the parameters may be followed as an indication of hemostasis response.50 If the patient is also receiving platelets, then it is important to remember that when platelets are stored in plasma, every plateletpheresis unit contains the equivalent of 1 bag of FFP. In this situation, either smaller doses or no additional doses of FFP may be required. In Europe and the United States, platelets may be stored in an additive solution of electrolytes instead of plasma.52 Because the additive solutions replace plasma, those platelet products cannot be considered a source of coagulation factors. National guidelines for the use of FFP exist both in the United States and abroad.50,51 The clinical indications for the therapeutic use of FFP include active bleeding before an invasive procedure in the presence of an inherited or acquired clotting factor deficiency, active bleeding in the setting of a consumptive coagulopathy or disseminated intravascular coagulation, massive transfusion, immediate reversal of warfarin effect in an actively bleeding patient, and thrombotic thrombocytopenic purpura.53 Patients with cancer may be at risk for abnormalities of hemostasis due to tumor pathology and evolution of the disease as well as treatment effect. Coagulation factor abnormalities may occur as a result of vitamin K deficiency from malnutrition, diarrhea, liver disease, biliary obstrucJanuary 2015, Vol. 22, No. 1 tion, use of vitamin K antagonists, and antibiotic therapy.54 In the setting of abnormal coagulation screening test results, invasive procedures such as surgery, line placement, indwelling catheter placement, among others, may result in significant blood loss. The use of FFP along with vitamin K and cryoprecipitate for additional fibrinogen replacement may be considered in these situations. However, the effectiveness of FFP used prophylactically in the nonbleeding patient prior to an invasive procedure or surgery in the setting of abnormal coagulation values has not been proven.55 A paucity of good randomized controlled trials have compared the use of FFP with no FFP. Two well-conducted randomized controlled trials reported a lack of evidence for the prophylactic use of FFP.55 Several issues exist when considering the use of FFP. Reversing a coagulopathy with FFP generally requires a large volume of transfused product. This could be a significant concern, particularly for patients who have blood volume status issues prior to transfusion and who are at risk for transfusion-associated volume overload. In addition, due to the relatively low concentration of clotting factors in a unit of FFP, the increase in factor activity after more than 1 L of transfused FFP may be modest. If immediate correction of coagulopathy is needed, then a product containing factors II, VII, IX, and X and proteins C and S and more concentrated forms of coagulation factors should be considered. The transfusion of plasma carries significant risk that should be weighed against its perceived benefit, especially when FFP is prophylactically used. Potential serious complications include transfusion-associated lung injury and volume overload as well as transfusion-transmitted infection. Allergic reactions to plasma are common and may, in rare cases, be life threatening. Pathogen inactivation is a process by which blood components are treated in a manner that damages nucleic acids, thus rendering the components free of infectious pathogens.56 One of these plasma components, Octaplas (Octapharma USA, Hoboken, New Jersey), is available for use in the United States. Octaplas is prepared from pools of about 1,000 donor units and then subjected to solvent detergent treatment for pathogen inactivation and reallocated into units of about 200 mL, which is similar to a standard unit of FFP.57 The solvent detergent treatment spares coagulation factors so that the product is considered to be similar to FFP. Granulocyte Transfusion Infections — particularly fungal infections — continue to be a source of morbidity and mortality in patients with neutropenia because of aggressive chemotherapy January 2015, Vol. 22, No. 1 or hematopoietic stem cell transplantation. With a granulocyte count below 1,000, the risk of infection is increased, and this risk is even further increased based on the duration of neutropenia. During the 1970s, several studies established that granulocyte transfusion was associated with improved survival rates in patients with gram-negative sepsis and granulocytopenia for at least 10 days.58-60 No carefully controlled studies of granulocyte transfusion exist in other clinical settings. However, as our ability to manage neutropenia and to treat gram-positive and gram-negative sepsis has improved with the use of newer antibiotics, the value of granulocyte transfusions has become questionable.61,62 Granulocyte transfusions in the 1970s up to the present contained about 1 × 1010 granulocytes and were obtained from donors, most of whom were stimulated with dexamethasone. The advent of granulocyte colony-stimulating factor (G-CSF) and its resultant use in patients to increase granulocyte counts and mobilize hematopoietic stem cells led to the possibility of using G-CSF stimulation of blood donors in order to obtain larger numbers of granulocytes for transfusion. When it is combined with dexamethasone, G-CSF can result in granulocyte counts of up to 40,000/µL with a yield of up to 8 × 1010 granulocytes.63,64 Small studies of these transfusions have suggested efficacy.65,66 However, no studies adequately establish the clinical value of granulocyte transfusions. A multicenter trial managed by the National Marrow Donor Program in 5 US centers studied 40 patients with infection and neutropenia.67 Survival rates with complete or partial response rates 4 weeks after initiating transfusions were 38% for invasive mold infection, 40% for bacteremia/candidemia, and 60% for severe bacterial infection.67 Thus, evidence suggests that granulocyte transfusion therapy is feasible and may be clinically effective. A recently completed large, multicenter clinical trial did not show benefit from granulocyte transfusions except in a small subgroup of patients who received very high doses of granulocytes.68 These result suggest it is possible that granulocyte transfusions may be clinically beneficial if very high doses of cells are given.68 Currently, if granulocyte transfusions are to be used, then cells obtained from dexamethasone and G-CSF–stimulated donors are recommended to obtain a substantial number of cells. These transfusions can provide an increased granulocyte count to more than 5,000/µL in many patients,66 and subsequent transfusions can maintain counts in this range.67 Indications for considering granulocyte transfusion include bacterial of fungal infections of the blood or proven tissue infections of bacteria of fungi unresponsive to antibiotics. Response to transfusion should not be evaluated on a daily basis, but granulocyte transfusions should be considered as a course of therapy similar to antibiotics. Therefore, Cancer Control 43 transfusions should be continued for a minimum of 5 days or until the infection has been resolved. Granulocytes should be transfused as soon as possible after collection because storage time is limited.69-71 Transfusion of a unit of granulocytes should not take more than 2 hours. Reactions to granulocyte transfusions are relatively common and generally similar to a febrile nonhemolytic transfusion reaction. Severe pulmonary reactions have been reported when granulocytes were infused in close proximity to amphotericin, but whether this represents a major risk or applies to other antifungals is not clear. It is best to separate the transfusion of granulocytes from amphotericin infusion by at least 2 hours. Outpatient Transfusion With improvements in medical treatments and longer survival rates among patients with cancer, the management of anemia and thrombocytopenia on an outpatient basis has become an important consideration. In patients with acute myeloid leukemia or high-risk myelodysplastic syndromes, the availability of highly effective antimicrobials and transfusion support has allowed a shift in care from inpatient to outpatient settings.72 In these patient populations, outpatient management of cytopenias has been shown to be safe and effective in both the postconsolidation and postinduction therapy periods.73,74 Outpatient treatment has several potential benefits, including reduced cost and resource utilization, improved quality of life, and decreased incidence of nosocomial infections.75 Important factors involved in outpatient management include establishing therapy guidelines, determining the location where the therapy takes place, and patient education. Communication with the local blood bank is also important, particularly with regard to special products. Indications and guidelines for inpatient transfusion are well established. However, it is not clear whether these should be applied or modified for outpatient transfusion. Thus, because no national guidelines exist for outpatient transfusions, each institution must determine its own indications. On one hand, the rational and physiology of the management of anemia or thrombocytopenia are the same for inpatients or outpatients, and, thus, possibly the guidelines and indications for transfusion should be the same. By contrast, patients are living in a different environment as outpatients. They are less acutely ill, less fragile, and more stable and thus should be more resilient. However, they are more removed from easy and quick access to medical care. Their care is provided by intermittent outpatient clinic visits that may involve travel and inconvenience. Thus, it is appropriate to manage transfusion to provide the stability and continuity that enables the patient 44 Cancer Control to function in the outpatient setting. It might also be appropriate to transfuse larger-than-usual inpatient doses of the component if doing so extends the time to the next clinic visit. For example, larger doses of platelets extend the time to the next transfusion,76,77 and, if the sole reason for a patient to return for a clinic visit is for a platelet count to determine the need for the next transfusion, then a larger dose can extend the time for the next clinic visit. Doing so provides a better quality of life for the patient and may also be more cost effective, although no such studies have been done to determine whether this is true. Another consideration is the laboratory value as the indication for the transfusion. For instance, if the hemoglobin level is slightly above 7 g/dL and the hospital’s guideline for RBC transfusion is 7 g/dL, then the transfusion might be considered to be inappropriate in the quality system monitoring. By contrast, if a return clinic visit is not needed for 1 or 2 weeks, then it would be inappropriate to have the patient return sooner simply to repeat the hemoglobin level to determine when the hemoglobin concentration is less than 7 g/dL so the transfusion would meet the hospital guideline. It seems that more appropriate care would be to transfuse the patient at that visit despite a hemoglobin concentration above the level recommended by the guideline. Thus, transfusing 2 units of RBCs or transfusing at a hemoglobin concentration of 8 g/dL or even 9 g/dL could be considered appropriate in the outpatient setting. The topic of indications for outpatient transfusions is not established and deserves considerable analysis and discussion because of different patient life situations. We also need to determine ways in which to offer the most cost-effective methods for providing care in the outpatient setting. Conclusions A hemoglobin level of 7 g/dL is a suitable indication for red blood cell transfusion in stable patients without complications. However, patients with cardiovascular disease or acute coronary syndrome should be transfused at a hemoglobin level of 8 g/dL. Indications for transfusion in patients with other types of complications have not been established. Patients with cancer have reported an increased feeling of wellbeing and stamina when maintained at hemoglobin levels at about 7 g/dL, but no structured studies have determined the optimal hemoglobin levels for patients with advanced cancer. Although coagulopathy is uncommon in patients with cancer, fresh frozen plasma is used as replacement therapy for moderate to severe coagulopathy. Fresh frozen plasma may also be used for increases in the international normalized ratio in preparation for invasive procedures, although no structured studies January 2015, Vol. 22, No. 1 have established the exact value. References 1. Shortt J, Polizzotto MN, Waters N, et al. Assessment of the urgency and deferability of transfusion to inform emergency blood planning and triage: the Bloodhound prospective audit of red blood cell use. Transfusion. 2009;49(11): 2296-2303. 2. Estrin JT, Schocket L, Kregenow R, et al. A retrospective review of blood transfusions in cancer patients with anemia. Oncologist. 1999;4(4):318-324. 3. Dicato M, Plawny L, Diederich M. Anemia in cancer. Ann Oncol. 2010;21(suppl 7):vii167-172. 4. Tas F, Eralp Y, Basaran M, et al. Anemia in oncology practice: relation to diseases and their therapies. Am J Clin Oncol. 2002;25(4):371-379. 5. National Institutes of Health (NIH). NIH Consensus Conference on Use of Red Cells. 3rd ed. Bethesda, MD: NIH; 1988. 6. Tas F, Eralp Y, Basaran M, et al. Anaemia in oncology practice: relation to diseases and their therapies. Am J Clin Oncol. 2004;(suppl 1):11-26. 7. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment [Erratum in J Natl Cancer Inst. 2000;92(6):497]. J Natl Cancer Inst. 1999;91(19):1616-1634. 8. Stasi R, Abriani L, Beccaglia P, et al. Cancer-related fatigue: evolving concepts in evaluation and treatment. Cancer. 2003;98(9):1786-1801. 9. Littlewood TJ, Bajetta E, Nortier JW, et al; Epoetin Alfa Study Group. Effects of epoetin alfa on hematologic parameters and quality of life in cancer patients receiving nonplatinum chemotherapy: results of a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2001;19(11):2865-2874. 10. Cabrales P, Tsai AG, Frangos JA, et al. Oxygen delivery and consumption in the microcirculation after extreme hemodilution with perfluorocarbons. Am J Physiol Heart Circ Physiol. 2004;287(1)H320-H330. 11. Wilkerson DK, Rosen AL, Sehgal LR, et al. Limits of cardiac compensation in anemic baboons. Surgery. 1988;103(6):665-670. 12. Weiskopf RB, Viele MK, Feiner J, et al. Human cardiovascular and metabolic response to acute, severe isovolemic anemia. JAMA. 1998; 279:217-221. 13. Toy P, Feiner J, Viele MK, et al. Fatigue during acute isovolemic anemia in healthy, resting humans. Transfusion. 2000;40(4):457-460. 14. Grimshaw K, Sahler J, Spinelli SL, et al. New frontiers in transfusion biology: identification and significance of mediators of morbidity and mortality in stored red blood cells. Transfusion. 2011;51(4):874-880. 15. Blumberg N. Deleterious clinical effects of transfusion immunomodulation: proven beyond a reasonable doubt. Transfusion. 2005;45(2 suppl):33S-40S. 16. Tinmouth A1, Fergusson D, Yee IC, et al; ABLE Investigators; Canadian Critical Care Trials Group. Clinical consequences of red cell storage in the critically ill. Transfusion. 2006;46(11):2014-2027. 17. Hébert PC, Wells G, Blajchman MA, et al; Transfusion Requirements in Critical Care Investigators; Canadian Critical Care Trials Group. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care [Erratum appears in N Engl J Med. 1999;340(13):1056]. N Engl J Med. 1999;340(6):409-417. 18. Vincent JL, Baron JF, Reinhart K, et al; Anemia and Blood Transfusion in Critical Care Investigators. Anemia and blood transfusion in critically ill patients. JAMA. 2002;288(12):1499-11507. 19. Corwin HL, Gettinger A, Pearl RG, et al. The CRIT Study: Anemia and blood transfusion in the critically ill--current clinical practice in the United States. Crit Care Med. 2004;32(1):39-52. 20. Carson JL, Terrin ML, Noveck H, et al. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med. 2011;365(26):2453-2462. 21. Hébert PC; Transfusion Requirements in Critical Care. The TRICC trial: a focus on the sub-group analysis. Vox Sang. 2002;(83 suppl):387-396. 22. Lacroix J, Hébert PC, Hutchison JS, et al; TRIPICU Investigators; Canadian Critical Care Trials Group; Pediatric Acute Lung Injury and Sepsis Investigators Network. Transfusion strategies for patients in pediatric intensive care units. N Engl J Med. 2007;356(16):1609-1619. 23. Nelson AH, Fleisher LA, Rosenbaum SH. Relationship between postoperative anemia and cardiac morbidity in high-risk vascular patients in the intensive care unit. Crit Care Med. 1993;21(6):860-866. 24. Whitman CB, Shreay S, Gitlin M, et al. Clinical factors and the decision to transfuse chronic dialysis patients. Clin J Am Soc Nephrol. 2013;8(11):1942-1951. 25. Berger MD, Gerber B, Arn K, et al. Significant reduction of red blood cell transfusion requirements by changing from a double-unit to a single-unit transfusion policy in patients receiving intensive chemotherapy or stem cell transplantation. Haematologica. 2012;97(1):116-122. 26. Carson JL, Grossman BJ, Kleinman S, et al. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Intern Med. 2012;157(1):49-58. 27. McCullough JJ. Complications in transfusion. In: McCullough JJ. Transfusion Medicine. 3rd ed. Chichester, West Sussex, UK: Wiley-Blackwell; 2011. 28. Lelubre C, Piagnerelli M, Vincent JL. Association between duration of storage of transfused red blood cells and morbidity and mortality in adult patients: myth or reality? Transfusion. 2009;49(7):1384-1394. 29. Opelz G, Sengar DP, Mickey MR, et al. Effect of blood transfusions on subsequent kidney transplants. Transplant Proc. 1973;5(1):253-259. 30. Blumberg N, Heal JM. Evidence for plasma-mediated immunomodulation--transfusions of plasma-rich blood components are associated with a January 2015, Vol. 22, No. 1 greater risk of acquired immunodeficiency syndrome than transfusions of red blood cells alone. Transplant Proc. 1988;20(6):1138-1142. 31. Vamvakas EC, Carven JH. Allogeneic blood transfusion, hospital charges, and length of hospitalization: a study of 487 consecutive patients undergoing colorectal cancer resection. Arch Pathol Lab Med. 1998;122(2):145-151. 32. Blajchman MA. Immunomodulatory effects of allogenic blood transfusions: Clinical manifestations and mechanisms. Vox Sang. 1988;74(suppl 2):315. 33. Kor DJ, Van Buskirk CM, Gajic O. Red blood cell storage lesion. Bosn J Basic Med Sci. 2009;(9 suppl 1):21-27. 34. Luten M, Roerdinkholder-Stoelwinder B, Schaap NP, et al. Survival of red blood cells after transfusion: a comparison between red cells concentrates of different storage periods. Transfusion. 2008;48(7):1478-1485. 35. Jabbour E, Kantarjian HM, Koller C, et al. Red blood cell transfusions and iron overload in the treatment of patients with myelodysplastic syndromes. Cancer. 2008;112(5):1089-1095. 36. American Association of Blood Banks (AABB). Standards for Blood Banks and Transfusion Services. 27th ed. Bethesda, MD: AABB; 2014. 37. Ratko TA, Cummings JP, Oberman HA, et al; University Health System Consortium. Evidence-based recommendations for the use of WBC-reduced cellular blood components. Transfusion. 2001;41(10):1310-1399. 38. Sharma RR, Marwaha N. Leukoreduced blood components: advantages and strategies for its implementation in developing countries. Asian J Transfus Sci. 2010;4(1):3-8. 39. Wenz B. Microaggregate blood filtration and the febrile transfusion reaction. A comparative study. Transfusion. 1983;23(2):95-98. 40. Blumberg N, Heal JM, Gettings KF, et al. An association between decreased cardiopulmonary complications (transfusion-related acute lung injury and transfusion-associated circulatory overload) and implementation of universal leukoreduction of blood transfusions. Transfusion. 2010;50(12):2738-2744. 41. Silliman CC, Paterson AJ, Dickey WO, et al. The association of biologically active lipids with the development of transfusion-related acute lung injury: a retrospective study. Transfusion. 1997;37(7):719-726. 42. Trial to Reduce Alloimmunization to Platelets Study Group. Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. N Engl J Med. 1997;337(26):1861-1869. 43. Bowden RA, Slichter SJ, Sayers M, et al. A comparison of filtered leukocyte reduced and cytomegalovirus (CMV) seronegative blood products for the prevention of transfusion-associated CMV infection after marrow transplant. Blood. 1995;86(9):3598-3603. 44. Miller WJ, McCullough J, Balfour HH, et al. Prevention of cytomegalovirus infection following bone marrow transplantation: a randomized trial of blood product screening. Bone Marrow Transplant. 1991;7(3):227-234. 45. Gilbert GL, Hudson IL, Hayes JJ. Prevention of transfusion-acquired cytomegalovirus infection in infants by blood filtration to remove leucocytes. Lancet. 1989;1(8649):1228-1231. 46. Hillyer CD, Emmens RK, Zago-Novaretti M, et al. Methods for the reduction of transfusion-transmitted cytomegalovirus infection: filtration versus the use of seronegative donor units. Transfusion. 1994;34(10):929-934. 47. McCullough JJ. Clinical uses of blood components. In: McCullough JJ. Transfusion Medicine. 3rd ed. Chichester, West Sussex, UK: Wiley-Blackwell; 2011. 48. Thaler M, Shamiss A, Orgad S, et al. The role of blood from HLA-homozygous donors in fatal transfusion-associated graft-versus-host disease after open-heart surgery. N Engl J Med. 1989;321(1):25-28. 49. Juji T, Takahashi K, Shibata Y. HLA-homozygous donors and transfusion-associated graft-versus-host disease. N Engl J Med. 1990;332(14):1004-1007. 50. O’Shaughnessy DF, Atterbury C, Bolton Maggs P; British Committee for Standards in Haematology, Blood Transfusion Task Force. Guidelines for the use of fresh-frozen plasma, cryoprecipitate and cryosupernatant. Br J Haematol. 2004;126(1):11-28. 51. Fresh-Frozen Plasma, Cryoprecipitate, and Platelets Administration Practice Guidelines Development Task Force of the College of American Pathologists. Practice parameter for the use of fresh-frozen plasma, cryoprecipitate, and platelets. JAMA. 1994;271(10):777-781. 52. Cohn CS, Stubbs J, Schwartz J, et al. A comparison of adverse reaction rates for PAS C versus plasma platelet units. Transfusion. 2014;54(8):1927-1934. 53. Goodnough LT, Levy JH, Murphy MF. Concepts of blood transfusion in adults. Lancet. 2013;381(9880):1845-1854. 54. Carlson KS, DeSancho MT. Hematological issues in critically ill patients with cancer. Crit Care Clin. 2010;26(1):107-132. 55. Stanworth S. The evidence based use of FFP and cryoprecipitate for abnormalities of coagulation tests and clinical coagulopathy. Hematology Am Soc Hematol Educ Program. 2007;179-186. 56. McCullough J. Pathogen inactivation: a new paradigm for blood safety. Transfusion. 2007;47(12):2180-2184. 57. Jilma-Stohlawetz P, Kursten FW, Horvath M, et al. Recovery, safety, and tolerability of a solvent/detergent-treated and prion-safeguarded transfusion plasma in a randomized, crossover, clinical trial in healthy volunteers. Transfusion. 2013;53(9):1906-1917. 58. Alavi JB, Roat RK, Djerassi I, et al. A randomized clinical trial of granulocyte transfusions for infection of acute leukemia. N Engl J Med. 1977;296(13):706-711. 59. Graw RH Jr, Herzig G, Perry S, et al. Normal granulocyte transfusion therapy. Treatment of septicemia due to gram-negative bacteria. N Engl J Med. 1972;287(8):367-371. Cancer Control 45 60. Herzig GP, Graw RG Jr. Granulocyte transfusions for bacterial infections. In: Brown EB, ed. Progress in Hematology. Vol 9. New York: Grune & Stratton; 1975:207. 61. Strauss RG. Granulocyte (neutrophil) transfusion therapy. In: Mintz PD, ed. Transfusion Therapy: Clinical Principles and Practice. 3rd ed. Bethesda, MD: AABB; 2010. 62. Strauss RG. Therapeutic neutrophil transfusions: are controlled studies no longer appropriate? Am J Med. 1978;65(6):1001-1006. 63. Liles WC, Juang JE, Llewellyn C, et al. A comparative trial of granulocyte-colony-stimulating factor and dexamethasone, separately and in combination, for the mobilization of neutrophils in the peripheral blood of normal volunteers. Transfusion. 1997;37(2):182-187. 64. Dale DC, Liles WC, Llewellyn C, et al. Neutrophil transfusions: kinetics and functions of neutrophils mobilized with granulocyte colony-stimulating factor (G-CSF) and dexamethasone. Transfusion. 1998;38(8):713-721. 65. Rutella S, Pierelli L, Piccirillo N, et al. Efficacy of granulocyte transfusions for neutropenia-related infections: retrospective analysis of predictive factors. Cytotherapy. 2003;5(1):19-30. 66. Price TH, Bowden RA, Boeckh M, et al. Phase I/II trial of neutrophil transfusions from donors stimulated with G-CSF and dexamethasone for treatment of patients with infections in hematopoietic stem cell transplantation. Blood. 2000;95(11):3302-3309. 67. Nichols WG, Strauss R, Ambruso D, et al. G-CSF stimulated granulocyte transfusions from unrelated community donors for severe infections during neutropenia: a phase II multi-center trial of feasibility and efficacy. Blood. 2003;102(11):978a. 68. Price TH, McCullough J, Ness P, et al. A randomized controlled trial on the efficacy of high-dose granulocyte transfusion therapy in neutropenic patients with infection. Paper presented at: 56th ASH Annual Meeting and Exposition; San Francisco, CA; December 6–9, 2014. https://ash.confex.com/ ash/2014/webprogram/Paper68775.html. Accessed November 13, 2014. 69. McCullough J, Weiblen BJ, Fine D. Effects of storage of granulocytes on their fate in vivo. Transfusion. 1983;23(1):20-24. 70. Glasser L. Effect of storage on normal neutrophils collected by discontinuous-flow centrifugation leukapheresis. Blood. 1977;50(6):1145-1150. 71. McCullough J. Liquid preservation of granulocytes. Transfusion. 1989;20(2):129-137. 72. Walter RB, Lee SJ, Gardner KM, et al. Outpatient management following induction chemotherapy for myelodysplastic syndromes and acute myeloid leukemia: a pilot study. Haematologica. 2011;96(6):914-917. 73. Girmenia C, Alimena G, Latagliata R, et al. Out-patient management of acute myeloid leukemia after consolidation chemotherapy. Role of a hematologic emergency unit. Haematologica. 1999;84(9):814-819. 74. Schrijvers D. Management of anemia in cancer patients: transfusions. Oncologist. 2011;16(suppl 3):12-18. 75. Crémieux PY, Barrett B, Anderson K, et al. Cost of outpatient blood transfusions in cancer patients. J Clin Oncol. 2000;18(14):2755-2761. 76. Slichter J, Kaufman R, Assmann SF, et al. Dose of prophylactic platelet transfusions and prevention of hemorrhage. N Engl J Med. 2010;362(17):600-613. 77. Goodnough LT, Kuter DJ, McCullough J, et al. Prophylactic platelet transfusions from healthy normal apheresis platelet donors undergoing treatment with thrombopoietin. Blood. 2001;98(5):1346-1351. 46 Cancer Control January 2015, Vol. 22, No. 1 Platelet transfusion has a well-defined role in treating patients with cancer. Ray Paul. Flowers for Phoebe, 2010. Acrylic, latex, enamel on canvas, 48" × 48". Private collection. Platelet Transfusion for Patients With Cancer Craig H. Fletcher, MD, Melkon G. DomBourian, MD, and Peter A. Millward, MD Background: Platelet transfusion is a critical and often necessary aspect of managing cancer. Low platelet counts frequently lead to bleeding complications; however, the drugs used to combat malignancy commonly lead to decreased production and destruction of the very cell whose function is essential to stop bleeding. The transfusion of allogeneic platelet products helps to promote hemostasis, but alloimmunization may make it difficult to manage other complications associated with cancer. Methods: The literature relating to platelet transfusion in patients with cancer was reviewed. Results: Platelet storage, dosing, transfusion indications, and transfusion response are essential topics for health care professionals to understand because many patients with cancer will require platelet transfusions during the course of treatment. The workup and differentiation of non–immune-mediated compared with immune-mediated platelet refractoriness are vital because platelet management is different between types of refractoriness. Conclusions: A combination of appropriate utilization of platelet inventory and laboratory testing coupled with communication between those caring for patients with cancer and those providing blood products is essential for effective patient care. Introduction Platelets are discoid anucleate cells that measure 3 to 5 µm at their greatest diameter. They are derived from megakaryocytes in the bone marrow and contain ABO antigens on their surface. Platelets are an essential component of hemostasis because they are From the Department of Clinical Pathology, Blood Bank and Transfusion Medicine, Beaumont Health, Royal Oak, Michigan. Address correspondence to Craig H. Fletcher, MD, Beaumont Health, 3601 West 13 Mile Road, Royal Oak, MI 48073. E-mail: Craig.Fletcher@ Beaumont.edu Submitted November 7, 2014; accepted November 14, 2014. No significant relationships exist between the authors and the companies/organizations whose products or services may be referenced in this article. The authors would like to thank the staff and their clinical colleagues at Beaumont Health for their ongoing support and teamwork. January 2015, Vol. 22, No. 1 responsible for forming a platelet plug, providing a framework for the formation of fibrin clots, and secreting cytokines and growth factors.1 Platelets express A and B red blood cell antigens, class I human leukocyte antigen (HLA), and platelet-specific antigens (eg, human platelet antigen [HPA]) on their surface.2,3 Platelets are available from 2 sources based on the method in which they are collected: apheresis platelets and whole blood–derived platelets. Apheresis platelets are obtained via an apheresis collection device from a single donor. Oftentimes, 2 or 3 apheresis platelet units can be acquired during this single collection event; each of these units is considered 1 adult dose. Whole blood–derived platelets are acquired from the platelet concentrate portion of a whole blood donation. Routinely, 4 to 6 platelet concentrates are pooled together to obtain a typical dose.4 Cancer Control 47 Both apheresis platelet and pooled whole blood– derived platelet units must contain a minimum of 3 × 1011 platelets per bag. These 2 products have similar clinical effects and can be interchangeably used.2,5 The leukoreduction of platelets provides several benefits, including the reduction of (1) the platelet alloimmunization rate, (2) cytomegalovirus transmission due to transfusion, and (3) febrile nonhemolytic transfusion reactions.2 Storage and Dosing Platelets are stored in the blood bank at room temperature (68–75°F [20–24°C]) on a platelet rotator to facilitate the exchange of oxygen. Primarily due to their risk of bacterial contamination (approximate risk: 1 per 1,000 units), platelets have a shelf life of 5 days; the day of collection is considered day 0.6 Volunteers who donate blood are tested for HIV, hepatitis B and C, and West Nile virus infections, and blood collection facilities must also screen all platelet products for bacteria,7,8 either via bacterial cultures or assessing bacterial growth by oxygen consumption measurement.1 One dose of platelets should increase the platelet count of an average-sized adult by 35,000 to 40,000/µL,9 and this increment can be measured with a post-transfusion platelet count or complete blood count. In adult patients, platelets are dosed in units. Dosing of platelets for pediatric patients may be done based on body weight (typical pediatric platelet dose, 5–10 mL/kg).5 Indications for Transfusion A platelet transfusion may be indicated for either a quantitative defect (thrombocytopenia) or a qualitative defect (dysfunctional platelets). The normal range for a platelet count is approximately 150,000 to 450,000/µL; however, the platelet count is but one aspect in determining a patient’s risk for bleeding. Many etiologies of thrombocytopenia exist in patients with cancer. The patient’s disease may directly cause thrombocytopenia via tumor involvement of the bone marrow, spleen, or both. Although myeloablative chemotherapeutic regimens may cause prolonged thrombocytopenias, nonmyeloablative chemotherapy produces variable degrees of thrombocytopenia based on drug selection, drug dosage, and number of cycles administered. Patients with cancer can develop microangiopathic conditions that may lead to platelet destruction, including disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, and vasculitis. Immune thrombocytopenia has been associated with patients with lymphoproliferative malignancies.10 Commonly used antibiotics, such as penicillins and cephalosporins, may also cause thrombocytopenia via an im48 Cancer Control mune-mediated, drug-induced mechanism known as hapten-dependent antibody formation.11 The AABB (formerly American Association of Blood Banks) recommends the following prophylactic platelet transfusion triggers: less than 10,000/µL in adult inpatients with therapy-induced hypoproliferative thrombocytopenia, less than 20,000/µL for central venous catheter placement, and less than 50,000/µL for either diagnostic lumbar puncture or major elective non-neuraxial surgery.12 Other common platelet transfusion triggers include less than 10,000/µL for stable, nonbleeding patients and less than 20,000/µL for febrile patients. A trigger of 100,000/µL is often used for neurosurgical patients or those patients experiencing ophthalmological bleeding.2,9 Despite these AABB recommendations, debate continues about the rationale, efficacy, and threshold of prophylactic platelet transfusions in patients with cancer. For example, Stanworth et al13 suggested that the effectiveness of prophylactic platelet transfusions may vary between specific groups of patients with cancer. When comparing chemotherapy and allogeneic hematopoietic stem cell transplantation among patients receiving prophylactic platelet transfusion versus not receiving prophylactic dosing, they reported decreased bleeding events in the prophylactic platelet transfusion group. However, when comparing patients who received autologous hematopoietic stem cell transplantation, the researchers saw no difference in bleeding events between the prophylactic and nonprophylactic platelet transfusion groups.13 Moreover, Schiffer14 emphasized the need for studies to evaluate platelet prophylaxis in patients with acute leukemia and protracted thrombocytopenia due to induction chemotherapy. Platelet Dysfunction Platelets become dysfunctional for many causes, including medication and herbal supplement use, renal failure (uremia), and genetic abnormalities (eg, Glanzmann thrombasthenia, Bernard–Soulier syndrome). In addition, the membranes used in cardiopulmonary bypass circuits and extracorporeal membrane oxygenation circuits can also cause platelet dysfunction.15 The most common medication to cause platelet dysfunction is aspirin, which irreversibly inhibits the enzyme cyclooxygenase I.15 A myriad of other medications inhibit platelet function, including nonspecific nonsteroidal anti-inflammatory drugs, adenosine diphosphate receptor inhibitors, adenosine reuptake inhibitors, glycoprotein IIB/IIIA inhibitors, thromboxane inhibitors, and β-lactam antibiotics.15 Response to Platelet Transfusion Several aspects of the platelet product can affect a patient’s response to transfusion, including dose of platelets received, type of product (apheresis or whole January 2015, Vol. 22, No. 1 blood–derived), ABO compatibility, and storage duration of the product. An analysis of the data collected by Triulzi et al16 demonstrated that post-transfusion platelet increments were higher in hematology/oncology patients who received (1) apheresis platelets, (2) ABO-identical platelets, and (3) fresher platelets (stored 3 days compared with 4 or 5 days).16 However, despite the differences in these 3 categories, none of these factors were predictive of World Health Organization grade 2 or higher bleeding.16 Many patient-specific factors also play a role in the response to platelet transfusion. In an analysis of data obtained from the Trial to Reduce Alloimmunization to Platelets (TRAP), Slichter et al17 concluded that the status of a patient’s spleen had the greatest impact on the post-transfusion platelet increment. Splenectomized patients had the highest increments, whereas patients with a palpable spleen had both lower platelet increments and a shorter time to their next platelet transfusion. Other factors that led to a lower platelet increment included increased weight and height, administration of amphotericin or heparin, bleeding, fever, and infection.17 One unit of platelets should increase the platelet count by 35,000 to 40,000/µL as measured within 1 hour following the transfusion. Two calculations exist to quantify the patient’s response to platelet transfusion: the corrected count increment (CCI) and the percent platelet recovery (PPR). The CCI incorporates the patient’s body surface area to evaluate response to platelet transfusion. An acceptable CCI is generally higher than 5,000/µL.18 The mathematical formula for calculating the CCI is: By contrast to the CCI calculation, the PPR utilizes the patient’s total blood volume instead of body (post-transfusion platelet count/µL – pretransfusion platelet count/µL) × body surface area (m2) CCI = ––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––– No. of platelets transfused × 1011 surface area to evaluate a patient’s response to platelet transfusion. An acceptable PPR is generally higher than 20%.19 The mathematical formula for calculating the PPR is: Platelet Refractoriness (post-transfusion platelet count/µL – pretransfusion platelet count/µL) × total blood volume × 100% PPR = –––––––––––––––––––––––––––––––––––––––––––––––––––––––––––– No. of platelets transfused × 1011 Platelet refractoriness can be defined as the failure to achieve a 1-hour post-transfusion platelet increment of 11,000/µL on 2 consecutive transfusions.17 Because transfusing ABO-incompatible platelets may also negatively impact the post-transfusion platelet January 2015, Vol. 22, No. 1 increment, many institutions require this failure to be with ABO-identical or ABO-compatible platelets. Platelet refractoriness can be broken down into 2 broad categories: non–immune-mediated and immune-mediated. Non–immune-mediated platelet refractoriness can be due to splenomegaly, sepsis, fever, medications, and active bleeding. Two-thirds of cases of platelet refractoriness are estimated to be nonimmune in nature and another one-fifth comprise both nonimmune and immune causes.20 Immune-mediated platelet refractoriness is due to alloantibody formation against the HLA system, the HPA system, or both. Risk factors for alloimmunization to these systems include prior transfusion, pregnancy, and transplantation.20 The 1-hour post-transfusion platelet increment helps to identify patients with platelet refractoriness, and it is also a key differentiating factor between the majority of nonimmune and immune causes. Although patients with nonimmune causes of refractoriness will typically show some platelet increment within 1 hour following the transfusion (likely a minimum increment of < 35,000/µL seen in nonrefractory patients), patients with immune-mediated refractoriness often do not demonstrate such an increment. One caveat to this differentiating factor is in patients with splenomegaly; in these patients, up to 90% of the total body platelet mass may be sequestered by the spleen.21 Nevertheless, an appropriate platelet transfusion strategy can still be pursued. For a nonimmune refractory patient, the underlying disease processes should be treated and more platelets should be transfused; by contrast, in an immune refractory patient, more appropriate platelets should be obtained. HLA-Matched and Crossmatched Platelets Transfusing patients who are refractory to platelets with anti-HLA or anti-HPA antibodies centers on identifying the antibody specificity and procuring antigen-negative platelets. If a screening test for anti-HLA antibodies is positive, then the first step in providing HLA-matched platelets is to determine the HLA type of the patient. The specificity of the anti-HLA antibodies may also be determined at this stage in the evaluation. The blood bank will then communicate with the blood supplier to obtain HLA-matched platelets (matching the HLA class I antigens, specifically the A and B loci). Some blood suppliers have databases of the HLA class I antigen types of apheresis platelet donors to facilitate the identification of potential donor–patient matches.22 Two strategies exist for the procurement of HLA-matched platelets. The first method involves identifying donors who are a high-grade match (grade A or B) with the HLA type of the patient. The second method, known as antibody specificity prediction, utilizes the antibody specificity to obCancer Control 49 tain HLA antigen–negative platelets. These 2 strategies are specific to each blood supplier and, thus, vary by region. The process to obtain HLA-matched platelets may take several days or even weeks based on the HLA type of the patient, the specificity of antibodies, and donor availability. The process to obtain HPA-matched platelets is similar to the process used to obtain HLA-matched platelets. If a screening test for anti-HPA antibodies is positive, then both the patient’s HPA type and the antibody specificity should be determined. The process to obtain HPA-matched platelets follows the same approach as obtaining HLA-matched platelets. An alternative to obtaining either HLA- or HPA-matched platelets for alloimmunized patients involves the use of crossmatched platelets. Typically, platelet crossmatching is performed at blood centers via solid-phase red cell adherence assay to assess the compatibility between the serum of the patient and the platelets of the donor (Wendy Enting, LPN, oral communication, November 2014). Crossmatch-compatible platelets are presumed to lack the antigen(s) to which the patient has formed antibodies. Benefits to platelet crossmatching include rapid turnaround time (hours), simultaneous screening of multiple platelet units, and the ability to obtain platelets without having to perform HLA/HPA typing on the patient and donor.23 In a systematic literature review, Vassallo et al24 found that while platelet crossmatching did not greatly improve failure rates (typically 20%–30% using HLA-matched platelets25) in alloimmunized refractory patients, platelet crossmatching did improve the availability of platelets for these patients. In an observational study of 114 patients who received 1,621 platelet transfusions, Petz et al26 concluded that all 3 methods for the selection of platelets for alloimmunized patients (HLA-matched, crossmatched, and antibody specificity prediction) were equally effective as measured by the PPR. Laboratory Assays Many different assays may be utilized to identify the presence and specificity of HLA and HPA antibodies. The various methodologies include lymphocytotoxicity testing, platelet immunofluorescence testing, lymphocyte immunofluorescence testing, enzyme-linked immunosorbent assays (ELISAs), antigen-capture ELISAs, monoclonal antibody-specific immobilization of platelet antigens, and flow cytometric assays.20 Each methodology has a unique set of benefits and drawbacks. In addition to variations in sensitivity and specificity rates, other factors that the health care professional must consider include assay complexity, turnaround time for obtaining results, the reproducibility of results, and concordance 50 Cancer Control rates across different platforms.27 The nuances and selection of these assays are determined by blood suppliers, reference laboratories, or both. Mitigating Platelet Transfusions The prevention of alloimmunization plays an important role in improving patient care and reducing the number of platelets transfused. Both leukoreduction and ultraviolet B irradiation were demonstrated in the TRAP study to be equally effective in preventing antibody-mediated platelet refractoriness during chemotherapy for acute myeloid leukemia.18 A total of 17% of study volunteers who received leukoreduced platelets became alloimmunized compared with 45% of those who received nonleukoreduced platelets.18 All platelets for patients with cancer should be leukoreduced; currently, nearly all platelets collected today are leukoreduced by blood suppliers. Many strategies have been proposed to mitigate the need for platelet transfusions, particularly in refractory patients.28 Medications such as antifibrinolytics (ε-aminocaproic acid), intravenous immunoglobulin, Rhesus immune globulin, and cyclosporine A have been used with varied success in small studies, as have other modalities such as plasma exchange, immunoadsorption, and massive platelet transfusion.28 Thrombopoietin receptor agonists (eg, eltrombopag, romiplostim) designed to increase platelet production have shown some effectiveness for the treatment of thrombocytopenia in patients with immune thrombocytopenia and chronic hepatitis C.29 Currently, clinical trials to expand these indications to treat thrombocytopenia in patients with cancer are ongoing (eltrombopag trials: NCT01656252, NCT02093325, NCT01147809, NCT01488565; romiplostim trials: NCT00299182, NCT02052882). Conclusions Platelet transfusion has a well-defined role in the treatment of patients with cancer. By understanding the requirements of storage, dosing, indications, and responses to platelet transfusion, health care professionals can provide the most appropriate care for their patients. Further knowledge of platelet refractoriness and how to care for refractory patients will enhance the proper utilization of laboratory testing and the allocation of scarce resources. References 1. American Association of Blood Banks (AABB). Circular of Information For the Use of Human Blood and Blood Components. Bethesda, MD: AABB; 2013. http://www.aabb.org/tm/coi/Documents/coi1113.pdf. Accessed November 16, 2014. 2. Slichter SJ. Evidence-based platelet transfusion guidelines. ASH Education Book. 2007;2007(1):172-178. 3. Yankee RA, Grumet FC, Rogentine GN. The selection of compatible platelet donors for refractory patients by lymphocyte HL-A typing. N Engl J Med. 1969;281(22):1208-1212. 4. Dumont LJ, Papari M, Aronson CA, et al. Whole-blood collection and January 2015, Vol. 22, No. 1 component processing. In: Fung MK, Grossman BJ, Hillyer CD, et al, eds. Technical Manual. 18th ed. Bethesda, MD: AABB; 2014:156. 5. Millward PA, Brecher ME. Therapeutic use of blood products. In: Bope ET, Rakel RE, Kellerman R, eds. Conn’s Current Therapy 2010. Philadelphia, PA: Saunders Elsevier; 2010:486-487. 6. Corash L. Bacterial contamination of platelet components: potential solutions to prevent transfusion-related sepsis. Expert Rev Hematol. 2011;4(5):509-525. 7. US Food and Drug Administration. 21 C.F.R. § 610.40. http://www. accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=610.40. Accessed November 16, 2014. 8. US Department of Health and Human Services; US Food and Drug Administration (FDA); Center for Biologics Evaluation and Research. Guidance for Industry and FDA Review Staff: Collection of Platelets by Automated Methods. Bethesda, MD: FDA. http://www.fda.gov/downloads/BiologicsBloodVaccines/ GuidanceComplianceRegulatoryInformation/Guidances/Blood/ucm062946. pdf. Accessed November 16, 2014. 9. McCullough J. Overview of platelet transfusion. Semin Hematol. 2010;47(3):235-242. 10. Liebman HA. Thrombocytopenia in cancer patients. Thromb Res. 2014;133(suppl 2):S63-S69. 11. Kenney B, Stack G. Drug-induced thrombocytopenia. Arch Pathol Lab Med. 2009;133(2):309-314. 12. Kaufman RM, Djulbegovic B, Gernsheimer T, et al. Platelet transfusion: a clinical practice guideline from the AABB. Ann Intern Med. 2014. [Epub ahead of print]. 13. Stanworth SJ, Estcourt LJ, Llewelyn CA, et al. Impact of prophylactic platelet transfusions on bleeding events in patients with hematologic malignancies: a subgroup analysis of a randomized trial. Transfusion. 2014;54(10):2385-2393. 14. Schiffer CA. They took a mulligan and mostly got it right…the issue of prophylactic platelet transfusion for patients receiving autologous stem cell transplantation. Transfusion. 2014;54(10):2372-2374. 15. Konkle BA. Acquired disorders of platelet function. Hematology Am Soc Hematol Educ Program. 2011;2011(1): 391-396. 16. Triulzi DJ, Assmann SF, Strauss RG, et al. The impact of platelet transfusion characteristics on posttransfusion platelet increments and clinical bleeding in patients with hypoproliferative thrombocytopenia. Blood. 2012;119(23):5553-5562. 17. Slichter SJ, Davis K, Enright H, et al. Factors affecting posttransfusion platelet increments, platelet refractoriness, and platelet transfusion intervals in thrombocytopenic patients. Blood. 2005;105(10):4106-4114. 18. The Trial to Reduce Alloimmunization to Platelets Study Group. Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. N Engl J Med. 1997;337(26):1861-1869. 19. Davis KB, Slichter SJ, Corash L. Corrected count increment and percent platelet recovery as measures of posttransfusion platelet response: problems and a solution. Transfusion. 1999;39(6):586-592. 20. Hod E, Schwartz J. Platelet transfusion refractoriness. Br J Haematol. 2008;142(3):348-360. 21. Aster RH. Pooling of platelets in the spleen: role in the pathogenesis of “hypersplenic” thrombocytopenia. J Clin Invest. 1966;45(5):645-657. 22. Bolgiano DC, Larson E, Slichter SJ. A model to determine required pool size for HLA-typed community donor apheresis programs. Transfusion. 1989;29(4):306-310. 23. Salama OS, Aladl DA, El Ghannam DM, et al. Evaluation of platelet cross-matching in the management of patients refractory to platelet transfusions. Blood Transfus. 2014;12(2):187-194. 24. Vassallo RR, Fung M, Rebulla P, et al; International Collaboration for Guideline Development, Implementation and Evaluation for Transfusion Therapies. Utility of cross-matched platelet transfusions in patient with hypoproliferative thrombocytopenia: a systematic review. Transfusion. 2014;54(4):1180-1191. 25. Slichter SJ. Platelet crossmatch testing for donor selection. Prog Clin Biol Res. 1982;88:153-164. 26. Petz LD, Garratty G, Calhoun L, et al. Selecting donors of platelets for refractory patients on the basis of HLA antibody specificity. Transfusion. 2000;40(12):1446-1456. 27.Fontão-Wendel R, Silva LC, Saviolo CBR, et al. Incidence of transfusion-induced platelet-reactive antibodies evaluated by specific assays for the detection of human leucocyte antigen and human platelet antigen antibodies. Vox Sanguinis. 2007;93(3):241-249. 28. Delaflor-Weiss E, Mintz PD. The evaluation and management of platelet refractoriness and alloimmunization. Transfus Med Rev. 2000;14(2):180-196. 29. Desmond R, Townsley DM, Dumitriu B, et al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood. 2014;123(12):1818-1825. January 2015, Vol. 22, No. 1 Cancer Control 51 Knowledge of transfusion complications related to HSCT can help with the early detection and treatment of patients before and after transplantation. Ray Paul. SP12-6796 × 40, 2013. Acrylic, latex, enamel on canvas printed with an image of myxofibrosarcoma with metastases to the artist’s lung, 26" × 36". Transfusion Support Issues in Hematopoietic Stem Cell Transplantation Claudia S. Cohn, MD, PhD Background: Patients receiving hematopoietic stem cell transplantation require extensive transfusion support until red blood cell and platelet engraftment occurs. Rare but predictable complications may arise when the transplanted stem cells are incompatible with the native ABO type of the patient. Immediate and delayed hemolysis is often seen. Methods: A literature review was performed and the results from peer-reviewed papers that contained reproducible findings were integrated. Results: A strong body of clinical evidence has developed around the common complications experienced with ABO-incompatible hematopoietic stem cell transplantation. These complications are discussed and the underlying pathophysiology is explained. General treatment options and guidelines are enumerated. Conclusions: ABO-incompatible hematopoietic stem cell transplantations are frequently performed. Immune-related hemolysis is a commonly encountered complication; therefore, health care professionals must recognize the signs of immune-mediated hemolysis and understand the various etiologies that may drive the process. Introduction Hematopoietic stem cell transplantation (HSCT) is used to treat a variety of hematological and congenital diseases. The duration and specificity of transfusion support for patients receiving HSCT depends on the disease, the source of the stem cells, the preparative regimen applied prior to transplantation, and patient factors during the post-transplantation recovery period. Human leukocyte From the Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota. Submitted March 27, 2014; accepted July 23, 2014. Address correspondence to Claudia S. Cohn, MD, PhD, D242 Mayo Memorial Building, MMC 609, 420 Delaware Street, South East, Minneapolis, MN 55455. E-mail: [email protected] Dr Cohn has received grants or research support funds and honorarium from Fenwal and Fresenius Kabi. 52 Cancer Control antigen (HLA) matching remains an important predictor of success with HSCT; however, the ABO barrier is often crossed when searching for the most appropriate HLA match between donor and patient. Crossing the ABO barrier has little or no effect on overall outcomes; however, complications can arise due to antigenic incompatibility between the transplanted cells and the patient.1 This review will discuss the transfusion support of patients receiving HSCT, common transfusion-related complications that health care professionals will likely encounter, and the measures required to safely deliver blood components. HSCTs can be broadly divided into related allogeneic, unrelated allogeneic, and autologous transplantation. Hematopoietic progenitor cells (HPCs) for allogeneic transplantation come from 3 sources: apheresis-derived, mobilized peripheral blood proJanuary 2015, Vol. 22, No. 1 genitor cells (HPC-A), bone marrow (HPC-M), and umbilical cord (HPC-C). HPC-A is commonly used for autologous transplantation. Pediatric patients receive more HPC-M, whereas adults receive more HPC-A. Use of HPC-C is on the rise in both populations.2 Patients undergoing HSCT remain dependent on red blood cell (RBC) and platelet transfusions until engraftment of these cell lines occurs. The platelet line is considered engrafted when a patient’s count is at least 20,000/µL after 3 consecutive days without platelet transfusion.3 RBC engraftment is more difficult to assess and may be defined by the appearance of 1% reticulocytes in the peripheral blood,4 or, on the day of the last RBC transfusion, with no transfusion given the following 30 days.5 Typically, neutrophil engraftment is defined as an absolute neutrophil count of more than 500/µL across 3 consecutive days.3 Engraftment is influenced by many factors, including the relationship of the donor to the patient, the stem cell source, and the dose of CD34+ cells in the transplantation.3 In general, engraftment time is shortest with HPC-A and greatest when HPC-C is used6; however, considerable patient-to-patient variability exists. One study that compared HPC-C and HPC-A noted roughly equivalent neutrophil recovery times but found a longer time to platelet and RBC engraftment for HPC-C.5 These prolonged engraftment times translated into higher transfusion rates for RBCs and platelets. Pretransplantation Support Prior to HSCT, patients may be immunocompetent or immunocompromised depending on their underlying disease. A patient who is immunocompetent (eg, aplastic anemia, hemoglobinopathies) is capable of mounting an immune response to transfusions, leading to alloimmunization against platelet antigens, HLAs present on the surface of leukocytes and platelets, or both. Antibodies against HLA contribute to delayed engraftment and graft rejection in some patient populations.7,8 As a result, pretransplantation transfusions in patients who are immunocompetent should be avoided because they are associated with increased graft failure rates.9,10 For patients who are stable, RBC transfusions can be minimized by using a hemoglobin trigger of 7 to 8 g/dL. Multiple studies have shown that using this hemoglobin threshold is at least as effective and results in similar outcomes as higher triggers unless a patient is symptomatically anemic.11,12 Platelet transfusions may also be minimized by adhering to evidence-based guidelines. Using a platelet count of 10,000/µL as a threshold for administering platelets to a nonbleeding patient has been well established.13 When transfusion is required, using leukoreduced components reduces the risk of alloimmunization.14 Patients who are immunocompromised, either beJanuary 2015, Vol. 22, No. 1 cause of their disease or due to chemotherapy, are less likely to become sensitized to foreign antigens. Nonetheless, using leukoreduced products to minimize the risk of alloimmunization is recommended. Extra care must also be taken if the stem cell donation comes from a blood relative. In this situation, family members should not give direct blood donations because doing so may lead to alloimmunization against major and/or minor HLAs present in the transplant.15 Post-Transplantation Support Chemotherapy regimens may be fully myeloablative or use reduced intensity conditioning to partially ablate the patient’s marrow. Either regimen will cause the patient to be dependent on RBC and platelet transfusions until engraftment of those cell lines occurs. Although granulocyte progenitor cells are also destroyed, granulocyte-colony stimulating factor may be given because granulocyte transfusions are reserved for specific scenarios. The need for plasma and cryoprecipitate transfusions is less frequent because HSCT does not typically interfere with the production of coagulation factors. Refer to the article by McCullough and colleagues in this issue for more detailed information on this topic. Although the frequency and extent of RBC and platelet transfusions are increased during the post-transplantation period, the indications for these transfusions do not change. Because no large prospective study specifically targets RBC transfusion triggers in patients undergoing HSCT, the more general guidelines from the AABB (formerly American Association of Blood Banks) may be used, which recommend adhering to a restrictive transfusion strategy (7.0–8.0 g/dL) in a stable patient who is hospitalized unless the patient is symptomatically anemic.16 Special care must be taken to transfuse irradiated RBC units alone, because the risk for transfusion-associated graft-vs-host-disease (TA-GVHD) is high in patients receiving HSCT.17 Because transplants often cross the ABO barrier, ABO compatibility may be complex in patients receiving HSCT. When the transplant creates an incompatibility issue (eg, group B transplantation into a group A patient), transfusing group O RBCs and AB plasma will be necessary (Table). The decision to switch a patient’s blood type is highly variable across institutions. At my institution, if a patient is independent of RBC transfusion for 100 days and no incompatible isohemagglutinins against the new RBC phenotype can be detected in 2 consecutive blood samples, then the patient’s native blood type is switched to the donor type for future transfusions. Patients receiving HSCT undergo a period of hypoproliferative thrombocytopenia that ends when the platelet line engrafts. To support patients through this Cancer Control 53 Table. — Component Type Selection for Hematopoietic Stem Cell Transplantation Crossing the ABO Barrier Type of Mismatch Major Minor Bidirectional Transplantation Donor Type Transfusion Recipient Type Red Blood Cell Plateletsa /Plasma A O O A AB B O O B AB AB O O AB AB A A O AB AB B B O AB O A O A AB O B O B AB O AB O AB A AB A O AB B AB B O AB A B O AB B A O AB Platelets stored in additive solution reduce the volume of incompatible plasma transfused. a aplasia, the general triggers for platelet transfusions apply. Because the patient is a chimera of donor and native blood types, ABO-incompatibility issues may arise. Because ABO antigens are present on the surface of platelets, one must consider the ABO type of the platelets and the anti-A and anti-B antibodies present in the donor plasma in which the platelets are stored. Choosing platelet units based on plasma compatible with both the donor and patient is necessary (see Table).18 However, if a group O patient is making high-titer ABO antibodies (> 512–1024), then attention must be paid to the ABO type of the platelet unit and care must be taken to avoid group A1 platelets. Using platelet additive solution (PAS), which removes 65% of donor plasma, is an option available when the isohemagglutinin titer is a concern.19 Washing the platelet unit will also reduce or eliminate problems with lysis. However, the procedure is labor intensive and may damage the platelets. Washing can also be logistically challenging because washed platelet units become outdated after 4 hours. Refer to the article in this issue by Fletcher and colleagues for more details on this topic. Recent studies have addressed questions of platelet dose and the comparative merits of therapeutic 54 Cancer Control compared with prophylactic platelet transfusions. One such study evaluated the effect of platelet dose on bleeding in patients with hypoproliferative thrombocytopenia.20 In this study, patients were randomly assigned to receive either low-dose (1.1 × 1011 platelets), medium-dose (2.2 × 1011), or high-dose (4.4 × 1011) prophylactic platelet transfusions when their first morning counts were 10,000/µL or lower. Overall, no significant difference was seen in World Health Organization grade 2 or higher bleeding events in the 3 groups.20 The low-dose group received significantly fewer platelets; however, this group also received transfusions more often. The authors concluded that using these different doses for prophylactic transfusion had no effect on the incidence of bleeding.20 However, a subgroup analysis of the study data showed that pediatric patients (range, 0–18 years of age) had a significantly higher risk of grade 2 or higher bleeding than adults across all platelet dose groups.21 This finding was most pronounced in the pediatric autologous transplant population.21 Two studies examined the efficacy and safety of prophylactic compared with therapeutic platelet transfusions.22,23 The Trial of Prophylactic Platelet Study (TOPPS) randomly assigned patients undergoing chemotherapy or stem cell transplantation into either a therapeutic or prophylactic arm.22 Patients in the prophylactic arm received a platelet transfusion in response to a first morning platelet count of less than 10,000/µL, whereas the therapeutic group received platelet transfusion when clinically indicated. Results showed that the therapeutic arm used significantly fewer platelets when compared with the prophylactic group; however, patients in the therapeutic arm had higher bleeding rates, more days with bleeding, and a shorter time to the initial bleeding episode than patients in the prophylactic cohort.22 It should be noted that 70% of the patients in this study were recipients of autologous stem cell transplants, which represents a group of people who have a lower risk of bleeding than those receiving allogeneic transplantation.20 When recipients of autologous transplantation were compared, the rate of bleeding was similar for both therapeutic and prophylactic groups.22 The second large prospective trial conducted by Wandt et al23 was performed under similar conditions as TOPPS. The researchers studied recipients of autologous stem cell transplantation and patients with acute myeloid leukemia undergoing chemotherapy. In this trial, higher rates of bleeding were seen in all patient groups receiving therapeutic platelet transfusions. In addition, the therapeutic group had 6 patients with head bleeds (2 of the 6 were fatal), whereas the prophylactic group had none.23 As with the TOPPS trial, a significant reduction in platelet transfusions was seen in the therapeutic arm. Of note, January 2015, Vol. 22, No. 1 the similar data in the 2 studies led to different conclusions. The TOPPS group concluded that the benefit of reduced bleeding made prophylactic transfusions a preferred practice for all patients,22 whereas Wandt et al23 made a distinction, stating that patients with acute myeloid leukemia undergoing chemotherapy should still receive prophylactic platelet transfusions but that the therapeutic strategy should become the new standard of care for patients receiving autologous stem cell transplantation. Complications Transfusion-related complications exist that are specific to, or more frequently seen in, the patient population receiving HSCT. Some of these complications arise when lymphocytes within the transplant are activated against the recipient, leading to TA-GVHD and passenger lymphocyte syndrome (PLS). Another complication, pure red cell aplasia (PRCA), occurs when a patient’s residual antibodies attack the transplant. Standard transfusion reactions, such as allergic or febrile nonhemolytic reactions, are frequently seen in this heavily transfused patient population. Refer to the article by Marques and colleagues in this issue for a more detailed discussion of standard transfusion reactions. Transfusion-Associated Graft-vs-Host Disease Graft-vs-host disease is seen in patients who are severely immunocompromised and have been exposed to immunocompetent lymphocytes that recognize the body as foreign due to differences in HLAs. TA-GVHD occurs when a susceptible patient is exposed to viable lymphocytes introduced via blood transfusion. The immunocompromised recipient is incapable of rejecting or mounting an attack against the lymphocytes in the graft. Although the basic underlying etiology is similar, TA-GVHD has a different presentation and natural history when compared with conventional graft-vs-host disease. Typically, TA-GVHD presents with a maculopapular rash, enterocolitis, and pancytopenia that begin 8 to 10 days following transfusion.17 As the attacking lymphocytes target the stem cells engrafting within the bone marrow, irreversible and complete bone marrow aplasia will result. TA-GVHD develops within 21 days of transfusion and is almost always fatal.24,25 Cellular blood components isolated from whole blood or collected by apheresis all contain some lymphocytes. RBC, platelet, and granulocyte units all carry risk for TA-GVHD; however, plasma and cryoprecipitate are acellular and do not pose a risk. To prevent TA-GVHD, lymphocytes within a blood component must be eliminated or disabled. Leukoreduction is not considered sufficient because the process reduces but does not completely eliminate white blood January 2015, Vol. 22, No. 1 cells.26,27 Frozen units may also carry risk because the lymphocytes may survive. Treating components with γ- or X-irradiation, or pathogen inactivation with UV irradiation, has been shown to be effective prophylaxis for TA-GVHD.28 A dose of at least 2500 cGy into the center of a cellular blood component and 1500 cGy throughout the unit leaves lymphocytes intact but unable to proliferate.29 This simple precaution prevents TA-GVHD. Irradiation at the indicated dose appears to damage the RBC membrane.30 The damage does not affect the oxygen-carrying capacity of the erythrocyte but does allow potassium to leak from the cell.31 The level of extracellular potassium has been shown to increase with storage time. As a result, RBCs may be stored for 28 days following irradiation.29 Because platelets are not affected by irradiation, their storage time of 5 days remains unchanged.32 All patients undergoing HSCT should receive irradiated components from the time of initiation of conditioning chemotherapy. The AABB suggests that HSCT recipients receive irradiated components for at least 1 year following transplantation,33 although many centers continue to provide irradiated products for the life of the patient.34 The British Committee for Standards in Haematology (BCSH) also recommends that irradiation begin with the initiation of conditioning chemotherapy; however, separate recommendations exist for patients receiving allogeneic compared with autologous HSCT.34 The BCSH recommends that patients receiving allogeneic HSCT should continue to receive irradiated components for 6 months following transplantation or until the lymphocyte count is greater than 1 × 109/L; however, if chronic graft vs host disease is present, then irradiated products should be indefinitely given.34 The BCSH states that patients receiving autologous HSCT should also receive irradiated components beginning from the time of initiation of conditioning chemotherapy, but this can revert to nonirradiated components 3 months after transplantation.34 If patients receiving autologous HSCT also received total body irradiation, then the BCSH recommends extending the use of irradiated products for 6 months following transplantation.34 Issues of ABO Compatibility Crossing the ABO barrier is not considered a contraindication with HSCT. A meta-analysis found no impact on overall survival rates when comparing ABO matched and mismatched HSCTs.35 Nonetheless, some complications may arise because of issues related to ABO incompatibility. The nature of the complication is often related to whether the incompatibility represents a major or minor mismatch (see Table), with a major mismatch occurring when the transplant contains RBCs incompatible with the plasma of the recipient. Conversely, a minor mismatch is present when plasma Cancer Control 55 from the donor contains isohemagglutinins against the RBCs of the recipient. Bidirectional transplantation (eg, group A transplant into group B recipient) carries both major and minor mismatches. Major ABO Mismatches Immediate and Delayed Hemolysis: When a major ABO mismatched transplantation is provided, immediate hemolysis may occur during the infusion. This complication is commonly seen when the HSCT is derived from bone marrow because more RBCs are present36; however, RBC depletion techniques have helped eliminate this complication.37 Because HSCTs derived from peripheral blood typically contain a minimal volume of RBCs (8–15 mL), clinically significant cases of immediate hemolysis have not been identified.36 Most HPC-C units are RBC-depleted prior to cryopreservation, and the residual erythrocytes lyse during cryopreservation; therefore, immediate hemolysis does not occur with the transplantation of cord blood. Preformed antibodies against non-ABO RBC antigens may remain in a recipient’s peripheral circulation for many weeks following transplantation. These antibodies may cause lysis when engrafted cells begin to produce new RBCs.38,39 In addition, chimeric patients may develop antibodies against ABO or non-ABO RBC antigens, thus resulting in delayed hemolysis.40 Pure Red Cell Aplasia: When recipients have isohemagglutinins specific for the ABO type of the transplant, delayed erythrocyte engraftment and PRCA may ensue. PRCA is seen frequently with group O patients receiving a group A transplantation or with bidirectional mismatches.41 The condition develops when antibodies against newly engrafted RBCs destroy erythrocyte progenitor cells in the bone marrow. This intramedullary destruction leads to severe anemia with no corresponding involvement of leukocyte or platelet cell lines. The incidence of PRCA is increased when reduced intensity conditioning regimens are used,42,43 likely due to residual recipient B lymphocytes, plasma cells, or both, thus producing isohemagglutinins. An increase in post-transplantation isohemagglutinin titers is also an important predisposing factor for PRCA.44-47 PRCA may spontaneously resolve,44,48 but treatment to reduce its duration is warranted to diminish the risk of iron overload from multiple RBC transfusions.49 Therapy for PRCA includes bolstering the graft-vs-host effect either through withdrawal of immunosuppression44 or with a donor infusion of leukocytes.42,50,51 Other treatments include erythropoietin,52 rituximab,53,54 bortezomib, or all 3 options in combination.43,55 Because PRCA is associated with high levels of isohemagglutinins,44-47 a direct reduction of titers by plasma exchange may be effective in some 56 Cancer Control patients.47,56 Although the reduction of titers before the transplantation has been attempted to prevent PRCA,57,58 knowing the actual effect of this approach is impossible. Some European centers use apheresis as standard care for reducing pretransplantation isohemagglutinin titers to fewer than 1:32.59 Minor Mismatches Passenger Lymphocyte Syndrome: If lymphocytes within the HSCT recognize the recipient RBCs as foreign, then antibodies may be produced that are specific for ABO or minor RBC antigens. PLS is seen most frequently in transplants that use a group O donor with a group A recipient,60 and it typically presents 7 to 14 days following transplantation with an abrupt onset of hemolysis. When the passenger lymphocytes produce antibodies against the ABO system, the hemoglobin level may precipitously drop. The laboratory signs of intravascular hemolysis (ie, hemoglobinemia, hemoglobinuria, elevated level of lactate dehydrogenase) should be used to follow the course of disease. In most cases, results on a direct antiglobulin test will be positive unless all antibody-bound cells have already lysed. Hemolysis can persist as long as incompatible RBCs are present, but it typically subsides within 5 to 10 days.61 Antibodies against minor RBC antigens have been less frequently reported. In these cases, hemolysis ranges from mild to severe and may be intravascular or extravascular depending on the nature of the antibody involved. The risk factors for PLS are similar to those seen in PRCA. A non-myeloablative-conditioning regimen carries greater risk than when full ablation is used.62 Because HPC-A preparations carry a greater lymphocyte load when compared with HPC-M and HPC-C collections, recipients of peripheral blood stem cells are at an increased risk for developing PLS.62-65 I am not aware of a PLS case reported with umbilical cord stem cell transplantation. Maintaining graft-vs-host disease prophylaxis with a T-cell inhibitor alone, such as cyclosporine A, without an accompanying B-cell inhibitor is also considered a risk factor.66,67 Alloimmunization Against Minor Red Blood Cell Antigens: The antibodies that cause PLS are temporary because they are derived from passenger lymphocytes that are not engrafted. When alloantibodies against RBCs are produced by the post-transplantation immune system, the antibodies may persist for several years,68 and they may be produced by the engrafted cells of the immune system of the donor69-71 or by the residual cells of the immune system of the recipient.68,72,73 The antibodies produced may be against donor RBCs, residual recipient RBCs, or, in some cases, both. The incidence of alloantibody formation against minor RBC antigens ranges from 2.1% to 3.7% in the published literature.74,75 These antibodies have not January 2015, Vol. 22, No. 1 been linked with significant complications.70,72 Prevention of Transfusion-Transmitted Cytomegalovirus Infection: Cytomegalovirus (CMV) infection continues to be a serious complication following HSCT.76-78 Most CMV infections may be due to a reactivation of the virus from a previous infection rather than due to the acquisition of a new strain.79 However, CMV antibody-negative persons are at risk for developing a transfusion-transmitted de novo CMV infection. To reduce this risk, one may use CMV-antibody negative blood or leukoreduced components. A large controlled trial and meta-analysis showed that leukoreduced components are as effective as antibody-negative components in the prevention of transfusion-transmitted CMV infection.80,81 However, a survey of AABB physician members showed wide variability in transfusion practice.82 Since then, 2 additional studies have been published with results that support the safety of using leukoreduced blood alone for the prevention of transfusion-transmitted CMV infection.83,84 These studies focused on transfusion and transmission in patients receiving allogeneic HSCT. A total of 123 patients who were CMV negative and who had received nearly 8,000 leukoreduced but unscreened blood products were analyzed. Both studies found no risk for transfusion-transmitted CMV infection. Anti-CMV immunoglobulin G was detected in some patients in both of the studies,83,84 but this effect was likely due to the passive transfer of antibodies during transfusions.84 Nonetheless, the overall risk of transfusion-transmitted CMV infection in leukoreduced components is not zero. A study of blood donors in Germany found CMV DNA in 44% of newly seropositive donors and the overall prevalence of CMV DNA was 0.13% in nearly 32,000 donations.85 The small risk of CMV-seronegative blood donors presenting in the window period of a new CMV infection has led to the suggestion that blood products for vulnerable patient groups be obtained from donors with a longstanding history of CMV-positive serology.86 An alternative suggestion may be to screen donated blood for CMV DNA or immunoglobulin M antibodies.86 Conclusions Transfusion support for patients receiving stem cell transplantation depends on many factors. The source of the transplant, the conditioning regimen, and the clinical status of the patient all must enter into the decision-making process regarding the safest component. Despite advances in knowledge, technology, and screening methodologies, complications may still occur and can lead to prolonged transfusion dependence. Knowledge of these complications can help with early detection and treatment, thus reducing the number of transfusions necessary in these patients. January 2015, Vol. 22, No. 1 References 1. Seebach JD, Stussi G, Passweg JR, et al; GVHD Working Committee of Center for International Blood and Marrow Transplant Research. ABO blood group barrier in allogeneic bone marrow transplantation revisited. Biol Blood Marrow Transplant. 2005;11(12):1006-1013. 2. Pasquini MC, Wang Z. Current use and outcome of hematopoietic stem cell transplantation: CIBMTR summary slides, 2013. http://www.cibmtr. org. Accessed October 16, 2014. 3. Simon TB, Snyder EL, Solheim BG, et al, eds. Rossi’s Principles of Transfusion Medicine. 4th ed. Oxford: Wiley-Blackwell; 2009. 4. Canals C, Muñiz-Díaz E, Martinez C, et al. Impact of ABO incompatibility on allogeneic peripheral blood progenitor cell transplantation after reduced intensity conditioning. Transfusion. 2004;44(11):1603-1611. 5. Solh M, Brunstein C, Morgan S, et al. Platelet and red blood cell utilization and transfusion independence in umbilical cord blood and allogeneic peripheral blood hematopoietic cell transplants. Biol Blood Marrow Transplant. 2011;17(5):710-716. 6. Danby R, Rocha V. Improving engraftment and immune reconstitution in umbilical cord blood transplantation. Front Immunol. 2014;5:68. 7. Storb R, Prentice RL, Thomas ED. Marrow transplantation for treatment of aplastic anemia. An analysis of factors associated with graft rejection. N Engl J Med. 1977;296(2):61-66. 8. Storb R, Prentice RL, Thomas ED, et al. Factors associated with graft rejection after HLA-identical marrow transplantation for aplastic anaemia. Br J Haematol. 1983;55(4):573-585. 9. Champlin RE, Horowitz MM, van Bekkum DW, et al. Graft failure following bone marrow transplantation for severe aplastic anemia: risk factors and treatment results. Blood. 1989;73(2):606-613. 10. Anasetti C, Doney KC, Storb R, et al. Marrow transplantation for severe aplastic anemia. Long-term outcome in fifty “untransfused” patients. Ann Intern Med. 1986;104(4):461-466. 11. Hébert PC, Wells G, Blajchman MA, et al; Transfusion Requirements in Critical Care Investigators; Canadian Critical Care Trials Group. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med. 1999;340(6):409-417. 12. Carson JL, Terrin ML, Noveck H, et al; FOCUS Investigators. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med. 2011;365(26):2453-2462. 13. Rebulla P, Finazzi G, Marangoni F, et al; Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto. The threshold for prophylactic platelet transfusions in adults with acute myeloid leukemia. N Engl J Med. 1997;337(26):1870-1875. 14. Trial to Reduce Alloimmunization to Platelets Study Group. Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. N Engl J Med. 1997;337(26):18611869. 15. Storb R, Weiden PL. Transfusion problems associated with transplantation. Semin Hematol. 1981;18(2):163-176. 16. Carson JL, Grossman BJ, Kleinman S, et al; Clinical Transfusion Medicine Committee of the AABB. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Intern Med. 2012;157(1):49-58. 17. Rühl H, Bein G, Sachs UJ. Transfusion-associated graft-versus-host disease. Transfus Med Rev. 2009;23(1):62-71. 18. Cooling L. ABO and platelet transfusion therapy. Immunohematology. 2007;23(1):20-33. 19. Surowiecka M, Zantek N, Morgan S, et al. Anti-A and anti-B titers in group O platelet units are reduced in PAS C versus conventional plasma units. Transfusion. 2014;54(1):255-256. 20. Slichter SJ, Kaufman RM, Assmann SF, et al. Dose of prophylactic platelet transfusions and prevention of hemorrhage. N Engl J Med. 2010;362(7):600613. 21. Josephson CD, Granger S, Assmann SF, et al. Bleeding risks are higher in children versus adults given prophylactic platelet transfusions for treatment-induced hypoproliferative thrombocytopenia. Blood. 2012;120(4):748-760. 22. Stanworth SJ, Estcourt LJ, Powter G, et al; TOPPS Investigators. A no-prophylaxis platelet-transfusion strategy for hematologic cancers. N Engl J Med. 2013;368(19):1771-1780. 23. Wandt H, Schaefer-Eckart K, Wendelin K, et al; Study Alliance Leukemia. Therapeutic platelet transfusion versus routine prophylactic transfusion in patients with haematological malignancies: an open-label, multicentre, randomised study. Lancet. 2012;380(9850):1309-1316. 24. Aoun E, Shamseddine A, Chehal A, et al. Transfusion-associated GVHD: 10 years’ experience at the American University of Beirut-Medical Center. Transfusion. 2003;43(12):1672-1676. 25. Williamson LM, Stainsby D, Jones H, et al. The impact of universal leukodepletion of the blood supply on hemovigilance reports of posttransfusion purpura and transfusion-associated graft-versus-host disease. Transfusion. 2007;47(8):1455-1467. 26. Kita T, Nei J, Matsu T, et al. GVHD after transfusion of stored RBC concentrates in a solution of mannitol, adenine, phosphate, citrate, glucose, and NaCl following trauma. Transfusion. 2000;40(3):297-301. 27. Smith DM, Kresie LA. Preventing transfusion-associated graft-versushost disease. Transfusion. 2007;47(1):173-174. 28. Moroff G, Leitman SF, Luban NL. Principles of blood irradiation, dose Cancer Control 57 validation, and quality control. Transfusion. 1997;37(10):1084-1092. 29. American Association of Blood Banks (AABB). Standards For Blood Banks and Transfusion Services. 29th ed. Bethesda, MD: AABB; 2014. 30. Rivet C, Baxter A, Rock G. Potassium levels in irradiated blood. Transfusion. 1989;29(2):185. 31. Hillyer CD, Tiegerman KO, Berkman EM. Evaluation of the red cell storage lesion after irradiation in filtered packed red cell units. Transfusion. 1991;31(6):497-499. 32. Read EJ, Kodis C, Carter CS, Leitman SF. Viability of platelets following storage in the irradiated state. A pair-controlled study. Transfusion. 1988;28(5):446-450. 33. Roback JD, Combs MR, Grossman BJ, et al, eds. Technical Manual. 16th ed. Bethesda, MD: American Association of Blood Banks (AABB); 2008. 34. Treleaven J, Gennery A, Marsh J, et al. Guidelines on the use of irradiated blood components prepared by the British Committee for Standards in Haematology blood transfusion task force. Br J Haematol. 2011;152(1):35-51. 35. Kanda J, Ichinohe T, Matsuo K, et al. Impact of ABO mismatching on the outcomes of allogeneic related and unrelated blood and marrow stem cell transplantations for hematologic malignancies: IPD-based meta-analysis of cohort studies. Transfusion. 2009;49(4):624-635. 36. Rowley SD. Hematopoietic stem cell transplantation between red cell incompatible donor-recipient pairs. Bone Marrow Transplant. 2001;28(4):315321. 37. Blacklock HA, Gilmore MJ, Prentice HG, et al. ABO-incompatible bone-marrow transplantation: removal of red blood cells from donor marrow avoiding recipient antibody depletion. Lancet. 1982;2(8307):1061-1064. 38. Yazer MH, Triulzi DJ. Immune hemolysis following ABO-mismatched stem cell or solid organ transplantation. Curr Opin Hematol. 2007;14(6):664670. 39. Cohn CS, Gaensler KL, Nambiar A. Engraftment associated complications: is it an allo- or an autoantibody? Presented at the AABB Annual Meeting; October 9–12, 2010; Baltimore, MD. 40. Chao MM, Levine JE, Ferrara JL, et al. Successful treatment of refractory immune hemolysis following unrelated cord blood transplant with Campath-1H. Pediatr Blood Cancer. 2008;50(4):917-919. 41. Malfuson JV, Amor RB, Bonin P, et al. Impact of nonmyeloablative conditioning regimens on the occurrence of pure red cell aplasia after ABO-incompatible allogeneic haematopoietic stem cell transplantation. Vox Sang. 2007;92(1):85-89. 42. Bolan CD, Leitman SF, Griffith LM, et al. Delayed donor red cell chimerism and pure red cell aplasia following major ABO-incompatible nonmyeloablative hematopoietic stem cell transplantation. Blood. 2001;98(6):1687-1694. 43. Griffith LM, McCoy JP, Jr, Bolan CD, et al. Persistence of recipient plasma cells and anti-donor isohaemagglutinins in patients with delayed donor erythropoiesis after major ABO incompatible non-myeloablative haematopoietic cell transplantation. Br J Haematol. 2005;128(5):668-675. 44. Gmür JP, Burger J, Schaffner A, et al. Pure red cell aplasia of long duration complicating major ABO-incompatible bone marrow transplantation. Blood. 1990;75(1):290-295. 45. Bär BM, Van Dijk BA, Schattenberg A, et al. Erythrocyte repopulation after major ABO incompatible transplantation with lymphocyte-depleted bone marrow. Bone Marrow Transplant. 1995;16(6):793-799. 46. Lee JH, Choi SJ, Kim S, et al. Changes of isoagglutinin titres after ABO-incompatible allogeneic stem cell transplantation. Br J Haematol. 2003;120(4):702-710. 47. Helbig G, Stella-Holowiecka B, Wojnar J, et al. Pure red-cell aplasia following major and bi-directional ABO-incompatible allogeneic stem-cell transplantation: recovery of donor-derived erythropoiesis after long-term treatment using different therapeutic strategies. Ann Hematol. 2007;86(9):677-683. 48. Benjamin RJ, Connors JM, McGurk S, et al. Prolonged erythroid aplasia after major ABO-mismatched transplantation for chronic myelogenous leukemia. Biol Blood Marrow Transplant. 1998;4(3):151-156. 49. Aung FM, Lichtiger B, Bassett R, et al. Incidence and natural history of pure red cell aplasia in major ABO-mismatched haematopoietic cell transplantation. Br J Haematol. 2013;160(6):798-805. 50. Bavaro P, Di Girolamo G, Olioso P, et al. Donor lymphocyte infusion as therapy for pure red cell aplasia following bone marrow transplantation. Br J Haematol. 1999;104(4):930-931. 51. Verholen F, Stalder M, Helg C, et al. Resistant pure red cell aplasia after allogeneic stem cell transplantation with major ABO mismatch treated by escalating dose donor leukocyte infusion. Eur J Haematol. 2004;73(6):441-446. 52. Heyll A, Aul C, Runde V, et al. Treatment of pure red cell aplasia after major ABO-incompatible bone marrow transplantation with recombinant erythropoietin. Blood. 1991;77(4):906. 53. Zecca M, De Stefano P, Nobili B, et al. Anti-CD20 monoclonal antibody for the treatment of severe, immune-mediated, pure red cell aplasia and hemolytic anemia. Blood. 2001;97(12):3995-3997. 54. Maschan AA, Skorobogatova EV, Balashov DN, et al. Successful treatment of pure red cell aplasia with a single dose of rituximab in a child after major ABO incompatible peripheral blood allogeneic stem cell transplantation for acquired aplastic anemia. Bone Marrow Transplant. 2002;30(6):405-407. 55. Poon LM, Koh LP. Successful treatment of isohemagglutinin-mediated pure red cell aplasia after ABO-mismatched allogeneic hematopoietic cell transplant using bortezomib. Bone Marrow Transplant. 2012;47(6):870-871. 58 Cancer Control 56. Daniel-Johnson J, Schwartz J. How do I approach ABO-incompatible hematopoietic progenitor cell transplantation? Transfusion. 2011;51(6):11431149. 57. Stussi G, Halter J, Bucheli E, et al. Prevention of pure red cell aplasia after major or bidirectional ABO blood group incompatible hematopoietic stem cell transplantation by pretransplant reduction of host anti-donor isoagglutinins. Haematologica. 2009;94(2):239-248. 58. Curley C, Pillai E, Mudie K, et al. Outcomes after major or bidirectional ABO-mismatched allogeneic hematopoietic progenitor cell transplantation after pretransplant isoagglutinin reduction with donor-type secretor plasma with or without plasma exchange. Transfusion. 2012;52(2):291-297. 59. Schwartz J, Winters JL, Padmanabhan A, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145-284. 60. Rowley SD, Donato ML, Bhattacharyya P.Red blood cell-incompatible allogeneic hematopoietic progenitor cell transplantation. Bone Marrow Transplant. 2011;46(9):1167-1185. 61. Petz LD. Immune hemolysis associated with transplantation. Semin Hematol. 2005;42(3):145-155. 62. Worel N, Greinix HT, Keil F, et al. Severe immune hemolysis after minor ABO-mismatched allogeneic peripheral blood progenitor cell transplantation occurs more frequently after nonmyeloablative than myeloablative conditioning. Transfusion. 2002;42(10):1293-1301. 63. Körbling M, Huh YO, Durett A, et al. Allogeneic blood stem cell transplantation: peripheralization and yield of donor-derived primitive hematopoietic progenitor cells (CD34+ Thy-1dim) and lymphoid subsets, and possible predictors of engraftment and graft-versus-host disease. Blood. 1995;86(7):2842-2848. 64. Bolan CD, Childs RW, Procter JL, et al. Massive immune haemolysis after allogeneic peripheral blood stem cell transplantation with minor ABO incompatibility. Br J Haematol. 2001;112(3):787-795. 65. Toren A, Dacosta Y, Manny N, et al. Passenger B-lymphocyte-induced severe hemolytic disease after allogeneic peripheral blood stem cell transplantation. Blood. 1996;87(2):843-844. 66. Hows J, Beddow K, Gordon-Smith E, et al. Donor-derived red blood cell antibodies and immune hemolysis after allogeneic bone marrow transplantation. Blood. 1986;67(1):177-181. 67. Gajewski JL, Petz LD, Calhoun L, et al. Hemolysis of transfused group O red blood cells in minor ABO-incompatible unrelated-donor bone marrow transplants in patients receiving cyclosporine without posttransplant methotrexate. Blood. 1992;79(11):3076-3085. 68. van Tol MJ, Gerritsen EJ, de Lange GG, et al. The origin of IgG production and homogeneous IgG components after allogeneic bone marrow transplantation. Blood. 1996;87(2):818-826. 69. Ting A, Pun A, Dodds AJ, et al. Red cell alloantibodies produced after bone marrow transplantation. Transfusion. 1987;27(2):145-147. 70.Zupańska B, Zaucha JM, Michalewska B, et al. Multiple red cell alloantibodies, including anti-Dib, after allogeneic ABO-matched peripheral blood progenitor cell transplantation. Transfusion. 2005;45(1):16-20. 71. Esteve J, Alcorta I, Pereira A, et al. Anti-D antibody of exclusive IgM class after minor Rh(D)-mismatched BMT. Bone Marrow Transplant. 1995;16(4):632-633. 72. Petz LD, Yam P, Wallace RB, et al. Mixed hematopoietic chimerism following bone marrow transplantation for hematologic malignancies. Blood. 1987;70(5):1331-1337. 73. Izumi N, Fuse I, Furukawa T, et al. Long-term production of pre-existing alloantibodies to E and c after allogenic BMT in a patient with aplastic anemia resulting in delayed hemolytic anemia. Transfusion. 2003;43(2):241-245. 74. de La Rubia J, Arriaga F, Andreu R, et al. Development of non-ABO RBC alloantibodies in patients undergoing allogeneic HPC transplantation. Is ABO incompatibility a predisposing factor? Transfusion. 2001;41(1):106-110. 75. Abou-Elella AA, Camarillo TA, Allen MB, et al. Low incidence of red cell and HLA antibody formation by bone marrow transplant patients. Transfusion. 1995;35(11):931-935. 76. Beck JC, Wagner JE, DeFor TE, et al. Impact of cytomegalovirus (CMV) reactivation after umbilical cord blood transplantation. Biol Blood Marrow Transplant. 2010;16(2):215-222. 77. Ljungman P, Griffiths P, Paya C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis. 2002;34(8):1094-1097. 78. Clark DA, Emery VC, Griffiths PD. Cytomegalovirus, human herpesvirus-6, and human herpesvirus-7 in hematological patients. Semin Hematol. 2003;40(2):154-162. 79. Marchesi F, Mengarelli A, Giannotti F, et al; Rome Transplant Network. High incidence of post-transplant cytomegalovirus reactivations in myeloma patients undergoing autologous stem cell transplantation after treatment with bortezomib-based regimens: a survey from the Rome Transplant Network. Transpl Infect Dis. 2014;16(1):158-164. 80. Bowden RA, Slichter SJ, Sayers M, et al. A comparison of filtered leukocyte-reduced and cytomegalovirus (CMV) seronegative blood products for the prevention of transfusion-associated CMV infection after marrow transplant. Blood. 1995;86(9):3598-3603. 81. Vamvakas EC. White-blood-cell-containing allogeneic blood transfusion and postoperative infection or mortality: an updated meta-analysis. Vox Sang. 2007;92(3):224-232. January 2015, Vol. 22, No. 1 82. Smith D, Lu Q, Yuan S, et al. Survey of current practice for prevention of transfusion-transmitted cytomegalovirus in the United States: leucoreduction vs. cytomegalovirus-seronegative. Vox Sang. 2010;98(1):29-36. 83. Nash T, Hoffmann S, Butch S, et al. Safety of leukoreduced, cytomegalovirus (CMV)-untested components in CMV-negative allogeneic human progenitor cell transplant recipients. Transfusion. 2012;52(10):2270-2272. 84. Thiele T, Krüger W, Zimmermann K, et al. Transmission of cytomegalovirus (CMV) infection by leukoreduced blood products not tested for CMV antibodies: a single-center prospective study in high-risk patients undergoing allogeneic hematopoietic stem cell transplantation. Transfusion. 2011;51(12):2620-2626. 85. Ziemann M, Krueger S, Maier AB, et al. High prevalence of cytomegalovirus DNA in plasma samples of blood donors in connection with seroconversion. Transfusion. 2007;47(11):1972-1983. 86. Ziemann M, Unmack A, Steppat D, et al. The natural course of primary cytomegalovirus infection in blood donors. Vox Sang. 2010;99(1):24-33. January 2015, Vol. 22, No. 1 Cancer Control 59 Therapeutic apheresis is an important treatment modality frequently used to manage specific complications in patients with underlying malignant disease. Ray Paul. Dark Shadows, 2010. Acrylic, latex, enamel on canvas, 30" × 30". Therapeutic Apheresis for Patients With Cancer Laura S. Connelly-Smith, MBBCh, DM, and Michael L. Linenberger, MD Background: Disease complications associated with certain malignancies may be mediated by cells or soluble molecules that traffic in the bloodstream. Because of this, therapeutic apheresis (TA) methodologies have been used to selectively remove or manipulate specific molecules, antibodies, or cellular elements to treat the underlying pathological process. For some disorders, TA is utilized as a rapid-acting and short-term adjunct to conventional chemotherapy or immunotherapy. For others, a series of scheduled treatments is recommended for optimal management. In all cases, the risks, benefits, and costs must be strongly considered. Methods: The current literature and published guidelines were reviewed to summarize the use of TA in the management of certain complications of cancer. Results: Although TA is relatively safe and useful as a first-line or salvage modality for some disorders, few prospective, randomized clinical trials exist and the majority of evidence is derived from observational studies. Expert-based, clinical practice guidelines have been developed to inform hematology/oncology professionals and apheresis physicians about the efficacy and limitations of TA for malignancy-related indications. Conclusions: Certain oncological conditions respond to TA and consensus guidelines are available to support clinical decision-making. However, well-designed, prospective intervention trials are needed to better define the role of TA for a variety of disorders. Introduction Therapeutic apheresis (TA) is used to treat certain disease complications in patients with cancer with the intention of removing or manipulating a specific molecule, antibody, or cellular element thought to be contributing to the underlying pathological process. From the Seattle Cancer Care Alliance (LSC-S, MLL), the Division of Hematology (LSC-S, MLL) of the School of Medicine at the University of Washington, and the Fred Hutchinson Cancer Research Center (MLL), Seattle, Washington. Address correspondence to Laura S. Connelly-Smith, MBBCh, DM, Seattle Cancer Care Alliance, 825 East Eastlake Avenue, Seattle, WA, 98109. E-mail: [email protected] Submitted June 15, 2014; accepted October 8, 2014. No significant relationships exist between the authors and the companies/organizations whose products or services may be referenced in this article. 60 Cancer Control In general, TA is a relatively safe procedure; however, insufficient clinical evidence to support the economic cost of TA can be a limitation to its use for some conditions. Evidence-based clinical practice guidelines have been developed and periodically updated by the American Society for Apheresis (ASFA) to help support the decision making of health care professionals regarding the use of TA.1 In this article, we provide a brief overview of TA and discuss considerations for its use as a treatment option. The apheresis modalities most commonly used to treat patients with cancer include the therapeutic plasma exchange (TPE), leukocytapheresis, extracorporeal photopheresis (ECP), thrombocytapheresis, and erythrocytapheresis. Herein, we review the known oncological diseases or associations for which specific TA modalities have been successfully January 2015, Vol. 22, No. 1 Table 1. — Indications for Therapeutic Apheresis in Patients With Cancer Therapeutic Apheresis Modality Therapeutic plasma exchange (TPE) Indication Hyperviscosity in monoclonal gammopathies Disease Condition Categorya Gradeb Symptomatic I 1B Prophylaxis for rituximab I 1C Myeloma kidney/myeloma cast nephropathy II 2B Paraneoplastic neurological syndromes (see also Table 4) Lambert Eaton myasthenic syndrome II 2C Other paraneoplastic neurological syndromes III 2C Hematopoietic stem cell transplantation– associated thrombotic microangiopathy Refractory III 2C Therapeutic leukocytapheresis Hyperleukocytosis With leukostasis clinical signs and symptoms I 1B Prophylaxis (asymptomatic) III 2C Extracorporeal photopheresis Cutaneous T-cell lymphoma, mycosis fungoides, Sézary syndrome Erythrodermic I 1B Nonerythrodermic III 2C GVHD Skin chronic GVHD II 1B Skin acute GVHD II 1C Non–skin acute and chronic GVHD III 2B Symptomatic II 2C Prophylactic III 2C I 1B Thrombocytapheresis Thrombocytosis with myeloproliferative neoplasm Erythrocytapheresis Polycythemia vera/primary erythrocytosis Denotes American Society for Apheresis category. Denotes American Society for Apheresis grade. For more information, refer to Tables 2 and 3. GVHD = graft-vs-host disease. Data from reference 1. a b employed. Table 1 summarizes these modalities, clinical conditions, and the most recent ASFA guideline recommendations.1 However, well-designed, prospective intervention trials are still required to fully define the role of TA for many of these disorders. TA plays an important role in the management of various oncological diseases. It is a procedure in which blood is separated from a patient, a portion of which is then removed or otherwise manipulated and the remainder is then returned to the patient. TA procedures include TPE (in which plasma is replaced with a colloid or crystalloid solution) and modalities that selectively remove and dispose of plasma solutes (plasmapheresis), white blood cells (WBCs; leukocytapheresis), or platelets (thrombocytapheresis). ECP is a type of leukocytapheresis procedure whereby the removed white cells are manipulated prior to being reinfused into the patient. Apheresis procedures can utilize centrifugation to separate blood components into layers within a rapidly rotating separation chamber based on their relative density — with red blood cells (RBCs) being the most dense, plasma the least dense — and intermediate layers, moving from the axis of rotation outward and consisting of platelet-rich plasma, lymphocytes, and granulocytes.1 Specific kits are designed to remove RBCs or plasma or cells of intermediate density from January 2015, Vol. 22, No. 1 anticoagulated blood during the procedure. The fluid returned back to the patient contains the undesired blood components along with anticoagulant, crystalloid, and/or colloid solutions. Membrane filtration systems separate and collect plasma on a principle similar to hemodialysis and ultrafiltration, namely using membranes permeable to high-molecular-weight proteins but not cellular elements. The predominant instruments and methodologies used for TA procedures in the United States utilize centrifugation.2 Clinical Adverse Events TA can be associated with minimal to potentially fatal adverse events, although the overall incidence is relatively low (5%–12%).3 Hypersensitivity reactions due to plasma or blood product replacement fluid can range from urticarial to anaphylactoid-type reactions. Hypocalcemia secondary to citrate anticoagulant can manifest as paresthesia, nausea, vomiting, lightheadedness, and twitching. Hypovolemia due to fluid shifts or vasovagal reaction may manifest as hypotension, muscle cramps, and headache. Rare, serious adverse events requiring the procedure to be interrupted or abandoned (0.8% incidence) or resulting in fatality (≤ 0.5%) due to cardiovascular events can include arrhythmia or ischemia, pulmonary edema, pulmonary embolism, and respiratory arrest; neurological Cancer Control 61 complications can also occur and may include tetany, seizures, and cerebrovascular accident.3 Hemorrhage, thrombosis, and infection are uncommon. The causes of death have included respiratory arrest, anaphylaxis, and catheter-associated sepsis.3 Vascular Access The majority of apheresis procedures are centrifugation based; therefore, they require withdrawal blood flow rates of 50 to 150 mL/minute.4-7 Peripheral antecubital veins that can be cannulated using 16- to 18-gauge polytetrafluoroethylene- or silicone-coated, dialysis-type steel needles will accommodate blood flow rates of 80 mL/minute and is adequate for centrifugation techniques. By contrast, filtration therapies require a blood flow rate of at least 150 to 200 mL/minute, which is unsuitable for antecubital veins.4-7 Other considerations specific to TA include whether the treatment relies on discontinuous, sequential blood exchange cycles (1 lumen is sufficient) or continuous processing (2 lumens are needed).4-6 When a central venous catheter (CVC) is necessary for a limited (< 2 weeks) course of TA, a nontunneled, semi-rigid polyethylene catheter should be considered.8 For a longer duration (> 2 weeks) of TA, a tunneled CVC is preferred over a nontunneled CVC due to less risk of infection.9 Typically, tunneled catheters designed for long-term use (weeks to months) are made of silicone and are more biocompatible, flexible, and have the least thrombogenicity. The preferred venous site of CVC insertion is the internal jugular vein, and both ultrasonographic guidance and fluoroscopy have been shown to be associated with a lower rate of complications during insertion.10 Central venous access is not always required.11-13 The Canadian Apheresis Study Group found that 67% of 5,234 TPE procedures could be completed with peripheral venous access alone.11 The frequency of complications due to CVC placement exceeds the frequency of complications directly related to the procedure.14 Central venous access has been identified as a major risk factor for complications of TPE in other studies.10,14-16 CVCs are associated with a higher total complication rate. These include infection (2%–28%), thrombosis (0.2%–11%), hemorrhage (2%–14%), and venous stenosis (10%–26%) with internal jugular catheters and up to 42% with subclavian vein catheters.17 In most series, the incidence of total adverse events associated with all vascular access is low at 1% to 2%.6,18,19 Data from the International Apheresis Registry 2007 report that peripheral veins are commonly used in Europe and Australia (66%–70% of apheresis treatments),7 whereas CVCs are the most common vascular access type used for TA procedures in North America, South America, and Asia (84%–98%).7 This regional difference in the use 62 Cancer Control of peripheral veins compared with CVCs has not been explained by differences in patient age, sex, the median number of treatments per patient, or the type of apheresis procedure.13 Nevertheless, peripheral venous access is underutilized in TA procedures and is the access of choice because it is associated with a lower risk of infection relative to CVCs and placement can be done immediately with a low risk of other serious complications.10 Complications of peripheral cannulation include risk of infection, venous infiltration, patient discomfort, thrombosis and sclerosis of veins, and the loss of future venous access. Peripheral vein access for TA is not a viable option in children due to their small venous caliber. Peripherally inserted central catheters are too small in caliber (4–5 Fr) to accommodate the negative pressure and blood flow rates required for TA procedures.10 Arteriovenous fistulas and grafts are viable options for long-term access when the treatment duration is expected to be over a period of several months or years.13 Evidence and Decision Making Hematologists and oncologists who may have incomplete knowledge of the indications, limitations, risks, and relative efficacy of the procedure might request TA. Because many procedures are for uncommon and infrequent indications, few randomized clinical trials or other high-level evidence studies are available to guide clinical decision-making. Therefore, the ASFA has undertaken a critical evaluation of published studies and observations, publishing periodic, evidence-based systematic reviews of TA applications since 2007. The ASFA clinical practice guidelines use the GRADE system, adopted from Guyatt et al,20 whereby each disease, including specific clinical presentations, is categorized for the role of TA and graded for the strength of recommendation and quality based on the published evidence (Tables 2 and 3).1,20 Table 2. — American Society for Apheresis Categories Category Description I Disorders for which apheresis is accepted as a first-line therapy, either primary stand-alone treatment or in conjunction with another mode of treatment. II Disorders for which apheresis is accepted as a second-line therapy, either as a stand-alone treatment or in conjunction with other modes of treatment. III Optimum role of therapeutic apheresis is not established. Decision making should be individualized. IV Disorders for which published evidence demonstrates or suggests apheresis to be ineffective or harmful. Institutional Review Board approval is desired if apheresis treatment is undertaken in these circumstances. From Schwartz J, Winters JL, Padmanabhan A, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145-284. Reprinted with permission from the American Society for Apheresis. January 2015, Vol. 22, No. 1 Table 3. — American Society for Apheresis Grading Recommendations Recommendation Description Quality of Evidence Application Grade IA Strong recommendation; highquality evidence RCTs without important limitations or overwhelming evidence from observational studies Strong recommendation; can apply to most patients in most circumstances without reservation Grade IB Strong recommendation; moderate quality evidence RCTs with important limitations (inconsistent results, methodological flaws, indirect, or imprecise) or exceptionally strong evidence from observational studies Strong recommendation; can apply to most patients in most circumstances without reservation Grade IC Strong recommendation; lowquality or very-low-quality evidence Observational studies or case series Strong recommendation; may change when higher-quality evidence becomes available Grade 2A Weak recommendation; high-quality evidence RCTs without important limitations or overwhelming evidence from observational studies Weak recommendation; best action may differ depending on circumstances or patient or societal values Grade 2B Weak recommendation; moderate-quality evidence RCTs with important limitations (inconsistent results, methodologic flaws, indirect, or imprecise) or exceptionally strong evidence from observational studies Weak recommendation; best action may differ depending on circumstances or patient or societal values Grade 2C Weak recommendation; lowquality or very-low-quality evidence Observational studies or case series Very weak recommendations; other alternatives may be equally reasonable RCT = randomized controlled trial. Adapted from Guyatt GH, Cook DJ, Jaeschke R, et al. Grades of recommendation for antithrombotic agents: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th ed). Chest. 2008;133(6 suppl):123S-31S. Reprinted with permission from the American College of Chest Physicians. ASFA category I and II indications are those for which TA is considered first-line or second-line therapy, respectively. Category III indications acknowledge the lack of high-level evidence to recommend the TA procedure as primary or second line; however, the treatment may be beneficial and, thus, individualized decision-making should be used to guide inclusion of TA in the treatment plan. Category IV reflects ineffectiveness or harm by TA with the risks outweighing benefits.21 The 6th edition published in 2013 is a compilation of 78 diseases or medical conditions assigned ASFA categories I to IV.1 All TA procedures discussed within this review are referenced according to the category and recommended grade per the ASFA 2013 guidelines with further updates as indicated.1 Therapeutic Plasma Exchange TPE involves the removal of a large volume of plasma and replacement with plasma, albumin, or both. The major mechanism of action of TPE is the removal of a pathological solute, such as autoantibodies, immune complexes, cryoglobulins, myeloma light chains, or cytokines. Of note, TPE may also have an immunomodulatory effect, including the modulation of the Th1/Th2 T-cell balance toward Th2,22 the suppression of interleukin 2 and interferon γ production,23,24 and an increase in suppressor T-cell function. A standard TPE procedure exchanges 1 to 1.5 plasma volumes resulting in the removal of 60% to 70% of intravascular large molecular weight solutes.25 Some large molecules (eg, immunoglobulin [Ig] G) distribute in both the intravascular and the January 2015, Vol. 22, No. 1 extravascular spaces; during TPE, the extravascular molecules can move into the intravascular space. Therefore, TPE may remove more total solute than might be predicted based on the pretreatment concentration because of re-equilibration occurring from the extravascular to the intravascular compartment. Because TPE removes normal plasma coagulation factors, the activities of factors V, VII, VIII, IX, and X, as well as von Willebrand factor (vWF), may significantly decline.26,27 Activities of factor VIII, factor IX, and vWF return to normal within 4 hours after TPE, whereas the remaining coagulation factors achieve pre-TPE activity levels within 24 hours.26 The exception to this is fibrinogen, which reaches 66% of preapheresis levels within 72 hours.28 TPE may also remove medications, especially those highly protein bound. The clinical impact of this effect is understood for relatively few drugs.29,30 Albumin is the most commonly used replacement fluid for TPE procedures. Normal plasma has the same oncotic pressure as 5% albumin.31,32 Thus, replacing plasma with 4% to 5% human serum albumin will maintain plasma volume and avoid hypotension. However, because albumin is expensive,33 some health care professionals may prefer to use albumin and saline, with the majority of the albumin being given at the end of the procedure. The combination of albumin and saline is hypo-oncotic and has been associated with a greater frequency of hypovolemic reactions and edema compared with using albumin alone.16 Another disadvantage is that albumin does Cancer Control 63 not replace coagulation factors, which may lead to significant post-treatment coagulopathy. Plasma is used as replacement fluid with TPE in a limited number of disorders. It avoids postprocedure coagulopathy and immunoglobulin depletion. Its disadvantages include transfusion reactions, citrate toxicity, and the potential for viral transmission. Plasma is indicated as replacement fluid to replace ADAMTS13 when treating thrombotic thrombocytopenic purpura (TTP) or when coagulopathy must be corrected.34 Neither cryosupernatant plasma nor solvent/ detergent–treated plasma has been shown to offer any advantage over standard plasma for any indication.34 A meta-analysis of 3 trials comparing fresh frozen plasma and cryosupernatant plasma for the initial treatment of TTP did not reveal any benefit for patients receiving cryosupernatant plasma.35 Similarly, controlled studies failed to establish superiority of solvent/detergent–treated plasma over fresh frozen plasma.34 The only cohort of patients with TTP who may benefit from the use of solvent/detergent–treated plasma are those with severe allergies to standard plasma.36 Hyperviscosity in Monoclonal Gammopathies Hyperviscosity syndrome (HVS) refers to the clinical sequelae caused by the altered physiology related to plasma hyperviscous states, most typically seen in Waldenström macroglobulinemia (WM; also known as lymphoplasmacytic lymphoma) associated with monoclonal IgM or, less frequently, with multiple myeloma associated with monoclonal IgA or IgG3. Specific signs and symptoms include mucosal bleeding, visual impairment with retinal hemorrhage or retinal detachment, headache, dizziness, vertigo, nystagmus, hearing loss, somnolence, coma, and seizure. Other manifestations include congestive heart failure (related to plasma volume overexpansion), respiratory compromise, coagulation abnormalities, anemia, fatigue, peripheral polyneuropathy (depending on the specific Ig properties), and anorexia. WM represents approximately 2% of all cases of non-Hodgkin lymphoma.37 When the IgM protein associated with WM exceeds a concentration of 4 g/dL, the relative plasma viscosity can exceed 4 centipoise (cp; relative to water: normal range, 1.4–1.8 cp) and HVS can occur.38 Unlike the situation with IgG, IgM is predominantly intravascular (> 80%) and increased viscosity with IgM can become exponential above a concentration of 3 g/dL. In turn, a small reduction in IgM concentration can have a significant effect on lowering serum viscosity. TPE is an effective, short-term treatment for complications of HVS.39-41 Because bleeding is the most common sign of HVS and retinal examination findings correlate with the symptomatic threshold for HVS in patients with WM, urgent TPE should be carried out 64 Cancer Control to reduce the likelihood of blindness from retinal hemorrhages or retinal detachment.42,43 TPE is a safe and well-tolerated procedure in this setting.44 It is not typically necessary to reduce the plasma viscosity to normal levels to relieve symptoms. However, some evidence suggests that patients with monoclonal IgM antibodies that produce neuropathy or other target organ dysfunction may benefit from a more aggressive effort to maintain serum viscosity near normal levels.45,46 Hyperviscosity with WM is an ASFA category I indication for TPE (grade 1B recommendation).1 Generally, 1 to 1.5 plasma volumes are exchanged per session, and fluid replacement usually consists of albumin and saline in various proportions. Plasma exchange reduces plasma viscosity by approximately 20% to 30% per session.47 Thus, 1 or 2 procedures can return the plasma viscosity to near normal levels and reduce the IgM concentration for several weeks. Concurrent chemotherapy is required to treat underlying disease and prevent rebound HVS. For asymptomatic patients with a serum viscosity level above 3 to 3.5 cp, an IgM concentration greater than 3 to 4 g/dL, or both, some experts suggest that TPE can be prophylactically used prior to starting rituximab therapy because significant transient increases in IgM levels can occur following single-agent rituximab therapy (considered a “flare”) in 30% to 70% of patients.38,48,49 Based on this concern, the ASFA guidelines have recommended TPE as prophylaxis treatment prior to rituximab to lower IgM concentrations of less than 5 g/dL (grade 1C recommendation).1 The flare phenomenon may be less with regimens that use chemotherapy prior to rituximab or regiments that omit rituximab for the first 1 or 2 cycles. Patients with myeloma and IgG3 subclass monoclonal paraproteinemia are more likely to develop HVS than other patients with myeloma.50,51 This usually occurs at higher than 4 g/dL of monoclonal IgG3 in the plasma. In cases of IgG-associated HVS, the increase in serum viscosity is approximately proportional to the concentration of the paraprotein.52 HVS may also occasionally occur in IgA and light-chain myeloma because of the formation of polymers; in the majority of these cases, it occurs when the concentration of monoclonal IgG exceeds 6 to 7 g/dL. Myeloma Cast Nephropathy Nearly 50% of patients with multiple myeloma develop renal disease.53,54 Acute kidney injury from cast nephropathy, also known as “myeloma kidney,” is the most common type and accounts for 30% to 80% of cases.55,56 The development of acute kidney injury is associated with worse 1-year survival rates and reduces the overall therapeutic options available to patients.53,54 Cast nephropathy is due to the interaction January 2015, Vol. 22, No. 1 and aggregation of filtered free light chains (FLCs) and Tamm–Horsfall protein, thus causing intratubular obstruction and damage. When the light chain production overcomes the capacity of the tubular cells to endocytose and catabolize the FLCs, the increased light chains in the tubular fluid of the distal tubule and thick ascending loop of Henle form tubular casts with the Tamm–Horsfall protein.57,58 As tubular obstruction progresses, the decline in renal function becomes irreversible. Other factors, such as dehydration, diuretics, hypercalcemia, hyperuricemia, and intravenous contrast media, may all potentiate cast formation and acute kidney injury. The key to treating cast nephropathy is the rapid lowering of FLCs. In addition to hydration and aggressive supportive care, antimyeloma chemotherapy is necessary, whether it be with an alkylating agent and prednisone therapy or one of the recent immune modulators (thalidomide, lenalidomide) and proteasome inhibitors (bortezomib). These latter agents have emerged as effective therapy and have been referred to as “renoprotective.”59 Supportive care with hemodialysis or peritoneal dialysis may also be needed. TPE has been used in hopes of reducing the delivery of plasma FLCs to the renal glomerulus for filtration. Two studies suggested that TPE was beneficial.60,61 In addition, a small prospective comparison of forced diuresis, melphalan, and prednisone (10 patients) vs forced diuresis, melphalan, prednisone, and TPE (11 patients) found a trend in favor of TPE, and a subgroup analysis of patients dependent on dialysis revealed that renal function recovered in 43% of the TPE group compared with 0% in the control group.62 These studies led to an endorsement of TPE for myeloma kidney by the Scientific Advisors of the International Myeloma Foundation.63 Subsequently, a large randomized trial of bortezomib-containing chemotherapy and supportive care, with or without TPE, failed to demonstrate a benefit for 5 to 7 TPE procedures over 10 days.64 However, this study has been criticized for the lack of FLC measurements, the lack of histological evidence of cast nephropathy, and the failure to consider early end points more specific to the recovery of renal function. In a report from the Mayo Clinic, plasma exchange in combination with bortezomib-based chemotherapy in 7 patients was associated with 6 patients (86%) having at least a partial response.65 Collectively, these observations suggest that a subgroup of patients with cast nephropathy might benefit from TPE, particularly those in nonoliguric renal failure who do not require dialysis.56,60 The severity of myeloma cast formation, including the need for dialysis, has been identified as the major factor associated with nonreversible renal failure, even in patients undergoing TPE.60,63,66,67 Moreover, biopsy findings that January 2015, Vol. 22, No. 1 indicate potential reversibility (eg, absence of fibrosis of all affected glomeruli) may be important predictors of success.60,62 The ASFA evidence-based guidelines lists TPE as a category II indication for myeloma kidney due to light-chain cast nephropathy.1 After initial management, especially in the case of nonoliguric patients, focus should be on fluid resuscitation (2.5–4 L/day), alkalinization of the urine, and chemotherapy. If serum creatinine remains elevated after several days, then renal biopsy should be considered to assess for cast nephropathy. If cast nephropathy is highly suspected or confirmed, then TPE can be initiated by processing 1 to 1.5 total plasma volumes every 1 to 2 days and using 5% albumin in saline as replacement fluid. Some studies support a course of 10 to 12 TPE procedures over 2 to 3 weeks and repeating this depending on patient response and clinical course.1 For patients who are oliguric, excrete at least 10 g of light chains per 24 hours, or whose serum creatinine level is at least 6 mg/dL, TPE may be included as adjunct therapy to initial chemotherapy and hemodialysis. If TPE and hemodialysis are to be performed on the same day, then the procedures can be performed in tandem without compromising the efficiency of the hemodialysis. Paraneoplastic Neurological Syndromes Paraneoplastic neurological syndromes (PNS) are symptoms or signs resulting from damage to the central or peripheral nervous system, including the neuromuscular junction and muscle, removed from the site of the malignancy or its metastases, and not due to remote effects caused by infection, ischemia, or metabolic disruptions.68 PNS can affect up to 1% of patients with cancer but may occur more frequently in those with non-Hodgkin lymphoma, small-cell lung cancer, and thymomas.69-72 In the majority of patients, PNS develop prior to the cancer diagnosis. The pathogenesis of PNS is thought to be immune-mediated as a result of a cross-reaction against antigens shared by the tumor and nervous system cells.68,73 Many antibodies are associated with paraneoplastic syndromes (Table 4).68,73,74 Their role in neuronal dysfunction is unclear and they can occur in fewer than 50% of patients with PNS.75 No studies have proven that these antibodies are pathogenic; however, these antibodies have become useful diagnostic markers, particularly in monitoring for relapse. The severity of the majority of PNS cases is due to the early and nonreversible destruction of neural structures by the inflammatory process; in many cases, the patient is severely debilitated within weeks to months.76,77 Prompt initiation of therapy following the diagnosis of PNS may stabilize symptoms and prevent PNS spreading to further areas in syndromes with Cancer Control 65 Table 4. — Paraneoplastic Neurological Syndromes and Their Associated Tumor Types, Symptoms, and Antibodies Syndrome Frequency of Paraneoplastic Origin (%) Major Symptoms Lambert–Eaton myasthenic syndrome 60 Muscle weakness, autonomic dysfunction SCLC VGCC antibodiesa Paraneoplastic cerebellar degeneration 50 Truncal, limb ataxia, dysarthria, saccadic gaze pursuit, nystagmus Ovary Breast SCLC Hodgkin lymphoma Anti-Yo Anti-Hu Anti-VGCC Anti-CV2/CRMP5 Paraneoplastic opsoclonus/ myoclonus 20 Saccades, ataxia, other cerebellar signs, generalized myoclonus, altered mental state, stupor, coma Neuroblastoma Breast SCLC Anti-Ri (ANNA-2) Sensory neuronopathy 20 Pain, paresthesias (arms > legs), numbness, ataxia SCLC Anti-Hu Anti-CV2/CRMP5 Anti-amphiphysin Limbic encephalitis 20 Seizures, short-term memory deficits, behavioral and psychiatric disturbances SCLC Testicular Anti-Hu ANNA-1 Paraneoplastic encephalomyelitis 10 Seizures, subacute dementia, personality changes (limbic encephalitis), subacute cerebellar signs, autonomic nervous system dysfunction SCLC ANNA-1 Anti-Hu Subacute visual loss, photosensitivity, night blindness, impaired color vision SCLC Cervical Melanoma Recoverin antibodies Stiffness predominantly upper limbs, painful spasms precipitated by sensory stimuli Breast Colon Lung Hodgkin lymphoma Malignant thymoma Antiamphiphysin Weight loss, constipation, abdominal distension, esophageal dysmotility, gastroparesis SCLC Anti-Hu Anti-CV2/CRMP5 Proximal myopathy, heliotrope rash, scaly plaques on dorsal hands Ovarian Lung Pancreatic Stomach Colorectal Non-Hodgkin lymphoma Cancer-associated retinopathy Paraneoplastic stiff-person syndrome 5–20 Chronic intestinal pseudo-obstruction Dermatomyositis 30 Associated Tumor Types Frequently Associated Paraneoplastic Antibodies Present in nearly all patients with the paraneoplastic and nonparaneoplastic form of Lambert–Eaton myasthenic syndrome. Data from references 68, 73, and 74. ANNA = antineuronal nuclear antibody, SCLC = small cell lung cancer, VGCC = voltage-gated calcium channel. a onconeural antibodies.78 For patients with an identified tumor, antitumor therapy should be rapidly instituted for stabilization or symptom improvement. The use of immunomodulatory therapy does not substantially modify the neurological outcome of patients whose tumors are successfully treated.76,79 For many paraneoplastic syndromes, removal of the tumor is the only effective treatment.80,81 The role and timing of immunotherapy for PNS is not well defined; however, many reports indicate its apparent benefit.82,83 No systematic studies exist concerning the type of immunosuppressive therapy, and no RCTs or quasi-RCTs exist on which to base treatment or practice.84 In patients without detectable tumor but with a prior history of malignancy and clinicopathological findings consistent with progressive PNS, it is appropriate to empirically start immunosuppressive therapy with or without antitumor treatment. 66 Cancer Control Initial therapies often include corticosteroids, TPE, intravenous immunoglobin (IVIG), immune adsorption, and/or rituximab. More aggressive second-line immunosuppression with cyclophosphamide, tacrolimus, mycophenolate, or cyclosporine may be used when no response to initial treatments is seen and the patient continues to lose neurological functions. More severe neurological deficits associated with antibodies against Yo, Hu, and CRMP5 are also the most refractory to immunosuppressive treatment. Survival from time of diagnosis is significantly worse in patients with anti-Yo (median, 13 months) or anti-Hu (median, 7 months) than in patients with anti-Tr (median, > 113 months) or anti-Ri (median, > 69 months).85 However, patients who receive antitumor treatment, with or without immunotherapy, live significantly longer than those who do not.76,84 The rationale for TPE with PNS is that plasma January 2015, Vol. 22, No. 1 antibody levels can be reduced and thereby ameliorate the damage to the peripheral nervous system in tissues. Plasma exchange can also reduce circulating levels of cytokines and other mediators of inflammation that may contribute to the effectiveness of TPE as immunomodulatory therapy. By comparison, PNS involving the central nervous system do not typically respond to TPE, a fact likely due to the inability of plasma therapy to decrease intrathecal antibody titers. Patients with acquired neuromyotonia and antibodies directed against voltage-gated potassium channels or paraneoplastic cerebellar degeneration with anti-Tr antibodies may be more likely to respond to TPE; however, many do not have malignancy. In 50% of cases, encephalitis associated with anti–N-methyl D-aspartate receptor antibodies responds to first-line treatment with corticosteroids, IVIG, or TPE. Although immunosuppression with corticosteroids, TPE, and/or IVIG may benefit those with LGI1- and CASPR2-antibody associated syndromes, residual memory impairment is common. However, large case series on long-term outcomes are currently lacking. Even less is known about the treatment and prognosis of other neuronal cell-surface antibody syndromes (eg, γ-aminobutyric acid [B], α-amino-3-hydroxy-5-methyl4-isoxazolepropionic acid receptor). Typically, they are treated similar to anti–N-methyl D-aspartate receptor encephalitis. Disorders such as paraneoplastic cerebellar degeneration are generally associated with neuronal loss; because they subacutely evolve and treatment is often delayed, the neurons die, thus making recovery impossible.85 Some central nervous system disorders, such as opsoclonus–myoclonus syndrome, may not involve cellular loss and, in fact, may have no identifiable pathological features. Thus, patients with these disorders, like those with the Lambert–Eaton myasthenic syndrome (LEMS), have the potential for recovery. LEMS is a syndrome that involves the neuromuscular junction and can typically respond well to immunosuppression and, subsequently, to treatment of the underlying tumor. TPE may be useful adjunct therapy for patients whose neurological deficit is severe or rapidly developing or among those who cannot tolerate treatment with IVIG (ASFA category II; grade 2C recommendation).1 Reports of benefit are tempered by the observation that responses can be slow and symptoms can worsen following the completion of TPE if additional immunosuppressive therapy is not employed. The reported TPE regimens for LEMS vary from 5 to 15 regiments of daily TPE over 5 to 19 days to 8 to 10 regimens of TPE carried out at 5- to 7-day intervals. Most reports employed 1.25 plasma volume exchanges.1 However, the peak effect is usually demonstrated after 2 weeks and largely subsides after January 2015, Vol. 22, No. 1 6 weeks.86 This may be due to the slower turnover of the presynaptic voltage-gated calcium channel compared with the postsynaptic acetylcholine receptor. The effectiveness of immunosuppressive therapy in non-LEMS PNS with onconeural antibodies is not supported by higher level evidence.84,87 Few studies prove efficacy, although several retrospective and small prospective studies support the benefit of immunosuppression for some patients and select syndromes.88-91 The ASFA guidelines have assigned a grade 2C recommendation for this category III indication.1 Procedures are performed daily or every other day for a total of 5 to 6 exchanges over 2 weeks, although the exact number of exchanges should be adjusted for each patient. Some patients will require maintenance therapy on a monthly or less frequent basis. TPE cannot be considered as standard therapy for PNS. Most patients treated with TPE have also received immunosuppressive drugs as well as specific anticancer therapy. Hematopoietic Stem Cell Transplantation– Associated Thrombotic Microangiopathy Thrombotic microangiopathy (TMA) refers to a histopathological appearance, describing arteriolar thrombi associated with intimal swelling and fibrinoid necrosis of the vessel wall.92 The microscopic injury results from a variety of insults that can cause the activation of intravascular platelets with the subsequent formation of platelet-rich thrombi within the microcirculation. TMA following allogeneic hematopoietic stem cell transplantation (HSCT), also called transplant-associated TMA, appears to be primarily triggered by mechanisms of endothelial cell injury, including conditioning chemotherapy,93-95 irradiation,96 immunosuppressive agents (eg, mammalian target of rapamycin, calcineurin inhibitor drugs),97-100 graftvs-host disease (GVHD),101-103 and opportunistic infections.104,105 The damaged endothelial cells release microparticles and vWF, which induce platelet adhesion/aggregation and a procoagulant state.106-108 This process consumes platelets and induces mechanical damage to RBCs as they impact microthrombi or fibrin strands obstructing the microcirculation. The clinical hallmarks of TMA include microangiopathic hemolytic anemia and thrombocytopenia, and the associated laboratory findings include schistocytes, increased serum lactate dehydrogenase, decreased serum haptoglobin, and indirect hyperbilirubinemia. Hemoglobinuria, either frank or microscopic, is frequent. Kidneys are the major target organs of transplant-associated TMA; thus, renal function abnormalities are common. Unlike idiopathic TTP, in which severe deficiency of the vWF-cleaving protease, ADAMTS13 (a disintegrin and metalloprotease with thrombospondin-1-like domains), leads to Cancer Control 67 the presence of ultra-large multimers of vWF and systemic platelet agglutination, multiple studies in post-transplantation TMA have failed to document a severe deficiency of ADAMTS13.109-111 TMA can occur within the first few weeks following transplantation or as a late complication, particularly in association with GVHD. One-year cumulative incidences of 13% and 15% were reported in patients undergoing nonmyeloablative conditioning and high-dose conditioning, respectively.112 Most large, retrospective studies report a prevalence of 10% to 25%.113 Transplant-associated TMA carries a poor prognosis. In a literature review of 35 published articles involving more than 5,423 allogeneic HSCT recipients, 447 study volunteers (8.2%) developed transplant-associated TMA and had a median mortality rate of 75% within 3 months of the diagnosis.114 Clinical risk factors associated with transplant-associated TMA include high-dose conditioning regimens, acute GVHD, female sex, older age, active infections, receiving transplantations from unrelated donors, and the combination of mammalian target of rapamycin and calcineurin inhibitor drugs.115 Currently, no consensus exists regarding the approach to treatment of transplant-associated TMA, and no randomized clinical trial data exist. Initial management involves the reduction or discontinuation of the mammalian target of rapamycin and calcineurin inhibitor drugs (especially if used in combination) along with aggressive treatment of underlying GVHD and infections. A role for TPE in this disorder remains unclear. Response rates of transplant-associated TMA to TPE are significantly lower (< 50%)116 than the high responses in idiopathic TTP (≤ 85%).117-119 A systematic review published in 2004 noted an 82% mortality rate among 176 study volunteers with transplant-associated TMA who underwent TPE compared with a 50% mortality rate among 101 study volunteers not treated with TPE, suggesting that the toxicity of the procedure outweighs the potential benefits.114 Similarly high cumulative mortality rates were cited by the Blood and Marrow Transplant Clinical Trials Network Toxicity Committee in a consensus statement recommending that TPE not be considered as standard of care for transplant-associated TMA.120 The difference seen in mortality rates may partly reflect the significant comorbidity of the post-transplantation state; however, it also supports the available data that indicate that transplant-associated TMA results from mechanisms distinct from those involved in idiopathic TTP. Because some patients with transplant-associated TMA appear to respond to treatment, a trial of TPE could be considered as salvage therapy for select patients with persistent, progressive, end-organ complications despite a resolution of infections and GVHD (ASFA category III; grade 2C recommenda68 Cancer Control tion).1 TPE for transplant-associated TMA is usually performed daily until a response is seen and is then either discontinued or tapered off, a process similar to treatment for idiopathic TTP. The therapeutic end point may be difficult to determine because the platelet count, schistocytes, and lactate dehydrogenase levels could be affected by incomplete engraftment and other post-transplantation complications. Based on the data from anecdotal reports, other salvage treatment options for transplant-associated TMA might include daclizumab, defibrotide, and rituximab.121-123 Such anecdotal reports of clinical response to eculizumab suggest that transplant-associated TMA could involve aberrant and autonomous complement activation and include some patients who may have an inherent defect in complement regulation.124,125 Therapeutic Leukocytapheresis The majority of leukocytapheresis procedures are carried out to treat hyperleukocytosis and complications of leukostasis associated with acute leukemias. Leukocytapheresis for Acute Leukemia and Leukostasis With Hyperleukocytosis Hyperleukocytosis is variably defined as a WBC or leukemic blast cell count above 50,000/μL or 100,000/μL. The incidence of hyperleukocytosis ranges between 5% and 13% in adult acute myeloid leukemia (AML) and between 10% and 30% in acute lymphoblastic leukemia (ALL).126 Although hyperleukocytosis does not appear to have a major impact in early mortality in ALL unless the WBC count is more than 250,000/μL, it is associated with an increased likelihood of induction death and reduced likelihood of achieving complete remission in cases of AML.127,128 Hyperleukocytosis with AML and ALL may be associated with disseminated intravascular coagulopathy, tumor lysis syndrome, and leukostasis. Leukostasis refers to end-organ complications due to microvascular leukoaggregates, hyperviscosity, tissue ischemia, infarction, and hemorrhage as a result of high numbers of leukocytes. The pathophysiology of leukostasis is based on the rheological properties of the blasts, which is a function of the deformability of the blasts (rigidity) and the volume of the blasts (cell fraction) in the blood,129 and the cytoadhesive interactions between the blasts and the endothelium.130 This second mechanism is based on the activation of the endothelium by blasts to secrete cytokines that in turn mediate the expression of specific receptors such as intercellular adhesion molecule 1, vascular cell adhesion protein 1, selectins, and others that promote blast adhesion.130 Leukostasis in ALL usually occurs with WBC counts higher than 400,000/μL.131 Compared with lymphoid blasts, myeloid blasts are larger, less deformable, and their cytokine products are more January 2015, Vol. 22, No. 1 prone to activate inflammation and the molecular expression of endothelial cell adhesion. A blast count above 100,000/μL is a good predictor of leukostasis in the myeloid phenotype AML (FAB M1, M2, M3v). The blast count is less reliable in monocytic lineage AML (FAB M4, M5) in which severe leukostasis can occur with WBC counts above 50,000/μL.132 Central nervous system manifestations of leukostasis can include confusion, somnolence, dizziness, headache, delirium, coma, and parenchymal hemorrhage, and pulmonary complications can include hypoxemia, diffuse alveolar hemorrhage, and respiratory failure with interstitial infiltrates, alveolar infiltrates, or both. Both pulmonary and neurological manifestations are associated with increased rates of mortality in adults and children. In cases of hyperleukocytosis in AML, the mortality rate has been reported to be between 5% and 30%.133 Definitive treatments for hyperleukocytosis in the setting of AML or ALL involve induction chemotherapy with aggressive supportive care. Hydroxyurea, cytarabine, or both are useful in temporizing cytoreductive agents for AML. Hyperuricemia and tumor lysis syndrome are treated with intravenous fluids, electrolyte replacement, allopurinol or rasburicase, alkalinization of the urine, and dialysis. Bleeding and coagulopathy are managed with plasma, cryoprecipitate, and/or platelet transfusions. However, RBC transfusions should be deferred to avoid augmenting hyperviscosity and promoting leukostasis. Leukocytapheresis allows for the rapid reduction of the intravascular leukemic cellular burden, thereby resolving leukostatic microvascular occlusion and improving tissue perfusion. No randomized prospective studies of leukocytapheresis for hyperleukocytosis or leukostasis have been published. Published data regarding the clinical value of therapeutic leukocytapheresis are limited, observational, and conflicting. This is partly due to different WBC thresholds prompting leukocytapheresis, patient selection, and therapeutic end points. Previous multiple retrospective cohort studies of AML demonstrate reduced early mortality; however, leukocytapheresis offers no benefit to overall outcome.134,135 Recently, a systematic review and meta-analysis using an intent-to-treat approach evaluated leukapheresis and low-dose chemotherapy interventions in patients with AML and WBC counts above 100,000/μL.136 Data were reviewed from 15 of the studies in which leukocytapheresis was used. In the analysis, the mean early mortality rate of 20.1% during the first month of induction chemotherapy in patients with hyperleukocytosis was not reduced by leukocytapheresis (or low-dose chemotherapy), suggesting limited benefit.136 The authors noted limitations of the primary studies: the studies were small, retrospective, January 2015, Vol. 22, No. 1 observational, and all had a moderate to high risk of confounding bias.136 Prophylactic leukocytapheresis remains a consideration for patients with AML and WBC counts above 100,000/μL without overt leukostasis manifestations as a means to rapidly reduce blood viscosity and facilitate safe RBC transfusion as well as to avoid leukostasis that might occur following the start of chemotherapy, particularly with the M4 or M5 subtype.137 Among children and adults with ALL, clinical symptoms of leukostasis develop in less than 3% at WBC counts lower than 400,000/μL.131 Therefore, prophylactic leukocytapheresis offers no advantage over aggressive induction chemotherapy and supportive care, including among those with tumor lysis syndrome. By comparison, pulmonary and central nervous system complications develop in more than 50% of children with ALL and WBC counts above 400,000/μL, suggesting that prophylactic leukocytapheresis might be beneficial in that setting.131 For patients with ALL or AML and clinical leukostasis complications, ASFA category I (grade 1B recommendation) has been assigned and is based on numerous reports and retrospective case series that describe the rapid reversal of pulmonary and central nervous system manifestations following cytoreduction with leukocytapheresis.1 However, improvement may not be observed if severe end-organ injury or hemorrhage has already occurred. The ASFA category III (grade 2C recommendation) for prophylactic leukocytapheresis probably reflects the limited and conflicting data available in the literature to guide treatment in patients who are asymptomatic.1 A single leukocytapheresis procedure can reduce the WBC count by 30% to 60%.1 Daily — or, on occasion, twice-daily — procedures for life-threatening cases can be performed by processing 1.5 to 2 blood volumes and using crystalloid or 5% albumin as the replacement fluid. RBC priming may be employed for adults with severe anemia; however, undiluted packed RBCs should be avoided in small children with hyperviscosity. For patients with AML and leukostasis complications, apheresis must be discontinued when the blast cell count is less than 50,000 to 100,000/μL and clinical manifestations are resolved or maximum benefit is achieved. Chemotherapy should not be postponed and is required to prevent the rapid reaccumulation of circulating blasts. Leukocytapheresis for Chronic Myeloid Leukemia With Hyperleukocytosis and Priapism The incidence of leukostasis as a result of hyperleukocytosis in adults presenting with CML has been estimated to be between approximately 12% and 60% among children with CML.138,139 The most recognized features of hyperleukocytosis in CML are constitutional (malaise and fever), cardiorespiratory, neurological, Cancer Control 69 or vascular, including retinal hemorrhage, myocardial ischemia, and priapism. Priapism occurs in 1% to 2% of males presenting with chronic phase CML and WBC counts above 500,000/μL.140-142 It is characterized by a prolonged, painful erection. Priapism in this setting is a urological emergency with a poor prognosis, and the risk of impotence in adults is 50% despite appropriate management.143 The primary mechanism is the aggregation of leukemic cells in the corpora cavernosa and the dorsal vein of the penis.144 A contributing factor is the venous congestion of the corpora cavernosa due to mechanical pressure on the abdominal veins by the enlarged spleen. Increased production of cytokines and adhesion molecules by leukemic cells can also be seen and will result in the activation of endothelial cells and lead to the increased sequestration of cells in the microvasculature.145 No standard treatment has been recommended for leukemic priapism. Systemic therapies include cytoreductive agents, such as high-dose hydroxyurea and tyrosine kinase inhibitors, with or without the addition of leukocytapheresis to reduce hyperviscosity.138,146 A review of the published literature revealed that 3 of 4 patients with ischemic priapism treated by leukocytapheresis had a resolution of priapism compared with 3 of 15 patients treated with chemotherapy alone.147 Some case series have reported on the successful use of therapeutic leukocytapheresis in combination with cytotoxic therapy to treat priapism.142,144,146,148,149 Although some of these studies indicated that a conservative approach may be successful in preserving erectile function, a combined modality approach is strongly recommended by the American Urological Association so that systemic treatment for the underlying disorder and intracavernous treatment be concurrently administered.147 Leukocytapheresis for Other Chronic Leukemias and Leukostasis With Hyperleukocytosis Leukostasis complications with other leukemias are rare but may occur with chronic myelomonocytic leukemia150 and WBC counts higher than 100,000/μL with a high level of lactate dehydrogenase. In cases of chronic lymphocytic leukemia, leukostasis is rare and is predominantly described in patients with WBC counts above 1,000,000/μL.151 Extracorporeal Photopheresis ECP is an immunomodulating cell therapy whereby a patient’s circulating WBCs are collected via a leukocytapheresis procedure, exposed ex vivo to photo-activatable 8 methoxypsoralen, irradiated with ultraviolet A light, and then reinfused into the patient. ECP was originally introduced in 1987 by Edelson et al152 for the treatment of Sézary syndrome, an aggressive form 70 Cancer Control of advanced cutaneous T-cell lymphoma (CTCL). In 1988, ECP was approved by the US Food and Drug Administration (FDA) for the treatment of advanced forms of CTCL, and has since become a recommended first-line therapy for selected patients with advanced stage CTCL (ASFA category I).1,153,154 ECP is also currently utilized for patients with acute and/or chronic skin and nonskin GVHD (ASFA categories II and III, respectively) and for solid organ transplant rejection (ASFA category II).1 Its use is also expanding into the treatment of select autoimmune diseases such as pemphigus vulgaris, scleroderma, inflammatory bowel disease, and nephrogenic systemic fibrosis (ASFA category III). The molecular mechanisms for the therapeutic effects of ECP are not fully understood. The cytotoxic effects and the role of other cell populations, including dendritic cells, T cells, and natural killer cells, continue to be investigated. Detailed discussions of all the cellular mediators in the process described below are beyond the scope of this article but have been reviewed elsewhere.155-158 Cell death by apoptosis appears to be a major mechanism of action that occurs within 24 to 72 hours of photoactivation; however, 5% to 15% of the total lymphocyte population is exposed to treatment during each procedure.159 Thus, additional and/or complementary mechanisms of action are also important. Exposed monocytes undergo apoptosis later than lymphocytes but a portion differentiate into immature dendritic cells.159 These dendritic cells have been identified as key mediators of peripheral tolerance157 and are found in patients treated with ECP for chronic GVHD.160 Together with macrophages, these immature dendritic cells are the antigen-presenting cells that recognize, engulf, and display cellular determinants from the apoptotic lymphocytes. After engulfing apoptotic cells, the immature dendritic cells differentiate into semi-mature dendritic cells, migrate to lymph nodes, and present antigenic peptides to T lymphocytes. This brings about a shift from a Th1 to a Th2 immune response, an increase in anti-inflammatory cytokines (eg, interleukin 10, transforming growth factor β), a decrease in proinflammatory cytokines, and the proliferation of T-regulatory cells. These T-regulatory cells down-regulate the GVHD process by inactivating T-effector cells158,161-163 and encouraging peripheral tolerance. In the treatment of CTCL, the apoptotic tumor debris is thought to provide target antigens for cytotoxic CD8+ lymphocytes.164 Cutaneous T-Cell Lymphoma Cutaneous lymphomas are characterized by the localization of malignant lymphocytes in the skin. Approximately two-thirds of these lymphomas are of T-cell origin. The most common form of CTCL is January 2015, Vol. 22, No. 1 mycosis fungoides, which makes up 60% of CTCL cases.153 By contrast, Sézary syndrome, which is an aggressive form of advanced CTCL, occurs in about 5% of patients.165 In Sézary syndrome, the prognosis is generally poor and has a median survival rate of less than 3 years.166 A meta-analysis of 19 studies that utilized ECP for patients at all stages of CTCL showed overall response rates of 55.5% and 55.7% with ECP alone and ECP in combination with other therapies, respectively.167 Scarisbrick et al 154 concluded that all patients with erythrodermic CTCL (major criteria) are candidates for ECP, and those with a peripheral blood T-cell clone and/or circulating Sézary cells comprising more than 10% of the lymphocytes and/or have a CD4:CD8 ratio higher than 10 (minor criteria) may also benefit from ECP. Response to ECP has been linked to a short duration of disease, the absence of bulky lymphadenopathy or internal organ involvement, a WBC count lower than 20,000/µL, fewer than 20% Sézary cells, normal or mildly abnormal natural killer cell activity, a level of CD8+ T cells above 15%, lack of prior intensive chemotherapy, and plaque-stage disease involving 10% to 15% of the skin surface.154 Fewer patients with nonerythrodermic CTCL have been treated with ECP and 1 randomized crossover study alone suggested no ECP benefit.168 Guidelines from the National Comprehensive Cancer Network recommend ECP as an option for advanced mycosis fungoides and Sézary syndrome (stage 2B, 3, or 4) when the disease is refractory to skin-directed treatment.169 However, ECP is not expected to increase survival; typically, the treatment delays the progression of disease and improves pruritus.170 The standard schedule of ECP for the treatment of CTCL consists of procedures performed on 2 consecutive days every 2 to 4 weeks and generally continued for up to 6 months to assess response.1,154 The median time for a response to ECP is 5 to 6 months, although response may take as long as 10 months in some patients. Those who respond after 6 to 8 cycles appear to have an improved long-term outcome. When maximal response is achieved, ECP treatments can be reduced to once every 6 to 12 weeks with the goal of discontinuation if relapses do not occur. If CTCL recurs in more than 25% of the skin, then ECP once or twice monthly should be reinstituted. If evidence exists of disease progression after 6 months of ECP alone, combination therapy should be considered. If minimal or no response is seen after 3 months of combination therapy, then ECP should be discontinued.1 Extracorporeal Cellular Therapy in Graft-vs-Host Disease GVHD remains a major complication of allogeneic January 2015, Vol. 22, No. 1 HSCT. Despite an overall improvement in human leukocyte antigen typing, conditioning regimens, supportive care, and post-transplantation immunosuppression, the overall incidence of GVHD has increased because an increasing number of older patients are undergoing allogeneic HSCT and the use of haploidentical, double-cord blood and human leukocyte antigen–mismatched donors are being used.171,172 GVHD following HSCT is classified as an acute, chronic, or overlap syndrome. Despite prophylactic therapy with immunosuppressive agents, 20% to 80% of patients develop acute GVHD following allogeneic HSCT. Acute GVHD results from the activation of donor T cells by host antigen–presenting cells, leading to T-cell– and cytokine-mediated tissue injury.171 Chronic GVHD is due to dysregulated allogeneic or autoreactive T cells, B cells, antigen-presenting cells, and natural killer cells, thus leading to fibrosis, inflammation, sclerosis, and atrophy of affected tissues.163 Moderate-to-severe GVHD is the leading cause of impaired immune function, compromised functional status, and transplantation-related deaths. High-dose corticosteroids are first-line therapy for moderate-to-severe acute and chronic GVHD with or without the use of calcineurin inhibitors.173 Patients with chronic GVHD require prolonged immunosuppressive treatment for an average of 2 to 3 years from the initial diagnosis, with 10% of those surviving for at least 7 years still requiring immunosuppressive treatment at that time or beyond.174 Severe GVHD unresponsive to treatment carries a high risk of death or severe morbidity due to end-organ complications, infections, or both, and the transplantation-related mortality rate exceeds 40%.175,176 To date, the US Food and Drug Administration has not approved a treatment option for GVHD. Therapies for steroid-refractory acute GVHD include mycophenolate mofetil, denileukin diftitox, sirolimus, infliximab, etanercept, pentostatin, horse vs rabbit antithymocyte globulin, and alemtuzumab.173,177,178 Evidence does not suggest that any one second-line agent is superior to another.173 As a result, decisions on which agent to use at individual treatment centers often vary according to the clinical experience of health care professionals, cost, and treatment availability. ASFA has reviewed the data available on the overall response rates to ECP for steroid-refractory acute GVHD and found that overall response rates in pediatric and adult patients ranged from 52% to 100%, with responses in cutaneous (66%–100%), gastrointestinal tract (40%–83%), and hepatic (27%–71%) acute GVHD.1 Higher response rates have been reported in early-onset GVHD179; however, the strongest predictor for response to ECP in a multivariate analysis was GVHD severity (100% response in grade 2 disease vs 30% in grade 3/4).180 Complete responses and Cancer Control 71 improved survival rates are often reported among acute GVHD cohorts; however, the nonrandomized and retrospective results for ECP are not superior to results reported for alternative salvage approaches for steroid-refractory acute GVHD. Therapies for steroid-refractory/dependent chronic GVHD include sirolimus, mycophenolate mofetil, azathioprine, thalidomide, ECP, total lymphoid irradiation, mesenchymal cells, imatinib, pentostatin, various monoclonal antibodies, and others.177,178 Approximately 30% to 65% of patients with chronic GVHD and dependent on steroids improve with ECP, but most experience partial responses alone.1 Skin, oral, and ocular chronic GVHD manifestations respond in 30% to 100% of cases, whereas liver, joint, and gastrointestinal complications improve in 30% to 80%, 50%, and 0% to 50% of cases, respectively.1 A review of 23 studies totaling 735 patients treated with ECP for steroid-resistant, intolerant, or dependent chronic GVHD noted that overall and complete response rates were observed in 64% and 35% of cases with skin involvement, in 56% and 27% cases of oral GVHD, and in 47% to 57% with gastrointestinal tract chronic GVHD.181 ECP has also been reported to stabilize lung function with bronchiolitis obliterans syndrome related to chronic GVHD182; however, response rates for lung involvement are typically lower, ranging from 0% to 66%.183,184 Patients responding to ECP also have a better probability of survival, both in children (96% vs 58% 5-year survival)185 and in adults (88% vs 18% at 2 years; relative risk, 11.6; P = .022).186 Maximal responses for chronic GVHD usually require 2 to 6 months of treatment. The single, randomized controlled trial using ECP for steroid-resistant skin chronic GVHD observed no statistically significant difference in total skin score at 12 weeks of ECP plus salvage GVHD therapy compared with salvage therapy alone.187 However, unblinded assessments recorded 40% complete and partial responses at 12 weeks in the ECP-treated group compared with 10% in the non-ECP group (P < .001).187 More rapid skin improvement was also observed at weeks 12 to 24 of ECP and corticosteroids could be more quickly tapered. Among 29 control patients from this study who crossed over to receive 24 weeks of ECP for refractory disease, objective responses occurred in the skin and extracutaneous tissue in 33% and up to 70%, respectively.187 No national consensus exists on the duration and discontinuation of ECP procedures. For acute GVHD, ECP is recommended on 2 consecutive days (1 cycle) per week until disease response and then tapered to alternate weeks before discontinuation.1 Some centers have recommended a minimum of 8 weeks.188 For chronic GVHD, 1 weekly cycle (or consider biweekly if treating mucocutaneous chronic GVHD alone) until 72 Cancer Control either a response or for 8 to 12 weeks, followed by a taper to every 2 to 4 weeks until maximal response.1 One author has proposed 2 to 3 procedures per week depending on disease severity for 4 weeks or more.189 Clinical response should be assessed weekly in acute GVHD and every 8 to 12 weeks in chronic GVHD; ECP should be discontinued in cases of no or minimal response. Some studies indicate that approximately 10% of patients with chronic GVHD given ECP may benefit from treatment longer than 12 to 24 months.181 Clinical practice guidelines and consensus statements addressing the use of ECP for GVHD collectively consider ECP as an established second-line therapy option for steroid-refractory chronic GVHD, particularly involving the skin.1,190-192 ECP has also been recommended as an adjunctive first-line modality for bronchiolitis obliterans syndrome and select pediatric patients with acute GVHD.1,190-192 More recently, a UK group has provided its consensus statement and guidance on the use of ECP in adult and pediatric patients with acute GVHD.188 The proven effectiveness of ECP in both acute and chronic GVHD cases is mirrored in the ASFA guidelines, which recommend ECP for chronic (category II; grade 1B recommendation) and acute (category II; grade 2C recommendation) GVHD.1 Thrombocytapheresis Thrombocytosis Associated With Myeloproliferative Neoplasms Thrombocytosis is defined as a peripheral blood platelet count above 350,000 to 400,000/μL. Reactive thrombocytosis is the most common cause of an elevated platelet count and can be caused by iron deficiency, inflammatory conditions, infections, malignancy, acute bleeding, hemolysis, and asplenia. Because the platelets in these conditions are functionally normal, the increased platelet count does not normally predispose to thrombosis or acute bleeding. However, functionally abnormal platelets are associated with the elevated platelet counts seen in patients with myeloproliferative neoplasms (eg, essential thrombocythemia, polycythemia vera, chronic myelogenous leukemia, primary myelofibrosis) and refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Functionally abnormal thrombocytosis is associated with an increased incidence of thrombohemorrhagic events.193,194 Accurate diagnoses of thrombocytosis are important for both prognostication and treatment.195 Diagnoses of essential thrombocythemia and polycythemia vera are currently in accordance with criteria from the World Health Organization and are based on a composite assessment of clinical and laboratory (hematological, morphological, and molecular) features.196 When evaluating thrombocytosis, the detection of the clonal mutation JAK2 V617F confirms January 2015, Vol. 22, No. 1 the presence of an underlying myeloproliferative neoplasm. However, the absence of this mutation does not rule out the possibility. Up to 50% of patients with essential thrombocythemia might be JAK2 V617F negative; however, finding a mutation in a newly described genetic marker, CALR, or, less commonly, MPL, can identify the majority of cases that are JAK2 mutation negative.197 Current risk stratification in essential thrombocythemia and polycythemia vera is designed to estimate the likelihood of thrombotic complications. High risk is defined by age older than 60 years or history of the presence of thrombosis, whereas low risk is defined by the absence of both of these 2 risk factors.198-200 Extreme thrombocytosis (platelet count > 1,000,000/μL) can be associated with acquired von Willebrand syndrome and, thus, a risk for bleeding.201 Risk factors for shortened survival rates in both polycythemia vera and essential thrombocythemia include advanced age, leukocytosis, and a history of thrombosis.198,199,202 Major thrombotic complications with essential thrombocythemia and polycythemia vera include stroke, transient ischemic attacks, myocardial infarction, peripheral arterial thrombosis, lower extremity deep venous thrombosis, pulmonary embolism, and venous thrombosis in unusual sites such as hepatic (Budd Chiari syndrome), portal, and mesenteric veins.203 The risk of thrombosis in essential thrombocythemia and polycythemia vera exceeds 20% and a substantial portion of patients experience microcirculation disturbances.204 The most frequent bleeding events are hemorrhages from the gastrointestinal tract followed by hematuria and other mucocutaneous hemorrhages. Hemarthrosis and large muscle hematomas are uncommon.203 Patients with essential thrombocythemia and low risk of thrombosis are given low-dose aspirin if microvascular symptoms are present but do not require cytoreductive therapy. High-risk patients are treated with cytoreductive therapy, such as hydroxyurea, interferon α, or, less commonly, anagrelide in conjunction with low-dose aspirin. Thrombocytapheresis has been used to treat acute thromboembolism or hemorrhage in select patients with essential thrombocythemia or polycythemia vera associated with uncontrolled thrombocytosis.205-207 The current ASFA guidelines are based on observational case studies or case reports (category II; grade 2C recommendation).1 Thrombocytapheresis should also be electively considered for cytoreduction in patients at increased risk of hemorrhage in whom hydroxyurea is contraindicated, such as in cases of pregnancy,208-210 or if cytoreductive therapy with hydroxyurea is likely to be too slow (eg, urgent surgery is required).211,212 Because the beneficial effect of platelet reduction is January 2015, Vol. 22, No. 1 generally quite brief, repeat procedures are often necessary, and it is generally recommended that platelet-lowering agents be given whenever possible to prevent rapid reaccumulation of circulating platelets.205,206 Each thrombocytapheresis procedure (treating 1.5–2 blood volumes) lowers the platelet count by about 30% to 60%. Pre- and post-platelet counts should be closely monitored to gauge the effectiveness of platelet removal and to guide subsequent treatments. The goal of thrombocytapheresis in acute thromboembolism or hemorrhage is the normalization of the platelet count and maintenance of a normal platelet count until pharmacological cytoreductive therapy takes effect. The goal for prophylaxis in highrisk patients who are pregnant or undergoing surgery or postsplenectomy should be based on the patient’s history of bleeding or thrombosis. Erythrocytapheresis Polycythemia Vera/Primary Erythrocytosis Polycythemia vera is characterized by bone marrow hypercellularity, atypical megakaryocyte hyperplasia, leukocytosis, thrombocytosis, splenomegaly, and a clinical predilection for thromboembolism, bleeding, hyperviscosity complications, and the evolution to myelofibrosis or AML. The JAK2 V617F mutation is found in more than 90% of cases.213,214 In polycythemia vera, whole blood viscosity increases significantly as the hematocrit level exceeds 50%. Malaise, headache, visual disturbances, pruritus, dizziness, confusion, slow mentation, and myalgia are the most common symptoms. Similar to essential thrombocythemia, 15% to 40% of patients with polycythemia vera may experience major arterial cerebrovascular or cardiovascular thromboembolic events, deep venous thrombosis, pulmonary embolism, or intra-abdominal venous thrombotic events. Thrombotic risk factors with polycythemia vera include uncontrolled erythrocytosis (hematocrit > 55%), age older than 60 years, history of prior thrombosis, cardiovascular comorbidities, immobilization, pregnancy, and surgery.215 RBC depletion by manual phlebotomy or by automated therapeutic erythrocytapheresis can correct hyperviscosity complications with uncontrolled polycythemia vera by lowering the hematocrit level, thereby reducing capillary shear and increasing microcirculatory blood flow and tissue perfusion. Classical manual phlebotomy is a simple, safe, and low-cost method. However, it can require a significant number of procedures to reach target values.216-218 Adverse events related to hypovolemia with manual phlebotomy occur in a substantial number of patients, and, thus, this treatment modality may not be tolerated in the elderly, those with small blood volumes, and those with cardiovascular compromise. However, with autoCancer Control 73 mated therapeutic erythrocytapheresis, up to 800 mL of RBCs per single procedure can be separated from other blood components and concurrently exchanged with a crystalloid or colloid solution, thus offering a far more efficient method in removing RBCs while maintaining isovolemic conditions. In the past 2 decades, 1 randomized trial and a number of small case series have described the advantages of automated therapeutic erythrocytapheresis for the treatment of hereditary hemochromatosis and erythrocytosis with improvements seen in treatment efficiency, morbidity, and patient experience.219-223 For patients with polycythemia vera and acute thromboembolism, severe microvascular complications, or bleeding, automated therapeutic erythrocytapheresis may be a useful alternative to emergent large-volume phlebotomy, particularly if the patient is hemodynamically unstable. Automated therapeutic erythrocytapheresis can also be successfully utilized with polycythemia vera complicated by thrombocytosis; during the same session, the hematocrit level can be lowered to 42% ± 45% and the platelets reduced to 500,000 to 600,000/μL.220,224 Therapeutic erythrocytapheresis may also be appropriate prior to surgery to reduce the high risk of perioperative thrombotic complications in a patient with polycythemia vera and a hematocrit level of more than 55%. A number of studies have been published supporting the use of therapeutic erythrocytapheresis as maintenance therapy. One study of 76 patients with polycythemia vera saw improvement in platelet function, as measured by thromboelastography, after therapeutic erythrocytapheresis, suggesting that the hemodilution achieved with the procedure may reduce thrombotic risk.225 A retrospective cohort analysis of 98 patients, including 6 with polycythemia vera and 92 with secondary erythrocytosis, observed that chronic automated therapeutic erythrocytapheresis allowed significantly greater treatment intervals (median, 135–150 days; range, 2–7 months) to maintain the target hematocrit level compared with chronic phlebotomy (median, 40 days; range, 20–60 days).226 The advantage of therapeutic erythrocytapheresis may be due to the relatively greater loss of iron that is associated with this modality that, in turn, limits the growth of hematopoietic cells.227 The ASFA guidelines designate polycythemia vera as a category I indication (grade 1B recommendation) for therapeutic erythrocytapheresis.1 Decisions to use an automated procedure over simple phlebotomy remain based on clinical urgency, necessity, cost, and consideration of the risk of adverse events that may be associated with automated procedures. Although the costs of a single therapeutic erythrocytapheresis procedure are substantially higher than phlebotomy, cost analysis has shown no significant difference in 74 Cancer Control maintenance treatment costs as a result of the fewer treatment procedures needed to reach recommended target values.221 One group developed a simple and practical mathematical model for predicting the efficiency of a single cycle of therapeutic erythrocytapheresis compared with a single phlebotomy procedure, which could in daily clinical practice aid in optimizing therapeutic erythrocytapheresis use and selecting a proper treatment modality for the individual patient.228 For example, the researchers determined that therapeutic erythrocytapheresis would not be optimal for patients with a small blood volume and/ or marginal achievable change in hematocrit level.228 For patients with polycythemia vera, the goal of therapeutic erythrocytapheresis is rapid normalization of hematocrit (ie, < 45%). A single procedure should be designed to achieve the desired postprocedure hematocrit level. Automated instruments allow the operator to choose a postprocedure hematocrit level and calculate the volume of blood removal necessary to attain the goal. Saline boluses may be required during the procedure to reduce blood viscosity in the circuit and avoid pressure alarms.1 Conclusions Therapeutic apheresis (TA) is an important treatment option utilized in patients to manage specific complications associated with malignancy. TA has been used as an emergent procedure, including as a therapeutic plasma exchange to treat symptomatic hyperviscosity or leukocytapheresis for the treatment of leukostasis. TA can be effective as first-line therapy — as seen in the use of extracorporeal photopheresis for erythrodermic cutaneous T-cell lymphoma — although often TA is attempted as salvage or adjunct therapy for conditions not responding to conventional chemotherapy or immunotherapy. Examples of such circumstances include therapeutic plasma exchange for the removal of antibodies associated with underlying paraneoplastic processes or the use of extracorporeal photopheresis for non–skin-associated graft-vs-host disease. TA modalities are relatively safe procedures; however, they are not without risk. In order for these modalities to be performed, experienced staff members are required. In all cases, the risks, benefits, and costs must be strongly considered before prescribing. The expert-based practice guidelines from the American Society for Apheresis have been developed to inform hematology/oncology professionals and apheresis physicians about the efficacy and limitations of TA for malignancy-related indications as well as to support clinical decision-making. However, well-designed, prospective intervention trials are still needed to better define the role of TA for a variety of disorders. January 2015, Vol. 22, No. 1 References 1. Schwartz J, Winters JL, Padmanabhan A, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145-284. 2. Burgstaler EA. Current instrumentation for apheresis. In: McLeod BC, Szczepiorkowski ZM, Weinstein R, et al, eds. Apheresis: Principles and Practice. 3rd ed. Bethesda, MD: AABB Press. 2010;95-130. 3. Okafor C, Ward DM, Mokrzycki MH, et al. Introduction and overview of therapeutic apheresis. J Clin Apher. 2010;25(5):240-249. 4. Madore F. Plasmapheresis. Technical aspects and indications. Crit Care Clin. 2002;18(2):375-392. 5. Linenberger ML, Price TH. Use of cellular and plasma apheresis in the critically ill patient: part 1: technical and physiological considerations. J Intensive Care Med. 2005;20(1):18-27. 6. Stegmayr B, Wikdahl AM. Access in therapeutic apheresis. Ther Apher Dial. 2003;7(2):209-214. 7. Malchesky PS, Koo AP, Skibinski CI, et al. Apheresis technologies and clinical applications: the 2007 International Apheresis Registry. Ther Apher Dial. 2010;14(1):52-73. 8.Schönermarck U, Bosch T. Vascular access for apheresis in intensive care patients. Ther Apher Dial. 2003;7(2):215-220. 9. Lok CE, Mokrzycki MH. Prevention and management of catheter-related infection in hemodialysis patients. Kidney Int. 2011;79(6):587-598. 10. Golestaneh L, Mokrzycki MH. Vascular access in therapeutic apheresis: update 2013. J Clin Apher.2013; 28(1):64-72. 11. Sutton DM, Nair RC, Rock G. Complications of plasma exchange. Transfusion. 1989;29(2):124-127. 12. Noseworthy JH, Shumak KH, Vandervoort MK; Canadian Cooperative Multiple Sclerosis Study Group. Long-term use of antecubital veins for plasma exchange. Transfusion. 1989;29(7):610-613. 13. Grishaber JE, Cunningham MC, Rohret PA, et al. Analysis of venous access for therapeutic plasma exchange in patients with neurological disease. J Clin Apher.1992;7(3):119-123. 14. Couriel D, Weinstein R. Complications of therapeutic plasma exchange: a recent assessment. J Clin Apher. 1994;9(1):1-5. 15. Basic-Jukic N, Kes P, Glavas-Boras S, et al. Complications of therapeutic plasma exchange: experience with 4857 treatments. Ther Apher Dial. 2005;9(5):391-395. 16. Shemin D, Briggs D, Greenan M. Complications of therapeutic plasma exchange: a prospective study of 1,727 procedures. J Clin Apher. 2007;22(5):270-276. 17. Allon M. Current management of vascular access. Clin J Am Soc Nephrol. 2007;2(4):786-800. 18. Mokrzycki MH, Kaplan AA. Therapeutic plasma exchange: complications and management. Am J Kidney Dis. 1994;23(6):817-827. 19. Mokrzycki MH, Balogun RA. Therapeutic apheresis: a review of complications and recommendations for prevention and management. J Clin Apheresis. 2011;26(5):243-248. 20. Guyatt GH, Cook DJ, Jaeschke R, et al. Grades of recommendation for antithrombotic agents: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th ed). Chest. 2008;133(6 suppl):123S-31S. 21. Shaz BH, Schwartz J, Winters JL. How we developed and use the American Society for Apheresis guidelines for therapeutic apheresis procedures. Transfusion. 2014;54(1):17-25. 22. Goto H, Matsuo H, Nakane S, et al. Plasmapheresis affects T helper type-1/T helper type-2 balance of circulating peripheral lymphocytes. Ther Apher. 2001;5(6):494-496. 23. Kambara C, Matsuo H, Fukudome T, et al. Miller Fischer syndrome and plasmapheresis. Ther Apher. 2002;6(6):450-453. 24. Shariatmadar S, Nassiri M, Vincek V. Effect of plasma exchange on cytokines measured by multianalyte bead array in thrombotic thrombocytopenic purpura. Am J Hematol. 2005;79(2):83-88. 25. Szczepiorkowski ZM, Winters JL, Bandarenko N, et al. Guidelines on the use of therapeutic apheresis in clinical practice: evidence-based approach from the Apheresis Applications Committee of the American Society for Apheresis. J Clin Apher. 2010;25(3):83-177. 26. Flaum MA, Cuneo RA, Appelbaum FR, et al. The hemostatic imbalance of plasma-exchange transfusion. Blood. 1979;54(3):694-702. 27. Chirnside A, Urbaniak SJ, Prowse CV, et al. Coagulation abnormalities following intensive plasma exchange on the cell separator. II. Effects on factors I, II, V, VII, VIII, IX, X and antithrombin III. Br J Haematol.1981;48(4):627-634. 28. Orlin JB, Berkman EM. Partial plasma exchange using albumin replacement: removal and recovery of normal plasma constituents. Blood. 1980;56(6):1055-1059. 29. Ibrahim RB, Liu C, Cronin SM, et al. Drug removal by plasmapheresis: an evidence-based review. Pharmacotherapy. 2007;27(11):1529-1549. 30. Kintzel PE, Eastlund T, Calis KA. Extracorporeal removal of antimicrobials during plasmapheresis. J Clin Apher. 2003;18(4):194-205. 31. Finlayson JS. Albumin products. Semin Thromb Hemost.1980;6:85-120. 32. Matejtschuk P, Dash CH, Gascoigne EW. Production of human albumin solutions: a continually developing colloid [Erratum appears in Br J Anaesth. 2001;86(2):301]. Br J Anaesth. 2000;85(6):887-895. January 2015, Vol. 22, No. 1 33. Winters JL, Brown D, Hazard E, et al. Cost-minimization analysis of the direct costs of TPE and IVIg in the treatment of Guillain-Barré syndrome. BMC Health Serv Res. 2011;11:101. 34. McLeod BC. Plasma and plasma derivatives in therapeutic plasmapheresis. Transfusion. 2012;52(suppl 1);38S-44S. 35. Michael M, Elliot EJ, Craig JC, et al. Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a systematic review of randomized controlled trials. Am J Kidney Dis. 2009;53(2):259-272. 36. McLeod B, Gregory SA. Solvent/detergent plasma replacement in a highly plasma allergic patient with TTP. J Clin Apher. 2001;16:98. 37. Wang H, Chen Y, Li F, et al. Temporal and geographic variations of Waldenstrom macroglobulinemia incidence: a large population-based study. Cancer. 2012;118(15):3793-3800. 38. Treon SP. How I treat Waldenström macroglobulinemia. Blood. 2009:114(12);2375-2385. 39. Fahey JL, Barth WF, Solomon A. Serum hyperviscosity syndrome. JAMA. 1965;192(6):120-123. 40. Schwab PJ, Fahey JL. Treatment of Waldenström’s macroglobulinemia by plasmapheresis. N Engl J Med. 1960;263(2):574-579. 41. Solomon A, Fahey JL. Plasmapheresis therapy in macroglobulinemia. Ann Intern Med. 1963;58(5):789-800. 42. Thomas EL, Olk RJ, Markman M, et al. Irreversible visual loss in Waldenström’s macroglobulinaemia. Br J Ophthalmol.1983;67(2):102-106. 43. Menke MN, Feke GT, McMeel JW, et al. Ophthalmologic techniques to assess the severity of hyperviscosity syndrome and the effect of plasmapheresis in patients with Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma. 2009;9(1):100-103. 44. Stone MJ, Bogen SA. Evidence-based focused review of management of hyperviscosity syndrome. Blood. 2012;119(10):2205-2208. 45. Stone MJ. Pathogenesis and morbidity of autoantibody syndromes in Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2011;11(1):157-159. 46. Frase LL, Stone MJ, Sammons CA. Long-term survival in Waldenström’s macroglobulinemia. Am J Med. 1998;104(5):507-508. 47. Ballestri M, Ferrari F, Magistroni R, et al. Plasma exchange in acute and chronic hyperviscosity syndrome: a rheological approach and guidelines study. Ann Ist Super Sanita. 2007;43(2):171-175. 48. Dimopoulos MA, Zervas C, Zomas A, et al. Treatment of Waldenström’s macroglobulinemia with rituximab. J Clin Oncol. 2002;20(9):2327-2333. 49. Ansell SM, Kyle RA, Reeder CB, et al. Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin Proc. 2010; 85(9):824-833. 50. Capra JD, Kunkel HG. Aggregation of gammaG3 proteins: relevance to the hyperviscosity syndrome. J Clin Invest. 1970;49(3):610-621. 51. Bloch KJ, Maki DG. Hyperviscosity syndromes associated with immunoglobulin abnormalities. Semin Hematol. 1973;10(2):113-124. 52. Fahey JL, Barth WF, Solomon A. Serum hyperviscosity syndrome. JAMA.1965;192(6):120-123. 53. Dimopoulos MA, Kastritis E, Rosinol L, et al. Pathogenesis and treatment of renal failure in multiple myeloma. Leukemia. 2008;22(8):1485-1493. 54. Knudsen LM, Hjorth M, Hippe E; Nordic Myeloma Study Group. Renal failure in multiple myeloma: reversibility and impact on the prognosis. Eur J Haematol. 2000;65(3):175-181. 55. Montseny JJ, Kleinknecht D, Meyrier A, et al. Long-term outcome according to renal histological lesions in 118 patients with monoclonal gammopathies. Nephrol Dial Transplant. 1998;13(6):1438-1445. 56. Pillon L, Sweeting RS, Arora A, et al. Approach to acute renal failure in biopsy proven myeloma cast nephropathy: is there still a role for plasmapheresis? Kidney Int. 2008;74(7):956-961. 57. Huang ZQ, Sanders PW. Localization of a single binding site for immunoglobulin light chains on human Tamm-Horsfall glycoprotein. J Clin Invest.1997;99(4):732-736. 58. Ying WZ, Sanders PW. Mapping the binding domain of immunoglobulin lightchains for Tamm-Horsfall protein. Am J Pathol. 2001;158(5):1859-1866. 59. Heher EC, Goes NB, Spitzer TR, et al. Kidney disease associated with plasma cell dyscrasias. Blood. 2010;116(9):1397-1404. 60. Pozzi C, Pasquali S, Donini U, et al. Prognostic factors and effectiveness of treatment in acute renal failure due to multiple myeloma: a review of 50 cases. Report of the Italian Renal Immunopathology Group. Clin Nephrol. 1987;28(1):1-9. 61. Zucchelli P, Pasquali S, Cagnoli L, et al. Controlled plasma exchange trial in acute renal failure due to multiple myeloma. Kidney Int. 1988;33(6):1175-1180. 62. Johnson WJ, Kyle RA, Pineda AA, et al. Treatment of renal failure associated with multiple myeloma. Plasmapheresis, hemodialysis, and chemotherapy. Arch Intern Med. 1990;150(4):863-869. 63. Durie BG, Kyle RA, Belch A et al. Myeloma management guidelines: a consensus report from the Scientific Advisors of the International Myeloma Foundation. Hematol J. 2003;4(6):379-398. 64. Clark WF, Stewart AK, Rock GA, et al; Canadian Apheresis Group. Plasma exchange when myeloma presents as acute renal failure: a randomized, controlled trial. Ann Intern Medicine. 2005;143(11):777-784. 65. Burnette BL, Leung N, Rajkumar SV. Renal improvement in myeloma with bortezomib plus plasma exchange. N Engl J Med. 2011;364(24):2365-2366. 66.Bladé J, Rosiñol L. Renal, hematologic and infectious complications Cancer Control 75 in multiple myeloma. Best Pract Res Clin Haematol. 2005;18(4):635-652. 67. Torra J, Bladé A, Cases A, et al. Patients with multiple myeloma and renal failure requiring long-term dialysis: presenting features, response to therapy, and outcome in a series of 20 cases. Br J Haematol. 1995;91(4):854-859. 68. Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Eng J Med. 2003;349(16):1543-1554. 69. Antoine JC, Camdessanché JP. Peripheral nervous system involvement in patients with cancer. Lancet Neurol. 2007;6(1):75-86. 70. Elrington GM, Murray NM, Spiro SG, Newsom-Davis J. Neurological paraneoplastic syndromes in patients with small cell lung cancer. A prospective survey of 150 patients. J Neurol Neurosurg Psychiatry. 1991;54(9):764-767. 71. Levy Y, Afek A, Sherer Y, et al. Malignant thymoma associated with autoimmune diseases: a retrospective study and review of the literature. Semin Arthritis Rheum. 1998;28(2):73-79. 72. Sculier JP, Feld R, Evans WK, et al. Neurologic disorders in patients with small cell lung cancer. Cancer. 1987;60(9):2275-2283. 73. Darnell RB. Onconeural antigens and paraneoplastic neurologic disorders: At the intersection of cancer, immunity, and the brain. Proc Natl Acad Sci USA. 1996;93(10):4529-4536. 74. Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn). 2012;18(2):366-383. 75. Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75(8):1135-1140. 76. Keime-Guibert F, Graus F, Fleury A, et al. Treatment of paraneoplastic neurological syndromes with antineuronal antibodies (anti-Hu, anti-Yo) with a combination of immunoglobulins, cyclophosphamide, and methylprednisolone. J Neurol Neurosurg Psychiatry. 2000;68(4):479-482. 77. Vernino S, O’Neill BP, Marks RS, et al. Immunomodulatory treatment trial for paraneoplastic neurological disorders. Neuro Oncol. 2004;6(1):55-62. 78. Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neuro. 2008;7(4):327-340. 79. Bradley WH, Dottino PR, Rahaman J. Paraneoplastic cerebellar degeneration in ovarian carcinoma: case report with review of immune modulation. Int J Gynecol Cancer. 2008;18(6):1364-1367. 80. Croteau D, Owainati A, Dalmau J, et al. Response to cancer therapy in a patient with a paraneoplastic choreiform disorder. Neurology. 2001;57(4):719-722. 81. Vigliani MC, Palmucci L, Polo P, et al. Paraneoplastic opsoclonus-myoclonus associated with renal cell carcinoma and responsive to tumour ablation. J Neurol Neurosurg Psychiatry. 2001;70(6):814-815. 82. Widdess-Walsh P, Tavee JO, Schuele S, et al. Response to intravenous immunoglobulin in anti-Yo associated paraneoplastic cerebellar degeneration: case report and review of the literature. J Neurooncol. 2003;63(2):187-190. 83. David YB, Warner E, Levitan M, et al. Autoimmune paraneoplastic cerebellar degeneration in ovarian carcinoma patients treated with plasmapheresis and immunoglobulin: a case report. Cancer. 1996;78(10):2153-2156. 84. Giometto B, Vitaliani R, Lindeck-Pozza E, et al. Treatment for paraneoplastic neuropathies. Cochrane Database Syst Rev. 2012;(12):CD007625. 85. Shams’ili S, Grefkens J, De Leeuw B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain. 2003;126(pt 6):1409-1418. 86. Newsom-Davis J, Murray NM. Plasma exchange and immunosuppressive drug treatment in the Lambert-Eaton myasthenic syndrome. Neurology. 1984;34(4):480-485. 87. Greenlee JE. Treatment of paraneoplastic cerebellar degeneration. Curr Treat Options Neurol. 2013;15(2):185-200. 88. Vernino S, O’Neill BP, Marks RS, et al. Immunomodulatory treatment trial for paraneoplastic neurological disorders. Neuro Oncol. 2004;6(1):55-62. 89. van Broekhoven F, de Graaf MT, Bromberg JE, et al. Human chorionic gonadotropin treatment of anti-Hu-associated paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2010;81(12):1341-1344. 90. Shams’ili S, de Beukelaar J, Gratama JW, et al. An uncontrolled trial of rituximab for antibody associated paraneoplastic neurological syndromes. J Neurol. 2006;253(1):16-20. 91. Widdess-Walsh P, Tavee JO, Schuele S, et al. Response to intravenous immunoglobulin in anti-Yo associated paraneoplastic cerebellar degeneration: case report and review of the literature. J Neuro Oncol. 2003,63(2):187-190. 92. Laszik Z, Silva F. Hemolytic-uremic syndrome, thrombotic thrombocytopenic purpura, and systemic sclerosis (systemic scleroderma). In: Jennet JC, Olson JL, Schwartz MM, et al, eds. Heptinstall’s Pathology of the Kidney. Philadelphia: Lippincott-Raven; 1998:1003-1057. 93. Chow AY, Chin C, Dahl G, Rosenthal DN. Anthracyclines cause endothelial injury in pediatric cancer patients: a pilot study. J Clin Oncol. 2006;24(6):925-928. 94. Nagaya S, Wada H, Oka K, et al. Hemostatic abnormalities and increased vascular endothelial cell markers in patients with red cell fragmentation syndrome induced by mitomycin C. Am J Hematol. 1995;50(4):237-243. 95. Oner AF, Gürgey A, Kirazli S, et al. Changes of hemostatic factors in children with acute lymphoblastic leukemia receiving combined chemotherapy including high dose methylprednisolone and L-asparaginase. Leuk Lymphoma. 1999;33(3-4):361-364. 96. Fajardo LF. The pathology of ionizing radiation as defined by morphologic patterns. Acta Oncol. 2005;44(1):13-22. 76 Cancer Control 97. Nacar A, Kiyici H, Oğüş E, et al. Ultrastructural examination of glomerular and tubular changes in renal allografts with cyclosporine toxicity. Ren Fail. 2006;28(7):543-547. 98. Burke GW, Ciancio G, Cirocco R, et al. Microangiopathy in kidney and simultaneous pancreas/kidney recipients treated with tacrolimus:evidence of endothelin and cytokine involvement. Transplantation. 1999;68(9):1336-1342. 99. Labrador J, López-Corral L, López-Godino O, et al. Risk factors for thrombotic microangiopathy in allogeneic hematopoietic stem cell recipients receiving GVHD prophylaxis with tacrolimus plus MTX or sirolimus. Bone Marrow Transplant. 2014;49(5):684-690. 100. Henry N, Li S, Kim HT, et al. Sirolimus and thrombotic microangiopathy after allogeneic stem cell transplantation. Blood. 2004;104:508a. 101. Fuge R, Bird JM, Fraser A, et al. The clinical features, risk factors and outcome of thrombotic thrombocytopenic purpura occurring after bone marrow transplantation. Br J Haematol. 2001;113(1):58-64. 102. Biedermann BC, Sahner S, Gregor M, et al. Endothelial injury mediated by cytotoxic T lymphocytes and loss of microvessels in chronic graft versus host disease. Lancet. 2002;359(9323):2078-2083. 103. Martinez MT, Bucher CH, Stussi G, et al. Transplant-associated microangiopathy (TAM) in recipients of allogeneic hematopoietic stem cell transplants. Bone Marrow Transplant. 2005;36(11):993-1000. 104. Matsuda Y, Hara J, Miyoshi H, et al. Thrombotic microangiopathy associated with reactivation of human herpesvirus-6 following high-dose chemotherapy with autologous bone marrow transplantation in young children. Bone Marrow Transplant. 1999;24(8):919-923. 105. Grigg A, Clouston D. Disseminated fungal infection and early onset microangiopathy after allogeneic bone marrow transplantation. Bone Marrow Transplant. 1995;15(5):795-797. 106. Cohen H, Bull HA, et al. Vascular endothelial cell function and ultrastructure in thrombotic microangiopathy following allogeneic bone marrow transplantation. Eur J Haematol. 1989;43(3):207-214. 107. Jimenez JJ, Jy W, Mauro LM. Endothelial microparticles released in thrombotic thrombocytopaenic purpura express von Willebrand factor and markers of endothelial activation. Br J Haematol. 2003;123(5):896-902. 108. Jy W, Jimenz J, Mauro LM, et al. Endothelial microparticles induce formation of platelet aggregates via a von Willebrand factor/ristocetin dependent pathway, rendering them resistant to dissociation. J Thromb Haemost. 2005;3(6):1301-1308. 109. Elliott MA, Nichols WL, Plumhoff EA, et al. Posttransplantation thrombotic thrombocytopenic purpura: a single-center experience and a contemporary review. Mayo Clin Proc. 2003;78(4):421-430. 110. Arai S, Allan C, Streiff M, et al. Von Willebrand-cleaving protease activity and proteolysis of von Willebrand factor in bone marrow transplant-associated thrombotic microangiopathy. Hematol J. 2001;2(5):292-299. 111. Allford SL, Bird JM, Marks DI. Thrombotic thrombocytopenic purpura following stem cell transplantation. Leuk Lymphoma. 2002;43(10):1921-1926. 112. Willems E, Baron F, Seidel L, et al. Comparison of thrombotic microangiopathy after allogeneic hematopoietic cell transplantation with high-dose or nonmyeloablative conditioning. Bone Marrow Transplant. 2010;45(4):689-693. 113. Laskin BL, Goebel J, Davies SM, et al. Small vessels, big trouble in the kidneys and beyond: hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Blood. 2011;118(6):1452-1462. 114. George JN, Li X, McMinn JR, et al. Thrombotic thrombocytopenic purpura-hemolytic uremic syndrome following allogeneic HPC transplantation: a diagnostic dilemma. Transfusion. 2004;44(2):294-304. 115. Batts ED, Lazarus HM. Diagnosis and treatment of transplantation-associated thrombotic microangiopathy: real progress or are we still waiting? Bone Marrow Transplant. 2007;40(8):709-719. 116. Daly AS, Xenocostas A, Lipton JH. Transplantation associated thrombotic microangiopathy: twenty-two years later. Bone Marrow Transplant. 2002;30(11):709-715. 117. Rock G, Shumak KH, Sutton DM, et al; Members of the Canadian Apheresis Group. Cryosupernatant as replacement fluid for plasma exchange in thrombotic thrombocytopenic purpura. Br J Haematol. 1996;94(2):383-386. 118. Korkmaz S, Keklik M, Sivgin S, et al. Therapeutic plasma exchange in patients with thrombotic thrombocytopenic purpura: a retrospective multicenter study. Transfus Apher Sci. 2013;48(3):353-358. 119. Brunskill SJ, Tusold A, Benjamin S, et al. A systematic review of randomized controlled trials for plasma exchange in the treatment of thrombotic thrombocytopenic purpura. Transfus Med. 2007;17(1):17-35. 120. Ho VT, Cutler C, Carter S, et al. Blood and marrow transplant clinical trials network toxicity committee consensus summary: thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11(8):571-575. 121. Wolff D, Wilhelm S, Hahn J, et al. Replacement of calcineurin inhibitors with daclizumab in patients with transplantation-associated microangiopathy or renal insufficiency associated with graft versus host disease. Bone Marrow Transplant. 2006;38(6):445-451. 122. Corti P, Uderzo C, Tagliabue A, et al. Defibrotide as a promising treatment for thrombotic thrombocytopenic purpura in patients undergoing bone marrow transplantation. Bone Marrow Transplant. 2002;29(6):542-543. 123. Au WY, Ma ES, Lee TL, et al. Successful treatment of thrombotic microangiopathy after haematopoietic stem cell transplantation with rituximab. Br J Haematol. 2007;137(5):475-478. January 2015, Vol. 22, No. 1 124. Jodele S, Fukuda T, Vinks A, et al. Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Biol Blood Marrow Transplant. 2014;20(4):518-525. 125. Peffault de Latour R, Xhaard A, Fremeaux-Bacchi V, et al. Successful use of eculizumab in a patient with post-transplant thrombotic microangiopathy. Br J Haematol. 2013;161(2):279-280. 126. Porcu P, Cripe LD, Ng EW et al. Hyperleukocytic leukemias and leukostasis: a review of pathophysiology, clinical presentation and management. Leuk Lymphoma. 2000;39(1-2):1-18. 127. Estey EH, Keating MJ, McCredie KB, et al. Causes of initial remission induction failure in acute myelogenous leukemia. Blood. 1982;60(2):309-315. 128. Eguiguren JM, Schell MJ, Crist WM, et al. Complications and outcome in childhood acute lymphoblastic leukemia with hyperleukocytosis. Blood. 1992;79(4):871-875. 129. Lichtman MA, Rowe JM. Hyperleukocytic leukemias: rheological, clinical and therapeutic considerations. Blood. 1982;60(2):279-283. 130. Stucki A, Rivier AS, Gikic M, et al. Endothelial cell activation by myeloblasts: molecular mechanisms of leukostasis and leukemic cell dissemination. Blood. 2001;97(7):2121-2129. 131. Lowe EJ, Pui CH, Hancock ML, et al. Early complications in children with acute lymphoblastic leukemia presenting with hyperleukocytosis. Pediatr Blood Cancer. 2005;45(1):10-15. 132. Novotny JR, Nückel H, Dührsen U. Correlation between expression of CD56/NCAM and severe leukostasis in hyperleukocytic acute myelomonocytic leukaemia. Eur J Haematol. 2006;76(4):299-308. 133. Marbello L, Ricci F, Nosari AM, et al. Outcome of hyperleukocytic adult acute myeloid leukaemia: a single-center retrospective study and review of literature. Leuk Res. 2008;32(8):1221-1227. 134. Bug G, Anargyrou K, Tonn T, et al. Impact of leukapheresis on early death rate in adult acute myeloid leukemia presenting with hyperleukocytosis. Transfusion. 2007;47(10):1843-50. 135. Giles FJ, Shen Y, Kantarjian HM, et al. Leukapheresis reduces early mortality in patients with acute myeloid leukemia with high white cell counts but does not improve long-term survival. Leuk Lymphoma. 2001;42(1-2):67-73. 136. Oberoi S, Lehrnbecher T, Phillips B, et al. Leukapheresis and lowdose chemotherapy do not reduce early mortality in acute myeloid leukemia hyperleukocytosis: a systematic review and meta-analysis. Leuk Res. 2014;38(4):460-468. 137. Porcu P, Farag S, Marcucci G, et al. Leukocytoreduction for acute leukemia. Ther Apher. 2002;6(1):15-23. 138. Adams BD, Baker R, Lopez JA, et al. Myeloproliferative disorders and the hyperviscosity syndrome. Emerg Med Clin North Am. 2009;27(3):459-476. 139. Rowe JM, Lichtman MA. Hyperleukocytosis and leucostasis: a common features of childhood chronic myelogenous leukemia. Blood. 1984;63(5): 1230-1234. 140. Shafique S, Bona R, Kaplan AA. A case report of therapeutic leukapheresis in an adult with chronic myelogenous leukemia presenting with hyperleukocytosis and leukostasis. Ther Apher Dial. 2007;11(2):146-149. 141. Ponniah A, Brown CT, Taylor P. Priapism secondary to leukemia: effective management with prompt leukapheresis. Int J Urol. 2004;11(9):809-810. 142. Rodgers R, Latif Z, Copland M. How I manage priapism in chronic myeloid leukaemia patients. Br J Haematol. 2012;158(2):155-164. 143. El Bahnasawy MS, Dawood A, Farouk A. Low-flow priapism: risk factors for erectile dysfunction. BJU. 2002;89(3):285-290. 144. Jameel T, Mehmood K. Priapism - an unusual presentation in chronic myeloid leukaemia:case report and review of the literature. Biomedica. 2009;25:197-199. 145. Stucki A, Rivier AS, Gikic M, et al. Endothelial cell activation by myeloblasts: molecular mechanisms of leukostasis and leukemic cell dissemination. Blood. 2001;97(7):2121-2129. 146. Shafique S, Bona R, Kaplan AA. A case report of therapeutic leukapheresis in an adult with chronic myelogenous leukemia presenting with hyperleukocytosis and leukostasis. Ther Apher Dial. 2007;11(2):146-149. 147. Montague DK, Jarow J, Broderick GA, et al. American Urological Association guideline on the management of priapism. J Urol. 2003;170(4 pt 1):1318-1324. 148. Castagnetti M, Sainati L, Giona F, et al. Conservative management of priapism secondary to leukemia. Pediatr Blood Cancer. 2008;51(3):420-423. 149. Ponniah A, Brown CT, Taylor P. Priapism secondary to leukemia: effective management with prompt leukapheresis. Int J Urol. 2004;11(9):809-810. 150. Stemmler J, Wittmann GW, Hacker U, Heinemann V. Leukapheresis in chronic myelomonocytic leukemia with leukostasis syndrome: elevated serum lactate levels as an early sign of microcirculation failure. Leuk Lymphoma. 2002;43(7):1427-1430. 151. Ganzel C, Becker J, Mintz PD, et al. Hyperleukocytosis, leukostasis and leukapheresis: practice management. Blood Rev. 2012;26(3):117-122. 152. Edelson R, Berger C, Gasparro F, et al. Treatment of cutaneous T-cell lymphoma by extracorporeal photochemotherapy. Preliminary results. N Engl J Med. 1987;316(6):297-303. 153. Trautinger F, Knobler R, Willemze R, et al. EORTC consensus recommendations for the treatment of mycosis fungoides/Sezary syndrome. Eur J Cancer. 2006;42(8):1014-1030. 154. Scarisbrick JJ, Taylor P, Holtick U, et al. U.K. consensus statement on the use of extracorporeal photopheresis for treatment of cutaneous T-cell lymphoma January 2015, Vol. 22, No. 1 and chronic graft-versus-host disease. Br J Dermatol. 2008;158(4):659-678. 155. Heshmati F. Updating ECP action mechanisms. Transfus Apher Sci. 2014;50(3):330-339. 156. Goussetis E, Varela I, Tsirigotis P. Update on the mechanism of action and on clinical efficacy of extracorporeal photopheresis in the treatment of acute and chronic graft versus host disease in children. Transfus Apher Sci. 2012;46(2):203-209. 157. Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol. 2010;11(1):21-27. 158. Alcindor T, Gorgun G, Miller KB, et al. Immunomodulatory effects of extracorporeal photochemotherapy in patients with extensive chronic graftversus-host disease. Blood. 2001;98(5):1622-1625. 159. Berger CL, Xu AL, Hanlon D, et al. Induction of human tumor-loaded dendritic cells. Int J Cancer. 2001;91(4):438-447. 160. Spisek R, Gasova Z, Bartunkova J. Maturation state of dendritic cells during the extracorporeal photopheresis and its relevance for the treatment of chronic graft-versus-host disease. Transfusion. 2006;46(1):55-65. 161. Yoo EK, Rook AH, Elenitsas R, et al. Apoptosis induction by ultraviolet light A and photochemotherapy in cutaneous T-cell lymphoma: relevance to mechanism of therapeutic action. J Invest Dermatol. 1996;107(2):235-242. 162. Heshmati F, Andreu G. Extracorporeal photochemotherapy: a historical perspective. Transfus Apher Sci. 2003;28(1):25-34. 163. Rook AH, Suchin KR, Kao DMF, et al. Photopheresis: clinical applications and mechanism of action. J Invest Dermatol Symp Proc. 1999;4(1):85-90. 164. Girardi M, Berger CL, Wilson LD, et al. Transimmunization for cutaneous T cell lymphoma: a phase I study. Leuk Lymphoma. 2006;47(8):1495-1503. 165. Zic JA. Photopheresis in the treatment of cutaneous T-cell lymphoma: current status. Curr Opin Oncol. 2012;(24 suppl 1):S1-S10. 166. Scarisbrick JJ. Staging and management of cutaneous T-cell lymphoma. Clin Exp Dermatol. 2006;31(2):181-186. 167. Zic JA. The treatment of cutaneous T-cell lymphoma with photopheresis. Dermatol Ther. 2003;16(4):337-346. 168. Child FJ, Mitchell TJ, Whittaker SJ, et al. A randomized cross-over study to compare PUVA and extracorporeal photopheresis in the treatment of plaque stage (T2) mycosis fungoides. Clin Exp Dermatol. 2004;29(3):231-236. 169. National Comprehensive Cancer Network. NCCN clinical practice guidelines: non-Hodgkin lymphoma. http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf. Accessed October 26, 2014. 170. Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28(31):4730-4739. 171. Ferrara JL, Levine JE, Reddy P, et al. Graft-versus-host disease. Lancet. 2009;373(9674):1550-1561. 172. Flowers ME, Inamoto Y, Carpenter PA, et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood. 2011;117(11):3214-3219. 173. Martin PJ, Rizzo JD, Wingard JR, et al. First- and second-line systemic treatment of acute graft-versus-host disease: recommendations of the American Society of Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2012;18(8):1150-1163. 174. Martin PJ, Inamoto Y, Carpenter PA, et al. Treatment of chronic graftversus-host disease: Past, present and future. Korean J Hematol. 2011;46(3):153-163. 175. Stewart BL, Storer B, Storek J, et al. Duration of immunosuppressive treatment for chronic graft-versus-host disease. Blood. 2004;104(12):3501-3506. 176. Vigorito AC, Campregher PV, Storer BE, et al. Evaluation of NIH consensus criteria for classification of late acute and chronic GVHD. Blood. 2009;114(3):702-708. 177. Inamoto Y, Flowers ME. Treatment of chronic graft-versus-host disease in 2011. Curr Opin Hematol. 2011;18(6):414-420. 178. Garnett C, Apperley JF, Pavlů J. Treatment and management of graftversus-host disease: improving response and survival. Ther Adv Hematol. 2013;4(6):366-378. 179. Perfetti P, Carlier P, Strada P, et al. Extracorporeal photopheresis for the treatment of steroid refractory acute GVHD. Bone Marrow Transplant. 2008;42(9):609-617. 180. Berger M, Pessolano R, Albiani R, et al. Extracorporeal photopheresis for steroid resistant graft versus host disease in pediatric patients: a pilot single institution report. J Pediatr Hematol Oncol. 2007;29(10):678-687. 181. Pierelli L, Perseghin P, Marchetti M, et al. Extracorporeal photopheresis for the treatment of acute and chronic graft-versus-host disease in adults and children: best practice recommendations from an Italian society of hemapheresis and cell manipulation (SIdEM) and Italian group for bone marrow transplantation (GITMO) consensus process. Transfusion. 2013;53(10):2340-2352. 182. Lucid CE, Savani BN, Engelhardt BG, at al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-SCT. Bone Marrow Transplant. 2011;46(3):426-429. 183. Perotti C, Del Fante C, Tinelli C, et al. Extracorporeal photochemotherapy in graft-versus-host disease: a longitudinal study on factors influencing the response and survival in pediatric patients. Transfusion. 2010;50(6):1359-1369. 184. Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment Cancer Control 77 of pulmonary chronic GvHD: report from the consensus conference on clinical practice in chronic GvHD. Bone Marrow Transplant. 2011;46(10):1283-1295. 185. Kanold J, Messina C, Halle P, et al; Paediatric Diseases Working Party of the European Group for Blood and Marrow Transplantation. Update on extracorporeal photochemotherapy for graft-versus-host disease treatment. Bone Marrow Transplant. 2005;(35 suppl 1):S69-S71. 186. Perseghin P, Galimberti S, Balduzzi A, et al. Extracorporeal photochemotherapy for the treatment of chronic graft-versus-host disease: trend for a possible cell dose-related effect? TherApher Dial. 2007;11(2):85-93. 187. Flowers ME, Apperley JF, van Besien K, et al. A multicenter prospective phase 2 randomized study of extracorporeal photopheresis for treatment of chronic graft-versus-host disease. Blood. 2008;112(7):2667-2674. 188. Das-Gupta E, Dignan F, Shaw B, et al. Extracorporeal photopheresis for treatment of adults and children with acute GVHD: UK consensus statement and review of published literature. Bone Marrow Transplant. 2014;49(10:1251-1258. 189. Heshmati F. Extra corporeal photo chemotherapy (ECP) in acute and chronic GVHD. Transfus Apher Sci. 2010;43(2):211-215. 190. Dignan FL, Clark A, Amrolia P, et al; Haemato-oncology Task Force of British Committee for Standards in Haematology; British Society for Blood and Marrow Transplantation. Diagnosis and management of acute graft-versus-host disease. Br J Haematol. 2012;158(1):46-61. 191. European Dermatology Forum. Guideline on extracorporeal photopheresis. http://www.euroderm.org/index.php/edf-guidelines. Accessed October 26, 2014. 192. Martin PJ, Rizzo JD, Wingard JR, et al. First and second-line systemic treatment of acute graft-versus-host disease: recommendations of the American Society of Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2012;18(8):1150-1163. 193. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011;117(22):5857-5859. 194. Finazzi G,Carobbio A, Thiele J, et al. Incidence and risk factors for bleeding in 1104 patients with essential thrombocythemia or prefibrotic myelofibrosis diagnosed according to the 2008 WHO criteria. Leukemia. 2012;26(4):716-719. 195. Barbui T, Thiele J, Passamonti F, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. 2011;29(23):3179-3184. 196. Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110(4):1092-1097. 197. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391-2405. 198. Passamonti F, Rumi E, Arcaini L, et al. Prognostic factors for thrombosis, myelofibrosis, and leukemia in essential thrombocythemia: a study of 605 patients. Haematologica. 2008;93(11):1645-1651. 199. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117(10):755-761. 200. Di Nisio M, Barbui T, Di Gennaro L, et al; Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) Investigators. The haematocrit and platelet target in polycythemia vera. Br J Haematol. 2007;136(2):249-259. 201. Budde U, Schaefer G, Mueller N, et al. Acquired von Willebrand’s disease in the myeloproliferative syndrome. Blood. 1984;64(5):981-985. 202. Gangat N, Wolanskyj AP, McClure RF, et al. Risk stratification for survival and leukemic transformation in essential thrombocythemia: a single institutional study of 605 patients. Leukemia. 2007;21(2):270-276. 203. Elliott MA, Tefferi A. Thrombosis and haemorrhage in polycythaemia vera and essential thrombocythaemia. Br J Haematol. 2005;128(3):275-290. 204. Tefferi A, Elliott M. Thrombosis in myeloproliferative disorders: prevalence, prognostic factors, and the role of leukocytes and JAK2V617F. Semin Thromb Hemost. 2007;33(4):313-320. 205. Grima KM. Therapeutic apheresis in hematological and oncological diseases. J Clin Apheresis. 2000;15(1-2):28-52. 206. Taft EG, Babcock RB, Scharfman WB, et al. Platelet pheresis in the management of thrombocytosis. Blood.1977;50(5):927-933. 207. Budde U, van Genderen PJJ. Acquired von Willebrand disease in patients with high platelet counts. Semin Thromb Hemost. 1997;23(5):425-431. 208. Tefferi A, Silverstein MN, Hoagland HC. Primary thrombocythemia. Semin Oncol. 1995;22(4):334-340. 209. Kaibara M, Kobayashi T, Matsumoto S. Idiopathic thrombocythemia and pregnancy: report of a case. Obstet Gynecol 1985;65(3 suppl):18S-19S. 210. Beard J, Hillmen P, Anderson CC, et al. Primary thrombocythaemia in pregnancy. Br J Haematol.1991;77(3):371-374. 211.Schött U. Essential thrombocythemia and coronary artery bypass surgery. J Cardiothorac Vasc Anesth 1994;8(5):552-555. 212. Hsiao HT, Ou SY. Successful microsurgical tissue transfer in a patient with postsplenectomy thrombocytosis treated with platelet-phoresis. J Reconstr Microsurg 1997;13(8):555-558. 213. Kralovics R, Passamonti F, Buser AS, et al. Again-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790. 214. James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 78 Cancer Control 2005;434(7037):1144-1148. 215. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood. 2013;121(23):4778-4781. 216. Brissot P, Ball S, Rofail D, et al. Hereditary hemochromatosis: patient experiences of the disease and phlebotomy treatment. Transfusion. 2011;51(6):1331-1338. 217. McDonnell SM, Grindon AJ, Preston BL, et al. A survey of phlebotomy among persons with hemochromatosis. Transfusion. 1999;39(6):651-656. 218. McDonnell SM, Preston BL, Jewell SA, et al. A survey of 2,851 patients with hemochromatosis: symptoms and response to treatment. Am J Med. 1999;106(6):619-624. 219. Kaboth U, Rumpf KW, Lipp T, et al. Treatment of polycythemia vera by isovolemic large-volume erythrocytapheresis. Klin Wochenschr. 1990;68(1):18-25. 220. Kaboth U, Rumpf KW, Liersch T, et al. Advantages of isovolemic large-volume erythrocytapheresis as a rapidly effective and long-lasting treatment modality for red blood cell depletion in patients with polycythemia vera. Ther Apher. 1997;1(2):131-134. 221. Rombout-Sestrienkova E, Nieman FH, Essers BA, et al. Erythrocytapheresis versus phlebotomy in the initial treatment of HFE hemochromatosis patients: results from a randomized trial. Transfusion 2012;52(3):470-477. 222.Vecchio S, Leonardo P, Musuraca V, et al. A comparison of the results obtained with traditional phlebotomy and with therapeutic erythrocytapheresis in patients with erythrocytosis. Blood Transfusion. 2007;5(1):20-23. 223. Wijermans P, Egmond van L, Ypma P, et al. Isovolemic erythrocytapheresis technique as an alternative to conventional phlebotomy in patients with polycythemia rubra vera and hemochromatosis. Transfus Apher Sci. 2009;40(2):137. 224. Gerson SL, Lazarus HM. Hematopoietic emergencies. Semin Oncol. 1989;16(6):532-542. 225. Rusak T, Ciborowski M, Uchimiak-Owieczko A, et al. Evaluation of hemostatic balance in blood from patients with polycythemia vera by means of thromboelastography: the effect of isovolemic erythrocytapheresis. Platelets. 2012;23(6):455-462. 226. Vecchio S, Leonardo P, Musuraca V, et al. A comparison of the results obtained with traditional phlebotomy and with therapeutic erythrocytapheresis in patients with erythrocytosis. Blood Transfus. 2007;5(1):20-23. 227. .Kabot U, Vehmeyer K, Liersch T. Polycythemia vera: reduced proliferative capacity of erythroid progenitor cells after large volume erythrocyte-apheresis apparently due to the massive loss of iron. J Clin Apheresis.1993;8:43. 228. Evers D, Kerkhoffs JL, Van Egmond L, et al. The efficiency of therapeutic erythrocytapheresis compared to phlebotomy: a mathematical tool for predicting response in hereditary hemochromatosis, polycythemia vera, and secondary erythrocytosis. J Clin Apher. 2014;29(3):133-138. January 2015, Vol. 22, No. 1 HLA assays have a role in pharmacogenomics, disease association, platelet transfusion, solid organ transplantation, and HSCT. Ray Paul. Currant, 2014. Acrylic, latex, enamel on canvas, 20" × 20". Using HLA Typing to Support Patients With Cancer Mark K. Fung, MD, PhD, and Kaaron Benson, MD Background: The human leukocyte antigen (HLA) system plays a crucial role in immune function, and HLA testing is often needed in the support of patients with cancer. Methods: We briefly review the published literature to clarify the nomenclature of the HLA system, currently available methods for HLA testing, and commonly used HLA assays. The uses of HLA testing in pharmacogenomics, disease association, platelet transfusion support, and in the management of both solid organ and hematopoietic stem cell transplantation are also reviewed. Results: HLA testing is commonly performed for select patient populations, including patients with cancer and in those requiring solid organ and hematopoietic stem cell transplantation. Conclusion: Newer molecular typing methods have helped improve patient outcomes following hematopoietic stem cell transplantation. Introduction The human leukocyte antigen (HLA) region encompasses a crucial set of genes that regulate immune function. It is the most polymorphic region of the human genome. HLA testing is often required in support of patients with cancer; for example, HLA testing is used in both solid organ and hematopoietic stem cell transplantation (HSCT), for selected pharmacogFrom the University of Vermont College of Medicine (MKF), the Department of Pathology (MKF), Blood Bank and HLA Laboratory, University of Vermont Medical Center, Burlington, Vermont, the H. Lee Moffitt Cancer Center & Research Institute (KB), and the University of South Florida Morsani College of Medicine (KB), Tampa, Florida. Submitted October 7, 2014; accepted October 27, 2014. Address correspondence to Mark K. Fung, MD, PhD, Department of Pathology, Blood Bank and HLA Laboratory, University of Vermont Medical Center, 111 Colchester Avenue, Burlington, VT 05401. E-mail: [email protected] No significant relationships exist between the authors and the companies/organizations whose products or services may be referenced in this article. January 2015, Vol. 22, No. 1 enomics testing for a personalized medical approach, and in support of immune-platelet refractory patients. The HLA nomenclature has been updated to address new information gained with molecular assays. Both serological and molecular HLA assays are available and the use of these tests will be addressed. Methods of Testing for HLA Antigens, Antibodies, and Genes A number of methods for determining HLA types were developed over the years as different technologies were discovered and used. Initially, the presence or absence of certain HLA types and the specific HLA type were determined with the use of antibodies via serological methods, including the microcytotoxicity method,1 whereby T or B lymphocytes from a patient or donor are incubated in vitro with serum containing HLA antibodies of a certain specificity (reactive against a particular HLA type), and, if the antigen is present, an in vitro activation of complement would occur, leading to detectable cell death in this assay. Cancer Control 79 Thus, an HLA type for an individual would be determined with the use of an array of sera with different HLA specificities on a plastic tray with multiple wells within which the lymphocytes of an individual would be added. The ability to identify different HLA types by this method is limited to the availability of sera containing the various HLA specificities. Therefore, this method is less commonly used as an initial method for HLA typing due to these limitations. However, it does have value in confirming the presence or absence of an antigen in rare instances in which molecular-based methods predict a certain HLA type but fail to recognize that the antigen is not produced due to mutations present in a gene or promoter not routinely tested. In addition, some laboratories continue to use this serological method for determining the presence or absence of certain disease-associated HLA types (eg, HLA-B27 for ankylosing spondylitis). Advancement in the use of antibodies to determine HLA type for a specific application has been seen in the use of flow cytometry with fluorescent conjugates added to anti-B27 to identify patients positive for HLA-B27. Otherwise, serological or antibody-based typing of HLA antigens is only used in applications in which knowledge of the HLA type at a broader serological grouping level (low-resolution HLA type) is sufficient (eg, HLA typing of platelet donors, for solid organ transplantation in certain countries where this is still permitted). It is rare to use serological HLA typing methods for patients receiving HSCT except perhaps for identifying HLA-matched siblings. Therefore, health care professionals should routinely confirm that the method used for patients receiving HSCT is via a molecular method, and that the method employed can generate sufficient specificity to identify the patient or donor HLA types to the allelic level where appropriate. The development of molecular methods for determining an individual’s HLA type was a significant advancement in avoiding the past technical challenges of needing serum with antibodies of all HLA types, including uncommon types present in only small groups of individuals. Although the HLA genes are highly polymorphic at multiple locations, the majority of these DNA sequence variations are contained within exons 2 and 3. With current molecular methods, once the specific nucleotide sequence polymorphisms unique to a particular HLA type are characterized, the necessary DNA probes or primers can be artificially synthesized and readily incorporated into commercial assay kits. The number of molecular methods that currently exist can be grouped into 3 categories, ie, sequence-based typing (SBT), sequence-specific primer (SSP) typing, and sequence-specific oligonucleotide (SSO) typing. SBT can be performed via the tradi80 Cancer Control tional Sanger nucleotide termination method or via next-generation sequencing methods for which several platforms have been developed for HLA typing. In general, SSP typing is performed through the use of a heat-stable DNA polymerase to generate detectable, specific DNA amplification products that can be produced and detected if HLA-type specific DNA primers properly bind to the individual’s template DNA. SSO typing is accomplished through the ability to detect more broadly generated, exon- and locus-specific DNA amplification products, but which are not HLA type-specific, that bind to SSO probes. By contrast, SSP typing requires multiple wells of reactions to determine the presence or absence of particular DNA polymorphisms, whereas SSO typing allows for the use of a single tube of locus-specific amplified products that must then bind to a specific location on a solid-phase surface or a uniquely identified bead associated with the particular polymorphism. With all 3 molecular methods, an HLA type is determined based on the collection of polymorphisms identified. With few exceptions, the majority of the molecular HLA typing assays commercially available and in use focus on identifying polymorphisms in exons 2 and 3 of the various HLA genes tested. As mentioned above with serological HLA testing, rare polymorphisms may exist outside of exons 2 and 3 that lead to lack of expression or altered expression of the HLA molecule, which is a specific limitation of the molecular method. Exon 4 testing may be added for HLA-A, HLA-B, and/or HLA-C typing for recipients of HSCT and their donors for more readily achievable high-resolution typing results. Future and ongoing developments in next-generation sequencing will eliminate these limitations when incorporating broader sequencing of the entire coding and noncoding regions. Nomenclature The nomenclature of the HLA system has significantly changed in recent years. Serological typing had to account for older “parent” and newer “split” antigens (eg, B12[44], B12[45]). Molecular methods required significant nomenclature changes; the latest major update from 2011 involved the reorganization of alleles so that they were properly aligned to allow similar antigens to be within the same group. Refer to Fig 1 for an example of the new molecular fields and what they represent.2 Molecular typing methods revealed that many serologically defined antigens were actually created by multiple alleles that could be individually defined. Individual alleles linked to the same antigen may behave differently and affect outcomes in areas like HSCT. Novel alleles are constantly being identified and the extensive polymorphism of the HLA system continues to be recognized. Applying the terms January 2015, Vol. 22, No. 1 HLA-A*02:101:01:02N HLA-A* 02: Locus* Field: 1 101: 01: 02N 2 3 4 Fig 1. — Nomenclature details for one single HLA allele at high resolution. HLA-A locus/gene, * separator: field 1/allele group (02): field 2/specific HLA protein (101): field 3/synonymous DNA substitution within the coding region (01): field 4/used to show differences in a noncoding region (02N). Oftentimes, the first 2 fields alone are reported by laboratories performing HLA typing because this information alone is clinically relevant (eg, HLA-A*02:101). The suffix N is added to denote a null allele, an allele that is not expressed. HLA = human leukocyte antigen. Data from reference 2. High-resolution molecular typing HLA-B*08:01, 51:01 Intermediate-resolution molecular typing HLA-B*08:01/02, 51:01/02/03 Low-resolution molecular typing HLA-B*08, 51 Serological typing HLA-B8, 51 Fig 2. — Serological and molecular low-, intermediate-, and high-resolution HLA typing results. The same HLA-B type is shown first by high-resolution molecular typing followed by intermediate-resolution, low-resolution, and serological typing. HLA = human leukocyte antigen. sponses. For example, by prospectively HLA typing patients with HIV-positive status to identify those with HLA-B*57:01, a significant reduction was seen in the risk of adverse events due to the strong association of hypersensitivity with the use of abacavir in these patients. In European populations, this allele is relatively common with a frequency of 6% to 7%.7 The highest frequency of HLA-B*57:01 has been reported in southwestern Asian populations in which up to 20% of the population are carriers.7 The US Food and Drug Administration recommends prescreening patients for B*57:01 prior to starting treatment with this antiretroviral medication.8,9 Refer to Table 1 for selected examples of drug-related adverse events and the identified HLA alleles associated with those events.10-13 Not all pharmacogenomics testing that held promise has been realized. Prescreening of patients before treatment with vitamin K antagonists such as warfarin for single-nucleotide polymorphisms in the genes VKORC1, CYP2C9, or both have not improved outcomes.14,15 With the advent of next-generation sequencing of the whole exome or genome, further research is likely to identify critical and non-HLA genes associated with both the beneficial and detrimental responses to select medications. low, intermediate, and high resolution helps explain the specificity of a given HLA type; low resolution is equivalent to serological typing results, and high resolution provides the specific allele pairs for a given locus. Intermediate resolution is the term for small numbers of allele pairs and indicates some additional testing beyond low resolution. Low-resolution typing is at the level of the digits comprising the first field in the DNA-based nomenclature and generally corresponds to the serological typing result (see Fig 1).2 An example is A*01; A*02, which would be A1, A2 by serology. Intermediate-resolution typing results include a subset of alleles sharing the digits in the first field of their allele name and for which some alleles sharing those digits are excluded. Examples include A*02:01 or A*02:02 or A*02:07 or A*02:20 but not other A*02 alleles and may be written as A*02:01/02/07/20. High-resolution typing is defined as a DNA-based typing result that includes a set of alleles that specifies and encodes the same protein sequence for the peptide-binding region of an HLA molecule and also excludes alleles not expressed as cell-surface proteins Disease Associations (Fig 2).3,4 Given the role of the immune system in many disThe letter G is a code that signifies a group of aleases, it is not surprising that certain polymorleles in which all have identical nucleotide sequences phisms in the HLA system, with specific HLA types, across the exons that encode for the peptide-binding are associated with an increased risk for disease region of the HLA molecule and includes exons 2 and 3 for class I and exon 2 Table 1. — Examples of Important Adverse Events and Their HLA Associations alone for HLA class II alleles. For exDrug HLA Allele HLA Frequency Adverse Event Study ample, HLA-B*27:05G represents alleles Abacavir B*57:01 6%–8% Caucasian Drug Shear10 B*27:05 and B*27:13 and these 2 alleles hypersensitivity 2.5% African American have the same clinical relevance.5,6 Allopurinol B*58:01 9%–11% Han Chinese 1%–6% Caucasian Drug hypersensitivity Hung11 Carbamazepine B*15:02 10%–15% Han Chinese < 0.1% Caucasian Stevens–Johnson syndrome/toxic epidermal necrolysis Chung12 Flucloxacillin B*57:01 6%–8% Caucasian Drug-induced liver injury Daly13 Pharmacogenomics Pharmacogenomics refers to specific genes in a given individual associated with particular responses, both beneficial and detrimental, to medications and other therapies. The HLA genes are well recognized as influencing select drug reJanuary 2015, Vol. 22, No. 1 HLA = human leukocyte antigen. Cancer Control 81 (Table 2). Despite these associations, the mechanism of the association with certain HLA types remains under investigation; the HLA type is one of multiple risk factors. The presence of a disease-associated HLA type alone is not sufficient to trigger disease. For example, HLA-B27 is commonly present in patients with ankylosing spondylitis, but many individuals who have HLA-B27 are without disease because HLA-B27 is a relatively common antigen and ankylosing spondylitis is an uncommon disease. Therefore, the health care professional should keep this in mind when obtaining and reviewing HLA typing results of a patient with cancer. Associations with HLA types for certain lymphoid malignancies have also been studied, including certain HLA types to chronic lymphocytic leukemia, multiple myeloma, acute lymphoblastic leukemia, and diffuse large B-cell lymphoma.16 In fact, the earliest association with HLA type and disease was with HLA-B types and Hodgkin disease.17 Findings have been more limited of associations of certain HLA types with solid Table 2. — HLA Alleles and Associated Diseases Disease HLA Allele Ankylosing spondylitis B*27:XX alleles (except B*27:06 and B*27:09) Antiglomerular basement membrane disease DRB1*15:01 DRB1*15:02 Birdshot retinochoroidopathy A*29:01 A*29:02 Celiac disease DQA1*05/DQB1*02 DQA1*03/DQB1*03:02 Idiopathic myopathy DQA1*04:01 DQA1*05:01 Narcolepsy DQB1*06:02 Pemphigus vulgaris DRB1*04:02 DRB1*04:03 DRB1*04:06 DRB1*08:02 DRB1*08:04 DRB1*14:04 DRB1*14:05 DRB1*14:08 Psoriasis C*06:XX Reiter syndrome B*40:01 Type 1 diabetes mellitus DRB1*03:01/02/11 Multiple alleles of DRB1*04:XX, including: DRB1*04:01/02/03/04/05/ 06/07/08/09/10/11/17/26 DQA1*03:01 DQB1*02:01 DQB1*03:02 HLA = human leukocyte antigen. 82 Cancer Control organ malignancies and include an increased risk of progression to hepatocellular carcinoma in patients with chronic hepatitis B18 and long-term survival rates among patients with gastric cancer.19 Despite the associations of HLA type with certain malignancies, HLA typing of patients with cancer is not routinely performed because these associations are relatively weak to date. However, HLA typing is routinely performed for patients and their potential donors when HSCT is considered. Platelet Support A poor response to platelet transfusion (ie, platelet transfusion refractoriness) can be due to nonimmune causes, immune causes, or both. Immune refractoriness is most often the result of HLA antibodies (either single or multiple specificities) and, much less commonly, antibodies directed against human platelet-specific antigens (HPAs). The risk of HLA alloimmunization can be significantly reduced with the use of leukoreduced red blood cells and platelets.20 Despite wide adoption of the leukoreduction of red blood cell and platelet components for patients with cancer, HLA alloimmunization remains a challenge. Parous women have been previously exposed to fetal HLA epitopes that may have elicited prior alloimmunization; the risk is greater with an increasing number of pregnancies.21 Men and nulliparous women may also have been HLA alloimmunized due to a prior blood transfusion that was either nonleukoreduced or was truly leukoreduced (leukoreduction is not 100% protective). Patients with poor responses to platelet transfusion at 10 minutes or 1 hour following transfusion should be investigated for possible immune causes for refractoriness.22 By using the correct count increment, one can calculate that the average-sized person receiving an average dose of platelets should increase his or her platelet count about 15,000/µL above the baseline platelet result at 1 hour following the transfusion if the precount was performed in close proximity to the transfusion.23 Qualitative screening assays are available to detect antiplatelet antibodies with HLA specificity, HPA specificity, or both. These specificities can also be identified when antigen-negative platelet transfusions are being considered (eg, HLA-A2-negative platelets for a patient with anti-HLA-A2 antibodies). The management of thrombocytopenia with HLA alloimmunization is covered in the article by Dr Fletcher and others in this issue. Care of these patients can often be quite difficult and preventive strategies are crucial. Hematopoietic Stem Cell Transplantation The outcomes of both related and unrelated donor HSCT are impacted by the extent of HLA matching January 2015, Vol. 22, No. 1 between the transplantation recipient and the donor. Several large studies have demonstrated that a greater degree of HLA match between donor and recipient improves overall survival rates,24 reduces both the incidence and severity of acute and chronic graft-vshost disease (GVHD)25 and improves rates of engraftment.26,27 When a suitable, related HLA-matched donor is unavailable, unrelated donor registries, such as the Be the Match Registry run by the National Marrow Donor Program, can often identify a perfect or wellmatched unrelated donor. The recipient’s racial and ethnic group will affect the likelihood of finding a high-resolution HLA-A, HLA-B, HLA-C, and HLA-DRB1 match, although whites of European descent have the highest probability (75%) and blacks of South or Central American descent have the lowest (16%).28 When these large, unrelated donor registries also fail to identify a matched unrelated donor, alternative donors such as mismatched adult unrelated donors, haploidentical-related donors, and umbilical cord blood (UCB) stem cell products are often used.29 The widespread use of DNA-based tissue typing methodologies has increased the accuracy and specificity of HLA typing, thus allowing for more precise HLA matching between recipients and donors. For most HSCT, a minimum of 4 HLA loci (HLA-A, HLA-B, HLA-C, HLA-DRB1) and, more often, 5 (HLA-A, HLA-B, HLA-C, HLA-DRB1, HLA-DQB1) are generally matched between recipient and donor pairs. Volunteer unrelated adult donors are selected to be closely matched to recipients at HLA-A, HLA-B, HLA-C, and HLA-DRB1 at the allele level when related HLA-matched donors are not available.30 High-level donor–recipient HLA matching is crucial for the success of unrelated adult donor HSCT.31,32 Additional loci considered by some HSCT programs include DPB1 and KIR. Although close HLA matching is crucial, it is not always possible and some mismatches fare better than others. Pidala et al33 identified certain amino acid substitutions that affected the peptide-binding site of the HLA class I antigen and increased the risk of severe acute GVHD and mortality. Some mismatches appear to have little to no increased risk.34 These “permissible” HLA mismatches have been perhaps most studied in the Japanese population.35,36 In Japan, fewer HLA haplotypes gives greater opportunity for studying isolated mismatches between recipient–donor pairs. The only potential curative measure for many patients with hematological malignancies is HSCT; however, about 70% of patients will not have an HLAmatched sibling donor considering the number of children per family in the United States and the likelihood of HLA identity being 25% with any 1 sibling.37 Therefore, the majority of recipients must turn to the unrelated volunteer donor pool. The National Marrow Donor Program has more than 20 million HLA-typed January 2015, Vol. 22, No. 1 donors in its database and affiliated registries.38 Many patients, particularly those of diverse racial and ethnic backgrounds, will not have a suitable matched, unrelated donor identified in the time period needed. UCB has helped to fill that gap for these patients, and more than 30,000 UCB transplants have been performed to date.39 UCB units are typically selected using low-resolution HLA typing (antigen level) for HLA-A and -B and high resolution (allele level) for HLA-DRB1. HLA-C matching was not generally considered in the past,40 but further study has shown that HLA-C matching with UCB may minimize mortality risks.41 While the degree of matching for UCB transplantation is not as extensive as it is for non-UCB sources, greater degrees of matching (eg, high resolution for HLA-A, HLA-B, HLA-C, and HLA-DRB1 vs low resolution for HLA-A and HLA-B) may also improve neutrophil recovery and reduce nonrelapse mortality rates when single cord blood units are transplanted.42 In general, for non-UCB HSCT, any single locus mismatch is associated with worse outcomes in overall survival, treatment-related mortality, and acute GVHD (ie, 9/10 worse than 10/10 match) with the exception of a mismatch at the DQB1 locus. There is no statistical difference if a single-locus mismatch occurs at the antigen or allele level, except perhaps for HLA-C with antigen mismatch worse than allele-level mismatch.30,43 Because the HLA-A, HLA-B, HLA-C, HLA-DRB1, and HLA-DQB1 loci influence the success of HSCT, investigators have also looked at the DPB1 locus to determine its impact. Early studies suggested that DPB1 matching does not impact overall survival rates, a fact that appeared fortunate because tight DPB1 linkage with other loci are lacking and would create difficulty in finding a DPB1 match.44 About 20% of 10/10 matched unrelated donor transplantations will be matched for DPB1.44 More recent work in grouping DPB1 mismatches based on T-cell epitopes has distinguished mismatches that might be tolerated (permissive) from those with increased risks (nonpermissive). Retrospective analyses in both 9/10 and 10/10-matched transplantations have shown that nonpermissive DPB1 mismatches were associated with a significantly increased risk of overall mortality, nonrelapse mortality, and severe acute GVHD than permissive mismatches.45 HLA alloantibodies directed against mismatched antigens are well established as a significant risk factor in solid organ transplantation (particularly for the kidney, heart, and pancreas); therefore, prescreening is required and frequently repeated. Increased risk of graft failure in HLA mismatched pairs with positive cytotoxicity crossmatch tests (39%) compared with those with negative compatible tests (10%) was reported by Anasetti et al.46 Cancer Control 83 Approximately 35% of patients receiving unrelated HSCT possess HLA antibodies, and the presence of donor-specific HLA antibodies (DSAs) against HLA-A, HLA-B, and/or DRB1 specificities, as determined by solid-phase immunoassay testing, is associated with graft failure.47 Therefore, HLA antibody evaluation in the recipient should be part of the routine workup for mismatched HSCTs.48 Both the National Marrow Donor Program and the Center for International Blood and Marrow Transplant Registry recommend the evaluation of HLA-DSAs in both adult and cord blood HLA mismatched HSCTs.24 The concept of considering noninherited maternal antigens (NIMAs) when selecting particular mismatched donors is an interesting one. Humans are exposed to NIMA HLA antigens in utero and the immature fetus appears to develop less reactivity to these non–self-antigens compared with non-NIMA alloantigens. Some treatment-related mortality associated with HLA-mismatched UCB HSCT may be alleviated with the use of NIMA-matched vs mismatched donor units and has been associated with improved rates of overall survival (Fig 3).44,49,50 The ability to provide NIMA-matched donors may prove difficult as the relative frequency of these donors may be low and searching may delay transplantation. In the Rocha et al49 and van Rood et al50 studies cited above, 7% to 10% of transplantations were NIMA matched. Natural killer cells have killer cell, immunoglobulin-like receptors (KIRs) on their surface that allow them to recognize HLA class I and, primarily, HLA-C surface molecules; they then can distinguish “self” from “non-self” and ultimately provide a benefit with such feats as destruction of virally infected cells or HLA-A HLA-B HLA-DRB1 NIMA Match Recipient A*02, 24 B*18, 35 DRB1*01:01, 11:04 UCB unit/donor A*02, 32 B*18, 35 DRB1*01:01, 11:04 UCB donor mother A*24, 32 B*07, 35 DRB1*01:01, 13:01 NIMA Mismatch Recipient A*02, 11 B*18, 35 DRB1*01:01, 11:04 UCB unit/donor A*02, 32 B*18, 35 DRB1*01:01, 11:04 UCB donor mother A*24, 32 B*07, 35 DRB1*01:01, 13:01 Fig 3. — Examples of NIMA-matched and mismatched UCB donors. HLA-A*24 is not carried by the UCB donor but is carried by the mother of the UCB donor and the recipient; thus, this represents a NIMA-matched UCB HSCT. HLA-A*11 is not carried by the UCB donor or the mother of the UCB donor; thus, this represents a NIMA-mismatched UCB HSCT. HLA = human leukocyte antigen, HSCT = hematopoietic stem cell transplantation, NIMA = noninherited maternal antigen, UCB = umbilical cord blood. Adapted from van Rood JJ, Stevens CE, Smits J, et al. Reexposure of cord blood to noninherited maternal HLA antigens improves transplant outcome in hematological malignancies. Proc Natl Acad Sci U S A. 2009;106(47):19952-19957. Republished with permission by Washington, DC: National Academy of Sciences. 84 Cancer Control cancer cells.51 All HLA-C alleles can be classified as either HLA C group 1 (C1) or group 2 (C2) and the KIR haplotypes are either grouped as A or B depending on which genes are present. Different pairings of the HLA and KIR molecules elicit either inhibitory or activating signals.52 Although early studies showed KIR mismatching could provide a survival advantage in acute myeloid leukemia, subsequent studies have had varied conclusions. Use of KIR data for donor selection should be considered within the framework of a clinical trial alone.53 The best approach to using HLA typing results when searching for an unrelated HSCT donor includes the following: • Look for a 7/8 or optimal 8/8 HLA-A, HLA-B, HLA-C, or DRB1 allele-matched donor. • Consider DQB1 allele-matched donors when multiple 7/8 or 8/8 matches are present for a preferred 9/10 or 10/10 HLA-A, HLA-B, HLA-C, DRB1, and DQB1 match. • Consider UCB HSCT when no 7/8 or 8/8 matches are identified. • Identify UCB units that are a minimum of 4/6 HLA-A, HLA-B, and DRB1 match with adequate cell dose. • A NIMA-matched donor may benefit the recipient and could be sought if there are multiple, similarly mismatched unrelated donor or UCB units and the HSCT is not delayed. • HLA antibody screening/matching should be performed when mismatched donors are considered. The National Marrow Donor Program recommends that high-resolution HLA typing be performed at the time of diagnosis for all adult patients with acute myeloid leukemia, acute lymphoblastic leukemia, myelodysplastic syndromes with an intermediate or high International Prognostic Scoring System, some with chronic myeloid leukemia, such as when an inadequate hematological or cytogenetic response occurs after a trial of tyrosine kinase inhibitors, and in certain chronic lymphocytic leukemia cases with, for example, high-risk cytogenetic or molecular features (eg, del[11q] or del[17p], ZAP70 or Cor CD38 positivity, unmutated immunoglobulin variable region heavy chain mutational status, short initial remission, resistant to fludarabine).54 The use of blood relatives as blood donors prior to stem cell infusion should be avoided because sensitization of the patient to donor minor histocompatibility antigens can increase the risk of allograft rejection.55 Solid Organ Transplantation Unlike stem cell transplantation, HLA matching is not initially required for solid organ transplantation (heart, liver, lung, kidney, pancreas, or bowel) if the January 2015, Vol. 22, No. 1 patient is not alloimmunized; rather, identifying and confirming ABO compatibility to avoid hyperacute rejection is more important. The use of HLA matching is associated with potentially improved allograft survival and reduced alloimmunization rates that might otherwise limit the availability of HLA-compatible organs. Furthermore, the preference for HLA matching does not require complete matching of all loci unless the patient is broadly alloimmunized against the majority of non-self HLA types. Typically, low-resolution HLA typing alone is required. No benefit has been identified with the use of allele-level or high-resolution typing in solid organ transplantations except in instances in which a patient might have allele-specific antibodies. In patients who are waiting for an available cadaveric allograft, those who are highly sensitized (> 90% of panel donors are reactive) will be eligible to receive an HLA-matched kidney from outside of their region. Whenever organ dysfunction is present in a patient with a history of solid organ transplantation (heart, liver, lung, kidney, pancreas, or bowel), an assessment of the possibility of allograft rejection should be considered. Allograft biopsy and testing for HLA antibody production would help assess for cellular and humoral allograft rejections, respectively. An examination of the biopsied tissue includes looking for evidence of lymphocytic infiltrates and thickening or fibrosis of vessel walls. Staining for the complement component C4d is used in kidney biopsies to look for evidence of humoral rejection. Testing for the presence of HLA antibodies against donor-mismatched antigens may be an initial noninvasive approach to identifying humoral graft rejection in patients with cancer and solid organ allografts. However, the absence of detectable DSAs does not exclude the possibility of humoral rejection because the allograft may adsorb most of the antibodies before any excess antibodies are detectable in the serum or plasma (termed silent alloimmunization). Furthermore, previously transplanted solid organ donor-mismatched HLA types should be avoided whenever possible in the selection of stem cell donors for subsequent stem cell transplantation due to the risk of prior alloimmunization and the increased risk of stem cell engraftment failure if a donor is chosen who expresses the same mismatched HLA type. In the instance of known humoral rejection, monitoring levels or titers of the DSA is commonly performed when detecting antibodies using a single-antigen bead flow cytometry method (eg, Luminex [Life Technologies, Carlsbad, California]) in addition to measuring fluorescence intensity. However, variation in the fluorescence intensity detected might be sufficiently high so that identification of an increasing or decreasing trend in antibody reactivity might require the use of normalization techniques or the use of January 2015, Vol. 22, No. 1 paired testing of a prior and current sample concurrently to minimize run-to-run variation and identify a true change in the level of reactivity. Conclusions Polymorphisms in the human leukocyte antigen (HLA) system influence the immune system in ways not yet completely understood, but associations are known to increase risk among patients with certain diseases and hypersensitivity to certain drugs. Knowledge of HLA type and whether alloimmunization has occurred may inform treatment and transfusion support plans. Numerous methods for HLA typing exist that include a single, multiple, or all clinically relevant HLA loci. In addition, these different methods may generate different degrees of detail regarding the HLA type depending on the specific treatment needed. HLA matching can be defined with different levels of stringency for different loci, thus balancing the increasing time needed to find the “perfect” allograft donor match and the risk of further disease progression/relapse prior to additional treatment with allogeneic stem cell transplantation versus less-stringent matching and an increased risk of allograft failure or life-threatening graft-vs-host disease. HLA antibody production through allosensitization may lead to more difficult, but not necessarily impossible, platelet transfusion support. However, HLA antibodies can also increase the risk of allograft failure for both stem cell and organ transplantations if patient antibodies are directed against donor HLA types. Therefore, it is critical for health care professionals to understand what HLA information (antigen typing, allele typing, or antibody testing) is needed for patient care and what impacts or risks are associated with that HLA type. References 1. Terasaki PI, McClelland JD. Microdroplet assay of human serum cytotoxins. Nature. 1964;204:998-1000. 2. Marsh SG, Albert ED, Bodmer WF, et al. Nomenclature for factors of the HLA system, 2010. Tissue Antigens. 2010;75(4):291-455. 3. Anthony Nolan Research Institute. Nomenclature for factors of the HLA system. http://hla.alleles.org. Accessed October 30, 2014. 4. College of American Pathologists. Definitions of histocompatibility typing terms. http://www.cap.org/apps/docs/laboratory_accreditation/histocompatibility_typing_terms.pdf. Accessed October 30, 2014. 5. Anthony Nolan Research Institute. G codes For reporting of ambiguous allele typings. http://hla.alleles.org/alleles/g_groups.html. Accessed October 30, 2014. 6. Mack SJ, Cano P, Hollenbach JA, et al. Common and well-documented HLA alleles: 2012 update to the CWD catalogue. Tissue Antigens. 2013;81(4):194-203. 7. Martin MA, Klein TE, Dong BJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for HLA-B genotype and abacavir dosing. Clin Pharmacol Ther. 2012;91(4):734-738. 8. US Food and Drug Administration. Ziagen (abacavir sulfate) tablets and oral solution: 2008. http://www.fda.gov/Safety/MedWatch/SafetyInformation/Safety-RelatedDrugLabelingChanges/ucm121838.htm. Accessed October 30, 2014. 9. Gatanaga H, Honda H, Oka S. Pharmacogenetic information derived from analysis of HLA alleles. Pharmacogenomics. 2008;9(2):207-214. 10. Shear NH, Milpied B, Bruynzeel DP, et al. A review of drug patch testing and implications for HIV clinicians. AIDS. 2008;22(9):999-1007. 11. Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Cancer Control 85 Natl Acad Sci U S A. 2005;102(11):4134-4139. 12. Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428(6982):486. 13. Daly AK, Donaldson PT, Bhatnagar P, et al; DILIGEN Study; International SAE Consortium. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816-819. 14. Kimmel SE, French B, Kasner SE, et al; COAG Investigators. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl J Med. 2013;369(24):2283-2293. 15. Verhoef TI, Ragia G, de Boer A, et al; EU-PACT Group. A randomized trial of genotype-guided dosing of acenocoumarol and phenprocoumon. N Engl J Med. 2013;369(24):2304-2312. 16. Alcoceba M, Sebastian E, Marin L, et al. HLA specificities are related to development and prognosis of diffuse large B-cell lymphoma. Blood. 2013;122(8):1448-1454. 17. Amiel JL. Study of the leukocyte phenotypes in Hodgkin’s disease. In: Curtoni ES, Mattiuz PI, Tosi RM, eds. Histocompatibility Testing. Munksgaard, Copenhagen: Consiglio Nazionale delle Ricerche; 1967:79-81. 18. Jin YJ, Shim JH, Chung YH, et al. Relationship of HLA-DRB1 alleles with hepatocellular carcinoma development in chronic hepatitis B patients. J Clin Gastroenterol. 2012;46(5):420-426. 19. Hayashi R, Ochiai T. HLA type and survival in gastric cancer. Cancer Res. 1986;46(7):3701-3703. 20. Trial to Reduce Alloimmunization to Platelets Study Group. Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. N Engl J Med. 1997;337(26):1861-1869. 21. Triulzi DJ, Kleinman S, Kakaiya RM, et al. The effect of previous pregnancy and transfusion on HLA alloimmunization in blood donors: implications for a transfusion-related acute lung injury risk reduction strategy. Transfusion. 2009;49(9):1825-1835. 22. O’Connell B, Lee EJ, Schiffer CA. The value of 10-minute posttransfusion platelet counts. Transfusion. 1988;28(1):66-67. 23. Davis KB, Slichter SJ, Corash L. Corrected count increment and percent platelet recovery as measures of posttransfusion platelet response: problems and a solution. Transfusion. 1999;39(6):586-592. 24. Spellman SR, Eapen M, Logan BR, et al; National Marrow Donor Program; Center for International Blood and Marrow Transplant Research. A perspective on the selection of unrelated donors and cord blood units for transplantation. Blood. 2012;120(2):259-265. 25. Petersdorf EW. Risk assessment in haematopoietic stem cell transplantation: histocompatibility. Best Pract Res Clin Haematol. 2007;20(2):155-170. 26. Petersdorf EW, Hansen JA, Martin PJ, et al. Major-histocompatibility-complex class I alleles and antigens in hematopoietic-cell transplantation. N Engl J Med. 2001;345(25):1794-1800. 27. Crocchiolo R, Ciceri F, Fleischhauer K, et al. HLA matching affects clinical outcome of adult patients undergoing haematopoietic SCT from unrelated donors: a study from the Gruppo Italiano Trapianto di Midollo Osseo and Italian Bone Marrow Donor Registry. Bone Marrow Transplant. 2009;44(9):571-577. 28. Gragert L, Eapen M, Williams E, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med. 2014;371(4):339-348. 29. Kekre N, Antin JH. Hematopoietic stem cell transplantation donor sources in the 21st century: choosing the ideal donor when a perfect match does not exist. Blood. 2014;124(3):334-343. 30. Lee SJ, Klein J, Haagenson M, et al. High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood. 2007;110(13):4576-4583. 31. Flomenberg N, Baxter-Lowe LA, Confer D, et al. Impact of HLA class I and class II high-resolution matching on outcomes of unrelated donor bone marrow transplantation: HLA-C mismatching is associated with a strong adverse effect on transplantation outcome. Blood. 2004;104(7):1923-1930. 32. Petersdorf EW, Anasetti C, Martin PJ, et al. Limits of HLA mismatching in unrelated hematopoietic cell transplantation. Blood. 2004;104(9):2976-2980. 33. Pidala J, Wang T, Haagenson M, et al. Amino acid substitution at peptide-binding pockets of HLA class I molecules increases risk of severe acute GVHD and mortality. Blood. 2013;122(22):3651-3658. 34. Fernandez-Viña MA, Wang T, Lee SJ, et al. Identification of a permissible HLA mismatch in hematopoietic stem cell transplantation. Blood. 2014;123(8):1270-1278. 35. Harkensee C, Oka A, Onizuka M, et al. Single nucleotide polymorphisms and outcome risk in unrelated mismatched hematopoietic stem cell transplantation: an exploration study. Blood. 2012;119(26):6365-6372. 36. Morishima S, Ogawa S, Matsubara A, et al; Japan Marrow Donor Program. Impact of highly conserved HLA haplotype on acute graft-versus-host disease. Blood. 2010;115(23):4664-4670. 37. McCullough J, Perkins HA, Hansen J. The National Marrow Donor Program with emphasis on the early years. Transfusion. 2006;46(7):1248-1255. 38. Appelbaum FR. Pursuing the goal of a donor for everyone in need. N Engl J Med. 2012;367(16):1555-1556. 39. Ballen KK, Gluckman E, Broxmeyer HE. Umbilical cord blood transplantation: the first 25 years and beyond. Blood. 2013;122(4):491-498. 40. Barker JN, Byam C, Scaradavou A. How I treat: the selection and acquisition of unrelated cord blood grafts. Blood. 2011;117(8):2332-2339. 41. Eapen M, Klein JP, Sanz GF, et al; Eurocord-European Group for Blood and Marrow Transplantation; Netcord; Center for International Blood and Mar86 Cancer Control row Transplant Research. Effect of donor-recipient HLA matching at HLA A, B, C, and DRB1 on outcomes after umbilical-cord blood transplantation for leukaemia and myelodysplastic syndrome: a retrospective analysis. Lancet Oncol. 2011;12(13):1214-1221. 42. Eapen M, Klein JP, Ruggeri A, et al; Center for International Blood and Marrow Transplant Research, Netcord, Eurocord, and the European Group for Blood and Marrow Transplantation. Impact of allele-level HLA matching on outcomes after myeloablative single unit umbilical cord blood transplantation for hematologic malignancy. Blood. 2014;123(1):133-140. 43. Woolfrey A, Klein JP, Haagenson M, et al. HLA-C antigen mismatch is associated with worse outcome in unrelated donor peripheral blood stem cell transplantation. Biol Blood Marrow Transplant. 2011;17(6):885-892. 44. Zino E, Frumento G, Marktel S, et al. A T-cell epitope encoded by a subset of HLA-DPB1 alleles determines nonpermissive mismatches for hematologic stem cell transplantation. Blood. 2004;103(4):1417-1424. 45. Fleischhauer K, Shaw BE, Gooley T, et al; International Histocompatibility Working Group in Hematopoietic Cell Transplantation. Effect of T-cell-epitope matching at HLA-DPB1 in recipients of unrelated-donor haemopoietic-cell transplantation: a retrospective study. Lancet Oncol. 2012;13(4):366-374. 46. Anasetti C, Amos D, Beatty PG, et al. Effect of HLA compatibility on engraftment of bone marrow transplants in patients with leukemia or lymphoma. N Engl J Med. 1989;320(4):197-204. 47. Yoshihara S, Taniguchi K, Ogawa H, et al. The role of HLA antibodies in allogeneic SCT: is the ‘type-and-screen’ strategy necessary not only for blood type but also for HLA? Bone Marrow Transplant. 2012;47(12):1499-1506. 48. Spellman S, Bray R, Rosen-Bronson S, et al. The detection of donor-directed, HLA-specific alloantibodies in recipients of unrelated hematopoietic cell transplantation is predictive of graft failure. Blood. 2010;115(13):2704-2708. 49. Rocha V, Spellman S, Zhang MJ, et al; Eurocord-European Blood and Marrow Transplant Group and the Center for International Blood and Marrow Transplant Research. Effect of HLA-matching recipients to donor noninherited maternal antigens on outcomes after mismatched umbilical cord blood transplantation for hematologic malignancy. Biol Blood Marrow Transplant. 2012;18(12):1890-1896. 50. van Rood JJ, Stevens CE, Smits J, et al. Reexposure of cord blood to noninherited maternal HLA antigens improves transplant outcome in hematological malignancies. Proc Natl Acad Sci U S A. 2009;106(47):19952-19957. 51. Kärre K. Immunology. A perfect mismatch. Science. 2002;295(5562): 2029-2031. 52. Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol. 2005;5(3):201-214. 53. Miller JS, Blazar BR. Control of acute myeloid leukemia relapse--dance between KIRs and HLA. N Engl J Med. 2012;367(9):866-868. 54. National Marrow Donor Program. HLA typing and matching. https:// bethematchclinical.org/transplant-therapy-and-donor-matching/hla-typing-andmatching. Accessed October 30, 2014. 55. Gajewski JL, Johnson VV, Sandler SG, et al. A review of transfusion practice before, during, and after hematopoietic progenitor cell transplantation. Blood. 2008;112(8):3036-3047. January 2015, Vol. 22, No. 1 Special Report Mobilization and Transplantation Patterns of Autologous Hematopoietic Stem Cells in Multiple Myeloma and Non-Hodgkin Lymphoma Luciano J. Costa, MD, PhD, Shaji Kumar, MD, Stephanie A. Stowell, MPhil, and Shari J. Dermer, PhD Background: The mobilization of hematopoietic stem cells can be a limiting factor for transplantation, yet little is known about how the availability of novel mobilizing agents has affected the practices of oncologists and transplant specialists. Methods: US-based oncologists (n = 48) and transplant specialists (n = 46) were separately surveyed with a partial overlap of assessed information. Results: More transplant specialists than oncologists believed that the time between referral and first consultation is adequate (89.1% vs 54.2%; P < .001). The presence of comorbidities was the most common reason for patients not being referred for transplantation. Among oncologists, 31.3% avoided cyclophosphamide and 16.7% avoided lenalidomide to prevent mobilization impairment in patients with multiple myeloma (MM). Chemotherapy mobilization for MM was used by 23.9% of transplant specialists due to higher CD34+ yields and by 21.7% due to its anti-MM effect. In non-Hodgkin lymphoma (NHL), 26.1% of transplant specialists used chemotherapy mobilization due to higher CD34+ yields, and 26.1% collected hematopoietic stem cells on the rebound prior to chemotherapy. With regard to plerixafor use in MM, 36.9% of transplant specialists reported that they did not use it, and 28.3% said they reserved it for second mobilization. In NHL, 4.3% of transplant specialists reported not using plerixafor, and 39.1% reserved it for second mobilization. Conclusions: Educational needs were identified to promote adequate referral for transplantation as well as successful and cost-effective methods for the mobilization of hematopoietic stem cells. Introduction Autologous hematopoietic stem cell transplantation From the Department of Medicine (LJC), Medical University of South Carolina, Charleston, South Carolina, the Division ofHematology (SK), Mayo Clinic, Rochester, Minnesota, and Med-IQ (SAS, SJD), Baltimore, Maryland. Dr Costa is now affiliated with the University of Alabama at Birmingham. Submitted July 22, 2014; accepted September 3, 2014. Address correspondence to Luciano J. Costa, MD, PhD, Department of Medicine and UAB-CCC, Bone Marrow Transplantation and Cell Therapy Program, University of Alabama at Birmingham, North Pavilion - NP 2554, 1802 6th Avenue South, Birmingham, AL 35294. E-mail: [email protected] Dr Costa has received consulting fees from sanofi-aventis and Onyx Pharmaceuticals. Dr Kumar has received consulting fees from Merck and has performed contracted research on behalf of the Mayo Clinic for Celgene Corporation, Cephalon, Genzyme, and Novartis. Ms Stowell’s spouse has conducted clinical research for Allergan, F. Hoffmann-La Roche, and Novo Nordisk and has received consulting fees from Bristol-Myers Squibb. Dr Dermer has nothing to disclose. This project was funded by an unrestricted educational grant provided by sanofi-aventis to Med-IQ. Sanofi-aventis was not involved in the elaboration of the survey, analysis of results, or writing of the manuscript. The authors thank Catherine Bretz Mullaney for grant oversight, Alyson Siclaire for project management, Robert Geist and Hollie Devine for assistance with survey development, Mary Catherine Downes and Kenny Khoo for outcomes management, Suzanne Jenkins for IT support, and Lisa Rinehart for editorial assistance. January 2015, Vol. 22, No. 1 (HSCT) is an increasingly important treatment option for several hematological malignancies, including multiple myeloma (MM) and non-Hodgkin lymphoma (NHL).1-5 The increasing use of autologous HSCT in individuals older than 65 years is partly due to the accumulation of data on the safety and efficacy of autologous HSCT for this age group.6,7 Furthermore, the emergence of novel mobilization agents has reduced the risk of mobilization failure, potentially extending the use of autologous HSCT to even more patients.8,9 The mobilization of hematopoietic stem cells fails in approximately 20% of patients with MM and up to 40% of patients with NHL.9,10 Poor mobilization can lead to poor engraftment, increased morbidity, greater resource utilization, and increased costs.10,11 The cause of poor mobilization can be partially explained by clinical variables (ie, age, underlying disease, prior therapies, underlying marrow function) and cannot be predicted.12 To ensure optimal mobilization, factors such as mobilization strategy, timing of hematopoietic stem cell collection, and identification of risk factors for poor mobilization must be considered; such factors have been summarized in published guidelines.13,14 Early communication between the primary oncologist and the transplant specialist is key to the critical timing of patient referral to a transplantation center.15 Cancer Control 87 Inconsistencies in practice approaches to hematopoietic stem cell mobilization and collection exist among health care professionals.16,17 To further understand perceptions and practices from the points of views of both oncologists and transplant specialists, a national survey was conducted. An accredited medical education company (Med-IQ, Baltimore, Maryland) collaborated with academic-based faculty to identify perceptions and practices affecting hematopoietic stem cell mobilization and transplantation as well as to identify barriers to successful transplantation in MM and NHL. Methods Two separate electronic survey tools assessed oncologist and transplant specialist perceptions and current practices related to autologous hematopoietic stem cell mobilization and transplantation. Eligible health care professionals were US-based, English-speaking physicians or nurse practitioners specializing in oncology or hematology. A random sample of 16,707 health care professionals meeting the inclusion criteria was e-mailed or faxed an invitation to complete the online surveys. Eligible participants expressing interest were provided a link to the Web-based surveys and asked to self-identify as either an oncologist (defined as one who did not perform autologous HSCT for patients with MM, lymphoma, or both) or a transplant specialist (defined as one who did). A total of 132 health care professionals responded to the invitation and, after self-identification, were directed to the appropriate survey. All participants were required to complete a consent form prior to beginning the survey. Those who completed both the consent form and a survey received a $50 honorarium as compensation for their time. The oncologist survey consisted of 17 multiple-choice questions; 4 questions required estimates of average number of patients and 1 question was open ended. The transplant specialist survey was composed of 21 multiple-choice questions and 1 open-ended question. Ten questions were common between the 2 surveys. The study protocol, including surveys, was submitted to an independent institutional review board (Chesapeake Review, Columbia, Maryland) and deemed exempt from oversight. For each survey, responses were anonymously pooled and data were downloaded from the online survey program and saved in an unidentified format. We described continuous numerical variables on the basis of median and interquartile range (IQR) and, where appropriate, categorical variables in terms of percentage with a 95% confidence interval (CI). Comparisons between proportions were performed using a chi-square test. All statistical analyses were 88 Cancer Control performed utilizing SPSS (IBM, Armonk, New York). In all inference analyses, 2-sided P values less than .05 were considered statistically significant. Survey Completion Forty-eight oncologists completed the survey (44 physicians and 4 nurse practitioners). The majority (n = 38) practiced in a nonacademic setting. The median number of new MM cases seen by oncologists each month was 2 (IQR, 1.37–4), and the median number of new NHL cases was 3.5 (IQR, 2–6). Oncologists managed a median of 20 patients with MM (IQR, 10–40) and 40 patients with NHL (IQR, 22.25–67) in their practices at the time they completed the survey. Overall, 46 transplant specialists completed the survey (44 physicians and 2 nurse practitioners). Eleven (23.9%) practiced in an academic setting and were fully dedicated to HSCT. Twenty-eight practiced in an academic setting dedicated to HSCT and also managed hematological malignancies. Seven transplant specialists were in nonacademic practices. The volume of HSCT procedures performed each year (including autologous and allogeneic) was self-reported to be less than 25 by 8.7% of transplant specialists, 25 to 49 by 4.3%, 50 to 99 by 41.3%, 100 to 200 by 17.4%, and more than 200 by 6.1%. Transplantation Referral We examined the possibility of delays in the referral process from the oncologist to the transplant specialist. Overall, oncologists perceived the process to be lengthier than transplant specialists (Fig 1), with 80.5% of transplant specialists stating that it took less than 2 weeks from referral to first encounter with a candidate for transplantation, whereas 39.6% of oncologists stated that patients referred to transplantation were typically seen within 2 weeks of referral (P < .001). The majority of oncologists (54.2%) believed the time between referral and first appointment was adequate (95% CI: 40.3–67.4), whereas 41.7% believed it was long but thought that the wait time had no detrimental effect on patient care (95% CI: 28.8–55.7). A total of 4.2% of oncologists believed that the time from referral to transplantation was too long and affected patient care (95% CI: 1.1–14.0). A higher proportion of transplant specialists (89.1% vs 54.2% of oncologists) believed that the time between referral and first appointment was adequate (95% CI: 76.9–95.2 vs 95% CI: 40.3–67.4, respectively; P < .001). Referral Patterns Multiple Myeloma When asked what percentage of their patients with MM younger than 65 years of age consulted with a transplant specialist in the first 6 months following diagnosis, 6.25% of oncologists indicated fewer January 2015, Vol. 22, No. 1 surance coverage (95% CI: 1.2–14.0), 4.2% cited lack of response to salvage therapy (95% CI: 1.2–14.0), and 70 27.1% cited none of the survey-sug60 gested reasons (95% CI: 16.6–41.0). 39.6 Similarity existed between the 33.3 50 opinions of oncologists and transplant specialists on when patients 40 19.6 with diffuse large B-cell lymphoma, 14.6 30 mantle cell lymphoma, and follicu13.0 lar lymphoma should be referred to 6.5 20 6.3 6.3 autologous HSCT (Fig 3). More trans0.0 plant specialists than oncologists 10 believed that patients with diffuse 0 large B-cell lymphoma and high>8 >8 <1 1–2 3–4 5–8 <1 1–2 3–4 5–8 risk disease should be evaluated for Week Weeks Weeks Weeks Weeks Week Weeks Weeks Weeks Weeks transplantation during initial therapy Transplant Clinicians Oncology Clinicians (P = not significant), and a larger proportion of transplant specialists Fig 1. — Perceptions of transplant specialists and oncologists on the time between referral and first believed that all patients with mantle appointment with a transplant physician. cell lymphoma should be considered than 5% (95% CI: 2.1–16.8), 25% indicated 6% to 20% for autologous HSCT while still undergoing first-line (95% CI: 14.9–38.8), 39.6% indicated 21% to 50% (95% therapy (P = .02). CI: 27.0–53.7), 16.7% indicated 51% to 80% (95% CI: 8.7–29.6), and 12.5% indicated more than 80% Prior Therapy and Mobilization (95% CI: 5.8–24.7). In regard to the reasons patients Because they were aware that some MM drugs influwith MM younger than 65 years of age would not be ence the efficacy of mobilization, 4.2% of oncologists referred for consultation with a transplant specialavoided the use of bortezomib in patients who might ist, 50% of oncologists cited comorbidities (95% CI: be eligible for transplantation (95% CI: 1.1–14.0), 36.9–63.6), 31.2% cited patient preference (95% CI: 31.3% avoided cyclophosphamide (95% CI: 19.9–45.3), 19.9–45.3%), 6.3% cited lack of insurance coverage 16.7% avoided lenalidomide (95% CI: 8.7–29.6), 8.3% (95% CI: 2.1–16.8), and 12.5% cited none of the above avoided liposomal doxorubicin (95% CI: 3.3–19.5), reasons (95% CI: 5.9–2.5). Among the oncologists, 18.7% considered the role of autologous HSCT in MM to 80 62.5 58.7 be in front-line therapy alone (95% 70 CI: 10.2–31.9), 66.7% felt that autologous HSCT should be employed in 60 both front-line and relapsed settings 32.6 (95% CI: 52.5–78.3), and 14.6% be50 lieved it should be used in relapsed 40 20.8 MM alone (95% CI: 7.3–27.2). Yet, little difference was seen between 30 10.4 opinions of when patients with MM 6.5 6.3 20 should be referred for transplantation consult (Fig 2). 0.0 10 60.9 Percent Percent 80 Non-Hodgkin Lymphoma When asked to identify reasons why patients younger than 65 years of age with relapsed diffuse large B-cell lymphoma would not have a transplantation consult, 41.7% of oncologists cited comorbidities (95% CI: 28.8–55.7), 16% cited patient preference (95% CI: 8.7–29.6), 4.2% cited lack of inJanuary 2015, Vol. 22, No. 1 0 A i td ag no si On s in iti t al On he ly py ra hi gh s -ri k pa tie nt s Up on l re ap se Transplant Clinicians A i td ag no si On s in iti t al On he ly py ra hi gh s -ri k pa tie nt s Up on l re ap se Oncology Clinicians Fig 2. — Opinions of transplant specialists and oncologists for which patients with multiple myeloma should be referred for a transplant evaluation. Cancer Control 89 80 A Fig 3A–C. — Opinion of transplant specialists and oncologists for which patients with (A) diffuse large B-cell lymphoma, (B) mantle cell lymphoma, and (C) follicular lymphoma should be referred for transplantation evaluation. AHSCT = autologous hematopoietic stem cell transplantation, CR = complete response, PR = partial response. 60.4 54.3 70 60 Percent 50 40 23.9 18.8 17.4 30 12.5 20 4.3 4.2 4.2 10 0 ne rapy rapy rapy ne rapy rapy rapy one t li t li N e e e e e e firs e th e th e th firs e th e th e th g g g g g g g g n n uri salva salva salva uri salva salva salva d d , , re re isk isk On After On After h r Befo h r Befo Hig Hig 80 B 58.7 70 60 39.6 33.3 Percent 50 23.9 40 22.9 15.2 30 20 4.2 2.2 10 0 e e y y e e y y rap alvag alvag rap rap rap alvag alvag the the the the s s s s e e e e e e n n r r n n n n o y, o t-li t-li t-li t-li efo efo ry, ,b irs irs tor firs firs y, b racto nf nf ory efrac t tor o o on on f c c e l l k k l l A A is is fra fra d/r d/r se se hr hr /re /re Hig lapse Relap Hig lapse Relap Re Re 50 C 30.4 45 33.3 28.3 23.9 40 22.9 22.9 35 18.8 Percent 30 13.0 25 20 15 4.3 2.1 10 5 0 a a e e R R in in rs rs se se R/C 2 yea ny tim relap SCT phom PR/C 2 yea ny tim relap SCT phom t tP H H a a m m s s < < d d A A r r e e ly ly n n Fi Fi se se ps ps co co for ar for ar lap Rela lap Rela Se role licul Se role licul Re Re l l o o o o f f N N Transplant Clinicians 90 Cancer Control Oncology Clinicians and 2.1% avoided thalidomide (95% CI: 0.4–10.9). A total of 54.2% of oncologists did not avoid any specific MM drug for this reason (95% CI: 40.3–67.4). When asked how the known impairment of lenalidomide on mobilization should be managed, similarity was seen between opinions: 4.2% of oncologists and 2.2% of transplant specialists believed that lenalidomide should not be used in induction therapy for MM, and 35.4% of oncologists and 28.3% of transplant specialists believed that lenalidomide could be used for induction, but patients would require chemomobilization to obtain an adequate number of CD34+ cells. The majority of responders (56.2% of oncologists and 63.0% of transplant specialists) believed that lenalidomide could be used, but hematopoietic stem cell mobilization and collection should occur after no more than 4 cycles of therapy. A small minority — 4.2% of oncologists and 6.5% of transplant specialists — answered that lenalidomide could be used for induction, but hematopoietic stem cell mobilization would be possible with the use of plerixafor alone. When oncologists were asked how the known effect of lenalidomide on mobilization affected their choice to use lenalidomide in patients with MM eligible for transplantation, 4.2% indicated that they did not use lenalidomide (95% CI: 1.1–14.0), 77.1% indicated that they used this agent but referred patients to transplantation before 4 completed cycles of therapy (95% CI: 63.5–86.7), and 18.8% used this agent without a specific cycle limit and were confident that the transplantation team could collect hematopoietic stem cells regardless of prior lenalidomide use (95% CI: January 2015, Vol. 22, No. 1 A 80 70 58.3 60 41.3 50 Percent 10.2–31.9). Similarly, because treatment for NHL can also influence the success of hematopoietic stem cell mobilization, 18.8% of oncologists avoided using bendamustine in patients eligible for transplantation (95% CI: 10.2–31.9), 4.2% avoided bortezomib (95% CI: 1.1–14.0), 60.4% avoided fludarabine (95% CI: 46.3–73.0), 16.7% avoided the hyperfractionated regimen of cyclophosphamide/vincristine/doxorubicin/dexamethasone (95% CI: 8.7–29.6), and 50.0% avoided radio immunotherapy (95% CI: 36.9–63.6). A total of 18.7% of oncologists did not avoid the use of any specific treatment in NHL for this reason (95% CI: 10.2–31.9). 35.4 28.3 40 19.6 30 20 8.7 10 4.2 2.2 2.1 0.0 0 <5 % % % % 0% –10 –20 –30 >3 5% 11% 21% <5 % % % % 0% –10 –20 –30 >3 5% 11% 21% 80 B 70 Mobilization Practices 52.1 60 Percent We explored how decisions regarding hematopoietic stem cell 50 mobilization were made at differ31.3 32.6 ent centers. Two decision-making 40 28.3 26.1 processes were equally common: 30 (1) the transplantation center had 12.5 a uniform method of mobilization 20 accepted and followed by all prac8.7 4.3 titioners, and (2) a mobilization 10 2.1 2.1 0.0 strategy was chosen based on strat0 ification for perceived risk of mobi% % N /A % % % % % % % % < 5 %–10 –20 –30 > 30 < 5 %–10 –20 –30 > 30 lization failure (34.8% of respond5 5 11% 21% 11% 21% ers for both; 95% CI: 22.7–49.2). Other frequently reported processTransplant Clinicians Oncology Clinicians es were that each individual physician at his or her site chose the Fig 4A–B. — Perceived risk of mobilization failure for (A) multiple myeloma and (B) non-Hodgkin mobilization method for his or her lymphoma. patients or that his or her center followed an algorithm to stratify patients to differtransplant specialists mentioned that their preferred ent strategies of mobilization based on peripheral method of hematopoietic stem cell mobilization for blood CD34+ enumeration (15.2% of responders for patients with MM was growth factor and planned or both; 95% CI: 7.5–28.3). “just-in-time” plerixafor, followed by high-dose cycloA notable discrepancy existed between oncolophosphamide and growth factor (26.1%) or growth gists and transplant specialists in terms of their perfactor alone (17.4%). For patients with NHL, 47.8% of ceived risk of inadequate mobilization in both MM and transplant specialists utilized growth factor following NHL (Fig 4). Although 83.4% of oncologists believed the last cycle of disease-appropriate chemotherapy as that the risk of mobilization failure in NHL was less a mobilization strategy, 34.8% utilized growth factor than 10%, 50.9% of the transplant specialists thought with or without planned or “just-in-time” plerixafor. the same (P = .02). Similarly, the risk of mobilization Growth factor following high-dose cyclophosphamide failure in MM was considered to be less than 5% by was the preferred method of hematopoietic stem cell 58.3% of oncologists, whereas 41.3% of the transplant mobilization in patients with NHL for 10.9% of transspecialist assessed this risk in the same way (P = .09). plant specialists. The preferred method of hematopoietic stem In regard to the use of chemotherapy mobilicell mobilization among transplant specialists varied zation in MM, 54.3% of transplant specialists indiaccording to underlying disease. Overall, 47.8% of cated that they did not use it because it is more January 2015, Vol. 22, No. 1 Cancer Control 91 toxic than other modalities and not necessary (95% CI: 40.2–67.8). By contrast, 23.9% indicated that they utilized chemomobilization primarily because of the higher CD34+ yields, and 21.7% indicated that they used chemomobilization because they believed the additional dose of chemotherapy would help the patient with better disease control. When asked when they utilized plerixafor for hematopoietic stem cell mobilization, 36.9% of transplant specialists indicated that they never used it because the drug is expensive or unnecessary (95% CI: 24.5–51.4), 28.3% indicated that they used it following mobilization failure (95% CI: 17.3–42.6), 39.1% indicated that they used it in patients with MM at risk of mobilization failure (95% CI: 26.4–53.5), and 17.4% indicated that they used it “just in time” for patients with a low number of CD34+ cells in peripheral blood following the use of a growth factor (95% CI: 9.1–30.7). For the use of chemomobilization in NHL, 39.1% of transplant specialists indicated that they did not use this modality (95% CI: 26.4–53.6), 26.1% used chemomobilization due to higher CD34+ yield (95% CI: 15.6–40.3), and 26.1% indicated that they used chemomobilization because the patient was already receiving chemotherapy for the management of NHL (95% CI: 26.4–53.6). A total of 8.7% indicated a preference for chemomobilization (95% CI: 3.4–20.3), citing the belief that the extra chemotherapy could help consolidate remission and prevent post-transplantation relapse. In regard to hematopoietic stem cell mobilization in NHL, 4.3% of the transplant specialists reported that they did not use plerixafor because it was expensive and unnecessary (95% CI: 1.2–14.5), 17.4% used plerixafor in all mobilizations (95% CI: 9.1–30.7), 26.1% used this agent in patients at risk of mobilization failure (95% CI: 15.6–40.3), 36.9% used a “just-in-time” approach with administration for patients with low CD34+ in the peripheral blood (95% CI: 24.5–51.4), and 39.1% used plerixafor following first mobilization failures (95% CI: 26.4–53.6). Discussion Surveys provide a fast and relatively inexpensive approach to gather information. However, a caveat of selection bias can be present such that the small proportion of responding health care professionals has dissimilar characteristics and opinions from all other practicing oncologists and transplant specialists. It is difficult to assess the representativeness of this sample because no detailed characterizations of these groups are available, particularly with regard to transplant specialists as discussed in a recent assessment of the transplant specialist workforce.18 Nevertheless, it appears that the survey captured the opinions of oncologists with intense activity in MM and NHL, as reflected in the high number of patients reported; in addition, 92 Cancer Control the transplant specialist group represented different practice types and volumes of patients treated. We identified that a high proportion of patients with MM younger than 65 years of age was not referred for transplantation consultation early in the course of disease, despite the fact that most oncologists supported a role for early HSCT in MM. This is in synchrony with data showing the increasing but low utilization of autologous HSCT among younger patients with MM.6 In terms of NHL, an overlap was seen in the opinion of oncologists and transplant specialists on the appropriate time for referral, although more transplant specialists believed that patients with mantle cell lymphoma should be referred for autologous HSCT while undergoing primary therapy. Even though oncologists and transplant specialists agreed on when patients should be referred for transplantation, a large number of oncologists perceived the referral process to be lengthy. This perception was not shared by a large majority of transplant specialists, who indicated that time from oncologist referral until first encounter with a transplant specialist may be a barrier and transplant centers may want to re-examine their processes to ensure that access is prompt and effective. Among oncologists, comorbidities were cited as the most frequent reason patients younger than 65 years of age with a clinical indication for autologous HSCT would not be referred. Even though some of these patients may have obvious contraindications (eg, end-stage heart failure, liver failure, dementia), we found it concerning that autologous HSCT may be ruled out as a therapeutic option without the recommended assessment of a transplant specialist.15 Transplant eligibility based on age, comorbidities, and underlying disease are best evaluated by a transplant specialist. Even though the age of 65 years is frequently cited as a limit for autologous HSCT, the safety and efficacy of this treatment in older individuals has been demonstrated.19,20 Similarly, common comorbidities may be compatible with autologous HSCT, including chronic kidney disease, stable coronary artery disease, controlled cardiac arrhythmias, hypertension, diabetes, and prior treated malignancies. Therefore, oncologists should be encouraged to refer patients for evaluation by a transplant specialist in accordance with accepted guidelines.15 Nearly one-third of oncologists avoided cyclophosphamide-containing regimens in patients with MM because they believed the drug would impair mobilization despite evidence suggesting otherwise.21,22 This proportion was even higher than the proportion of oncologists who avoided lenalidomide, a drug shown to impair mobilization.23,24 More than one-half of health care professionals from both specialties acknowledged that lenalidomide can be used January 2015, Vol. 22, No. 1 in induction as long as patients proceed with early hematopoietic stem cell mobilization. However, 1 in 3 oncologists and 1 in 4 transplant specialists believed that patients who received lenalidomide induction should be mobilized with chemotherapy despite data that demonstrate these patients can be effectively mobilized with growth factor or growth factor plus plerixafor, particularly when referred early for mobilization.25,26 In regard to NHL, oncologists seemed concerned about the use of fludarabine and radioimmunotherapy in patients who are candidates for transplantation. Several important observations were made in regard to mobilization practices. Oncologists perceived mobilization failure to be a less frequent problem than transplant specialists. This lack of awareness may be a contributing factor for late referral for autologous hematopoietic stem cell collection. In approximately one-third of the cases, we found that transplantation programs have a standard mobilization strategy; in another one-third of cases, mobilization strategy is chosen based on perceived risk of inadequate mobilization. A minority of transplant specialists adapted mobilization strategies based on the CD34+ enumeration, a practice that can lead to resource rationalization and a high rate of mobilization.27-29 Transplant specialists indicated more frequent use of chemomobilization in NHL, partly because such patients are already receiving disease-specific chemotherapy and mobilization can often be performed during recovery from the previous cycle of chemotherapy. In MM, most transplant specialists who indicated a preference for chemomobilization believed that its use would offer better long-term disease control, which is an unconfirmed hypothesis.30,31 A large proportion of transplant specialists indicated that they do not use plerixafor mobilization due to its high cost or reserve its use for a second attempt in patients who have failed initial mobilization. However, some studies suggest that, although plerixafor is costly, its judicious use based on individualized risk of poor mobilization avoids the expense of remobilization and medical care for the complications of chemotherapy mobilization, thereby neutralizing the cost difference and potentially allowing more patients to proceed to transplantation.27,32,33 Conclusions This survey suggests that several areas are in need of improvement. Oncologists may benefit from additional education on the importance of referring potential candidates for autologous hematopoietic stem cell transplantation early on as well as the implication of their therapy choices on the collection of hematopoietic stem cells. Because hematopoietic stem cell mobilization methods remain diverse, more education January 2015, Vol. 22, No. 1 and broader discussions involving transplant specialists may improve patient outcomes. In addition, more prospective trials and clinical data are needed to further delineate mobilization practices. Oncologists and transplant specialists should be encouraged to work together to streamline processes that promote easy and fast access for referred patients. References 1. Dreyling M, Hiddemann W; European MCL Network. Current treatment standards and emerging strategies in mantle cell lymphoma [Erratum appears in Hematology Am Soc Hematol Educ Program. 2011;2011:562]. Hematology Am Soc Hematol Educ Program. 2009:542-551. 2. Oliansky DM, Gordon LI, King J, et al. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the treatment of follicular lymphoma: an evidence-based review. Biol Blood Marrow Transplant. 2010;16(4):443-468. 3. Child JA, Morgan GJ, Davies FE, et al; Medical Resesarch Council Adult Leukaemia Working Party. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. N Engl J Med. 2003;348(19):1875-1883. 4. Attal M, Harousseau JL, Stoppa AM, et al; Intergroupe Francais du Myelome. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. N Engl J Med. 1996;335(2):91-97. 5. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N Engl J Med. 1995;333(23):15401545. 6. Costa LJ, Zhang MJ, Zhong X, et al. Trends in utilization and outcomes of autologous transplantation as early therapy for multiple myeloma. Biol Blood Marrow Transplant. 2013;19(11):1615-1624. 7. McCarthy PL Jr, Hahn T, Hassebroek A, et al. Trends in use of and survival after autologous hematopoietic cell transplantation in North America, 1995-2005: significant improvement in survival for lymphoma and myeloma during a period of increasing recipient age. Biol Blood Marrow Transplant. 2013;19(7):1116-1123. 8. Gertz MA. Current status of stem cell mobilization. Br J Haematol. 2010;150(6):647-662. 9. DiPersio JF, Micallef IN, Stiff PJ, et al. Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin’s lymphoma. J Clin Oncol. 2009;27(28):4767-4773. 10. Pusic I, Jiang SY, Landua S, et al. Impact of mobilization and remobilization strategies on achieving sufficient stem cell yields for autologous transplantation. Biol Blood Marrow Transplant. 2008;14(9):1045-1056. 11. Gertz MA, Wolf RC, Micallef IN, Gastineau DA. Clinical impact and resource utilization after stem cell mobilization failure in patients with multiple myeloma and lymphoma. Bone Marrow Transplant. 2010;45(9):1396-1403. 12. Costa LJ, Nista EJ, Buadi FK, et al. Prediction of poor mobilization of autologous CD34+ cells with growth factor in multiple myeloma patients: implications for risk-stratification. Biol Blood Marrow Transplant. 2014;20(2):222228. 13. Duong HK, Savani BN, Copelan E, et al. Peripheral blood progenitor cell mobilization for autologous and allogeneic hematopoietic cell transplantation: guidelines from the American Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2014;20(9):1262-1273. 14. Giralt S, Costa L, Schriber J, et al. Optimizing autologous stem cell mobilization strategies to improve patient outcomes: consensus guidelines and recommendations. Biol Blood Marrow Transplant. 2014;20(3):295-308. 15. National Morrow Donor Program. Recommended timing for transplant consultation. https://bethematchclinical.org/WorkArea/DownloadAsset.aspx?id=3545. Accessed October 8, 2014. 16. Lee SJ, Joffe S, Artz AS, et al. Individual physician practice variation in hematopoietic cell transplantation. J Clin Oncol. 2008;26(13):2162-2170. 17. Bevans M, Tierney DK, Bruch C, et al. Hematopoietic stem cell transplantation nursing: a practice variation study. Oncol Nurs Forum. 2009;36(6):E317-E325. 18. Gajewski JL, LeMaistre CF, Silver SM, et al. Impending challenges in the hematopoietic stem cell transplantation physician workforce. Biol Blood Marrow Transplant. 2009;15(12):1493-1501. 19. Sharma M, Zhang MJ, Zhong X, et al. Older patients with myeloma derive similar benefit from autologous transplantation. Biol Blood Marrow Transplant. 2014. Epub ahead of print. 20. Buadi FK, Micallef IN, Ansell SM, et al. Autologous hematopoietic stem cell transplantation for older patients with relapsed non-Hodgkin’s lymphoma. Bone Marrow Transplant. 2006;37(11):1017-1022. 21. Mikhael JR, Reeder CB, Libby EN III, et al. Results from the phase II dose expansion of cyclophosphamide, carfilzomib, thalidomide and dexamethasone (CYCLONE) in patients with newly diagnosed multiple myeloma. Blood. 2012:120(21):445. Cancer Control 93 22. Reeder CB, Reece DE, Kukreti V, et al. Cyclophosphamide, bortezomib and dexamethasone induction for newly diagnosed multiple myeloma: high response rates in a phase II clinical trial. Leukemia. 2009;23(7):1337-1341. 23. Kumar S, Dispenzieri A, Lacy MQ, et al. Impact of lenalidomide therapy on stem cell mobilization and engraftment post-peripheral blood stem cell transplantation in patients with newly diagnosed myeloma. Leukemia. 2007;21(9):2035-2042. 24. Paripati H, Stewart AK, Cabou S, et al. Compromised stem cell mobilization following induction therapy with lenalidomide in myeloma. Leukemia. 2008;22(6):1282-1284. 25. Costa LJ, Abbas J, Hogan KR, et al. Growth factor plus preemptive (‘just-in-time’) plerixafor successfully mobilizes hematopoietic stem cells in multiple myeloma patients despite prior lenalidomide exposure. Bone Marrow Transplant. 2012;47(11):1403-1408. 26. Sinha S, Gertz MA, Lacy MQ, et al. Majority of patients receiving initial therapy with lenalidomide-based regimens can be successfully mobilized with appropriate mobilization strategies. Leukemia. 2012;26(5):1119-1122. 27. Micallef IN, Sinha S, Gastineau DA, et al. Cost-effectiveness analysis of a risk-adapted algorithm of plerixafor use for autologous peripheral blood stem cell mobilization. Biol Blood Marrow Transplant. 2013;19(1):87-93. 28. Costa LJ, Alexander ET, Hogan KR, et al. Development and validation of a decision-making algorithm to guide the use of plerixafor for autologous hematopoietic stem cell mobilization. Bone Marrow Transplant. 2011;46(1):6469. 29. Li J, Hamilton E, Vaughn L, et al. Effectiveness and cost analysis of “just-in-time” salvage plerixafor administration in autologous transplant patients with poor stem cell mobilization kinetics. Transfusion. 2011;51(10):2175-2182. 30. Gertz MA, Kumar SK, Lacy MQ, et al. Comparison of high-dose CY and growth factor with growth factor alone for mobilization of stem cells for transplantation in patients with multiple myeloma. Bone Marrow Transplant. 2009;43(8):619-625. 31. Dingli D, Nowakowski GS, Dispenzieri A, et al. Cyclophosphamide mobilization does not improve outcome in patients receiving stem cell transplantation for multiple myeloma. Clin Lymphoma Myeloma. 2006;6(5):384-388. 32. Costa LJ, Miller AN, Alexander ET, et al. Growth factor and patient-adapted use of plerixafor is superior to CY and growth factor for autologous hematopoietic stem cells mobilization. Bone Marrow Transplant. 2011;46(4):523-528. 33. Shaughnessy P, Islas-Ohlmayer M, Murphy J, et al. Cost and clinical analysis of autologous hematopoietic stem cell mobilization with G-CSF and plerixafor compared to G-CSF and cyclophosphamide. Biol Blood Marrow Transplant. 2011;17(5):729-736. 94 Cancer Control January 2015, Vol. 22, No. 1 Special Report Functional Health Literacy, Chemotherapy Decisions, and Outcomes Among a Colorectal Cancer Cohort Evan L. Busch, MPH, Christopher Martin, MSPH, Darren A. DeWalt, MD, and Robert S. Sandler, MD Background: Functional health literacy is essential for the self-management of chronic diseases and preventive health behaviors. Patients with cancer who have a low level of health literacy may be at greater risk for poor care and poor outcomes. Methods: We assessed health literacy using the Short Test of Functional Health Literacy in Adults in 347 participants with colorectal cancer who were nested within a prospective observational study of system, health care provider, and participant characteristics influencing cancer outcomes. Results: Having adequate health literacy increased the likelihood that participants with stage 3/4 disease received chemotherapy (odds ratio, 3.29; 95% confidence interval, 1.23–8.80) but had no effect on cancer stage at diagnosis or vital status at last observation during postenrollment follow-up. No difference was seen in health literacy status regarding participant beliefs and preferences about chemotherapy among those with stage 3/4 disease, nor in participant roles in deciding whether to receive chemotherapy. Conclusions: Patients with lower levels of health literacy were less likely to receive chemotherapy compared with participants with higher levels of health literacy. Therefore, clear communication related to key health care decisions may lead to fewer disparities due to a patient’s level of health literacy. Introduction Functional health literacy, including the ability to read and understand medication labels, educational materials, hospital directional signs, and appointment slips, is essential for the self-management of chronic diseases and preventive health behaviors.1 However, individuals with the greatest health care needs may have the least ability to read and comprehend information needed to successfully function as patients.1,2 Inadequate health literacy could decrease the likelihood that these individuals at high risk will have beneficial health outcomes. Having inadequate or marginal functional health literacy places patients at increased risk for medication nonadherence, hospital admission, poor health status, and worse clinical outcomes than their counterparts with higher levels of health literacy.1,3-5 From the Departments of Epidemiology (ELB) and Medicine (CM, DAD, RSS), University of North Carolina, Chapel Hill, North Carolina. Submitted July 2, 2014; accepted August 18, 2014. Address correspondence to Robert S. Sandler, MD, Department of Medicine, University of North Carolina at Chapel Hill, 4157 Bioinformatics Building, Campus Box 7555, Chapel Hill, NC 27599-7555. E-mail: [email protected] No significant relationships exist between the authors and the companies/organizations whose products or services may be referenced in this article. This work was supported in part by grants from the National Institutes of Health (P30 DK034987, U01 CA93326). January 2015, Vol. 22, No. 1 A lower level of health literacy has also been associated with lower patient information seeking.6 Patients with poor reading ability may have issues accessing the health care system, understanding recommended treatments, and following the instructions of health care professionals. The American Medical Association has recognized that limited patient literacy impedes diagnosis and treatment, so it has adopted policies to increase the recognition of — and effect change in — functional health literacy.1 In particular, health literacy levels may influence cancer outcomes. New patients may receive large amounts of unfamiliar technical information about their diagnosis. Oftentimes, health care professionals invite patients to participate in choosing among complicated treatment options. Adhering to chosen treatments can be a Byzantine process of understanding and complying with surgery, radiation therapy, multiple and varying chemotherapy regimes, and follow-up visits involving several health care professionals. To evaluate the role of health literacy in decisions related to cancer treatment and to estimate the impact of health literacy on patient outcomes, we assessed health literacy in a set of patients with colorectal cancer (CRC) enrolled in a cohort study of health care processes. CRC has one of the largest disease burdens of any form of cancer, with approximately 140,000 new cases and 50,000 deaths in 2014 in the United States,7 making it a good test case of the effects of health literacy on cancer outcomes. We hypotheCancer Control 95 sized that greater levels of health literacy would be associated with early-stage disease, increased patient participation in treatment decisions, receipt of more appropriate treatment, and improved rates of survival. Methods Study Population Participants were enrolled in the Cancer Care Outcomes Research and Surveillance Consortium (CanCORS), a prospective, population-based, multisite observational study of participants with lung and colon cancers that has been previously described.8 The population was diverse with respect to race, socioeconomic status, and geography. The purpose of the study was to assess the impact of system, care provider, and patient factors on cancer outcomes. Participants were at least 21 years of age at the time of CRC diagnosis and were enrolled within 3 months of diagnosis during 2003 to 2006. The study collected participant surveys, surrogate surveys for participants who were deceased or too ill to participate, and medical records data. Abstractors at each site collected information on tumor characteristics and cancer treatments received. Participant and, when necessary, surrogate surveys were completed using computer-assisted telephone interviews. Surveys included items about demographic and socioeconomic factors (age, insurance coverage, income), communication with health care professionals, and beliefs and preferences regarding cancer treatments. The surveys have been previously described.9 CanCORS included patients with colorectal and lung cancers who were enrolled by 7 groups of investigators.8 North Carolina recruited 990 patients with CRC, and it was the only site to administer health literacy assessments. The study population was a random sample of 347 participants from the North Carolina–based CanCORS study. The sample was stratified by self-reported years of education with oversampling of lower strata to achieve similar-sized strata of adequate vs inadequate or marginal health literacy, ultimately with the goal of enhancing power for planned analyses. We used the following sampling fractions for the respective ranges of years of education: 0 to 8 years (100%), 9 to 11 years (100%), 12 years (70%), and more than 12 years (40%). The sample included all cancer stages. The Institutional Review Board at the University of North Carolina at Chapel Hill approved the protocol. All participants provided informed consent. Measure of Health Literacy Functional health literacy was assessed using the Short Test of Functional Health Literacy in Adults 96 Cancer Control (S-TOFHLA).10,11 A trained interviewer visited participant homes to administer the assessment in person. The assessment tested reading comprehension using 36 questions in response to 2 prose passages. Across the entire sample, 4 different interviewers were used, but 1 interviewer alone administered the assessment for any given participant. All interviewers were trained in how to administer the S-TOFHLA. The interviewer read a scripted introduction and instructions to the participant, and then remained silent while the participant completed the questionnaire. Participants were given up to 7 minutes to complete the questionnaire, but they were not told beforehand that the assessment would be timed. S c o r e s we r e c a t e g o r i z e d a s i n a d e q u a t e (0–16 correct), marginal (17–22 correct), or adequate (23–36 correct). For analysis, we combined marginal and inadequate scores into 1 category. Chemotherapy Decisions As part of the surveys, participants in CanCORS answered questions about whether they received adjuvant chemotherapy, how the chemotherapy decision was made, and their beliefs and preferences regarding chemotherapy. Chemotherapy is generally recommended for patients with colon cancer diagnosed with stage 3 or 4 disease.12 We examined responses about chemotherapy decisions among participants with CRC in whom health literacy was assessed and who were diagnosed with either stage 3 or 4 disease. Survey responses of “Do not know,” “Declined to answer,” “Not applicable,” or were missing were considered noninformative. Noninformative responses were excluded when conducting Fisher exact tests to compare survey responses by level of health literacy. Outcomes CanCORS tumors were staged according to the TNM classification system. We considered stage 1/2 to be early-stage disease and stage 3/4 to be late-stage disease. Participants were followed for survival after baseline data collection. Vital status for all participants was verified using the Social Security Death Index on May 4, 2010, providing at least 42 months of follow-up observation time for each person. We defined participant survival as dichotomous vital status (alive or dead) at last observation. Statistical Analysis Among participants in the literacy sample with any stage of cancer (N = 347), we calculated overall and health literacy–stratified descriptive statistics for demographic and socioeconomic characteristics. Chi-square tests of association were conducted to examine differences in participant characteristics January 2015, Vol. 22, No. 1 by level of health literacy. We performed logistic regression analyses to estimate associations between health literacy and (1) whether participants received chemotherapy (for stage 3/4 disease), (2) cancer stage at diagnosis (for all participants), and (3) all-cause mortality at last observation after baseline (for all participants). Across all stages, we estimated the marginal effect of health literacy on survival as well as its conditional effect on demongraphic and socioeconomic covariates. Among 130 participants with stage 3/4 disease in the health literacy sample, we calculated overall and health literacy–stratified descriptive statistics for responses to survey questions about their beliefs and preferences regarding chemotherapy, communication with health care professionals about chemotherapy, and their role in making the decision whether to receive chemotherapy. We used Fisher exact tests to evaluate whether survey responses differed by participant health literacy level. For all tests of association, P values less than .05 were considered statistically significant. Analyses were performed at the University of North Carolina at Chapel Hill, which was the CanCORS site for all participants included in this study. We used CanCORS core data (version 1.16), medical record data (version 1.12), and participant survey data (version 1.12). All analyses were performed using SAS version 9.3 for Windows (SAS Institute, Cary, North Carolina). Results Table 1 presents descriptive statistics among the 347 participants for whom health literacy was assessed. Despite our goal of a sample with approximately equal numbers with adequate January 2015, Vol. 22, No. 1 Table 1. — Participant Characteristics Characteristic Overall (N = 347)a n % Adequate Literacy (n = 242)a n Marginal/Inadequate Literacy (n = 105)a % n % 43 58 57 57 44 43 P valueb Sex Male 159 47 101 Female 178 53 134 .02 Race White 264 78 201 86 63 62 Nonwhite 73 22 34 14 39 38 < .0001 Age, years < 65 160 47 130 55 30 29 ≥ 65 177 53 105 45 72 71 < .0001 Education Less than high school 72 23 36 16 36 43 High school/GED 112 36 80 35 32 39 Above high school 82 26 76 33 6 7 Other 44 14 35 15 9 11 < .0001 Marital Status Currently married/living with partner 211 63 158 67 53 52 Widowed, divorced, separated, or never married 126 37 77 33 49 48 .008 Household Income ($), past year < 20,000 75 24 42 19 33 40 20,000–39,999 82 27 56 25 26 31 40,000–59,999 52 17 42 19 10 12 ≥ 60,000 78 25 73 32 5 6 Refused/ do not know 22 7 13 6 9 11 < .0001 Cancer Stage 1/2 187 59 3/4 130 41 132 60 55 57 89 40 41 43 .7 Received Chemotherapy Yes 164 No 146 53 129 57 35 42 47 98 43 48 58 .02 Vital Status at Last Observation Alive 260 77 187 80 73 72 Dead 77 23 48 20 29 28 .1 Differences between numbers of patients for each column and number of patients for each characteristic are representative of missing data. Percentages are representative for nonmissing data for the characteristic. b P values were based on chi-square tests of each characteristic by levels of health literacy.” GED = general educational development. a Cancer Control 97 and marginal/inadequate levTable 2. — Participant Preferences and Role in Receipt of Chemotherapy by Literacy Levela els of health literacy, 105 (30%) Total Number of Patients (%) P were categorized as marginal/ Survey Question b (N = 130) value inadequate. Compared with Adequate Marginal/ Literacy Inadequate those with an adequate level (n = 89), % Literacy (n = 41), % of health literacy, participants Did any of your doctors tell you not to have chemotherapy? with marginal/inadequate health literacy were more Yes 2 1 (1) 1 (4) .4 frequently men, nonwhite, at No 108 84 (99) 24 (96) least 65 years of age, not curAfter talking with your doctors about chemotherapy, how likely did you think it was that rently married or living with chemotherapy would help you live longer? a partner, had not completed 9 9 (11) 0 (0) .1 high school, and had annu- Not likely/a little likely 95 71 (89) 24 (100) al household incomes below Somewhat likely/very likely $40,000. After talking with your doctors about chemotherapy, how likely did you think it was that Among participants with chemotherapy would help you with problems you were having because of your (cancer)? stage 3/4 disease having an Not likely/a little likely 9 9 (18) 0 (0) .1 adequate level of health lit- Somewhat likely/very likely 61 42 (82) 19 (100) eracy increased the odds Which statement best describes the role you played when the decision was made about of receiving chemotherapy chemotherapy? compared with those with You made the decision with little 5 4 (5) 1 (4) .2c a marginal/inadequate level or no input from your doctors. of health literacy (odds ratio You made the decision after 27 (33) 5 (20) [OR], 3.29; 95% confidence in- considering your doctors’ opinions. 32 terval [CI], 1.23–8.80). HowYou and your doctors made the 58 44 (53) 14 (56) ever, across all stages, having decision together. an adequate level of health Your doctors made the decision 5 5 (6) 0 (0) literacy did not increase after considering your opinion. the odds of presenting with Your doctors made the decision 8 3 (4) 5 (20) early-stage compared with with little or no input from you. late-stage disease (OR, 1.11; If you had to make a choice now, would you prefer treatment that extends life as much as possi95% CI, 0.68–1.80). ble, even if it means having more pain and discomfort, or would you want treatment that focuses For participants with on relieving pain and discomfort as much as possible, even if it means not living as long? stage 3/4 disease, Table 2 Extend life as much as possible 58 45 (58) 13 (52) .6 presents responses to survey Relieve pain or discomfort as much questions about participant 44 32 (42) 12 (48) as possible beliefs and preferences regarding chemotherapy, their If you had to make a choice now, would you prefer treatment that extends life as much as possible, even if it means using up all of your financial resources, or would you want treatroles in deciding whether to ment that costs you less, even if means not living as long? receive chemotherapy, and Extend life as much as possible 65 46 (61) 19 (73) .3 their communication with Treatment that costs less 37 30 (39) 7 (27) health care professionals about chemotherapy. Of the aThis analysis is limited to patients with stages 3 and 4 (ie, those for whom chemotherapy is recommended). For each item, differences between presented frequencies and total number of participants represent missing or noninformative 130 participants with stage responses (eg, “Do not know”). b 3/4 disease, 89 (68%) had P values were based on 2-sided Fisher exact tests and excluded missing and noninformative responses. an adequate level of health cFor the Fisher exact test, responses dichotomized as “Patient principally made the decision/patient and doctors made decision together” vs “Doctors principally made the decision.” literacy and 41 (32%) had inadequate/marginal levels of health literacy — percentages comparable with those with marginal/inadequate level of health literacy) and for the sample across all stages. We found no statistito help them with issues related to their cancers (82% cally significant differences in participant responses of those with an adequate level of health literacy and by level of health literacy. Participants of all levels of 100% of those with a marginal/inadequate level of health literacy thought that, after discussing chemohealth literacy). therapy with a health care professional, the treatment Although the differences were not statistically was likely to help them live longer (89% of those with significant, participants with stage 3/4 disease an adequate level of health literacy and 100% of those and an adequate level of health literacy played 98 Cancer Control January 2015, Vol. 22, No. 1 63% of patients with 9 to 11 years of education and 34% of patients who Variablea Univariate Multivariateb graduated from high school had a marginal/inadequate level of functionOR 95% CI OR 95% CI al health literacy.13 Furthermore, low Health literacy (inadequate/marginal 1.55 0.91–2.64 0.90 0.42–1.94 rates of adequate levels of functional vs adequate) health literacy are common, particularSex (male vs female) 1.93 1.15–3.24 2.16 1.09–4.28 ly among the elderly and those with Race (nonwhite vs white) 1.50 0.84–2.70 1.45 0.66–3.19 less formal education.4,10,14-17 Among Age (≥ 65 vs < 65 years) 1.46 0.87–2.45 1.59 0.82–3.08 elderly patients (> 60 years of age) Education (less than high school 1.63 0.90–2.97 1.46 0.70–3.03 at urban public hospitals, one study vs completed high school/GED) found that 81% of English speakers and Marital status (not living with anyone 1.55 0.92–2.59 1.40 0.68–2.88 83% of Spanish speakers had marginal/ vs married/living with a partner) inadequate levels of functional health Household income (< $40,000 1.46 0.82–2.59 1.23 0.57–2.67 literacy.17 Functional health literacy is vs ≥ $40,000) lower among older age groups even Cancer stage (3/4 vs 1/2) 2.62 1.52–4.52 2.25 1.21–4.20 after adjusting for differences in mental a For each variable (a vs b), a = index group and b = reference. status, frequency of reading the news, b Results from a model with all variables in the table simultaneously included as independent variables. health status, and visual acuity.18 The CI = confidence interval, OR = odds ratio. GED = general educational development. physical health of participants with lower reading levels has been found a more prominent role in deciding whether to to be poor compared with that of participants with receive chemotherapy than those with inadehigher reading levels even after adjusting for conquate/marginal levels of health literacy (see Table founding sociodemographic variables.2,19 Individuals 2). Among those with informative responses, 75 of with an inadequate level of health literacy are also 83 (90%) participants with an adequate level of health more likely to report depressive symptoms, explained literacy reported either making the decision to receive in part by their worse health status.2,20 chemotherapy themselves or together with their Health care professionals must be sensitive to health care professional (in constrast with the the level of functional health literacy of their patients health care professional alone making the deciwhen they provide information regarding treatment sion) compared with 20 of 25 (80%) among those options and prognoses. Analyses of the readability of with an inadequate/marginal level of health litpatient education materials, discharge instructions, eracy. Three of the 83 participants (4%) with an and consent forms have found that these materials adequate level of health literacy and informative are typically written at too complex a level for many responses reported that their health care profesor most patients.21,22 Some evidence suggests that taisional made the decision about chemotherapy loring communications for adults with low literacy with little or no patient input compared with 5 of can be effective.23 However, patients with a variety 25 (20%) among those with a marginal/inadequate of health literacy levels may have difficulty underlevel of health literacy and informative responses. standing health information; therefore, improving In the unadjusted regression of survival on health communication may help patients across all levels literacy, those with a marginal/inadequate level of of health literacy. health literacy had increased odds of being deceased To mitigate barriers to health literacy, health care at last observation compared with those with an adprofessionals should take steps when meeting with equate level of health literacy, but the effect was not patients to ensure that communication is clear and statistically significant and disappeared when conthat patients understand what is being taught to them. ditioned on cancer stage as well as demongraphic One recommended strategy involves the health care and socioeconomic covariates (Table 3). Male sex and professional asking the patient questions toward the stage 3/4 disease were associated with greater odds end of a clinical encounter to assess whether the of being deceased at last observation, but no other patient recalls and understands the information or notable effects were detected. instructions provided.24 For example, the health care professional might ask the patient about the name, Discussion dose, and frequency of a medication that was just Functional health literacy in patients with cancer may prescribed. This approach, which is often called the play a crucial role in successful treatment and out“teach-back” method, provides health care profescomes. Importantly, health literacy is distinct from sionals with an opportunity to confirm patient underformal education. For example, one study found that standing and gives patients the opportunity to solidify Table 3. — Factors Associated With Death at Last Observation (n = 347) January 2015, Vol. 22, No. 1 Cancer Control 99 their understanding. Previous studies of health literacy found that lower levels of literacy were correlated with being male,16 elderly,4,15-17 and having less formal education15,16 and income.16 The demongraphic and socioeconomic characteristics of our sample followed these patterns (see Table 1). In evaluating our hypotheses, we found that an adequate level of health literacy increased the likelihood of receiving chemotherapy among patients with stage 3/4 disease, a finding that suggests greater levels of health literacy might help patients receive better care. However, we detected no other clear differences by level of health literacy in patient beliefs, preferences, or decision-making about chemotherapy. We did not find an association between level of health literacy and either cancer stage at diagnosis or vital status at last observation. Limitations Our study had several limitations. First, our small sample limited our power to detect differences by health literacy status. Second, the S-TOFHLA might not precisely capture the desired construct of health literacy. Instead, it could be better regarded as a test of reading comprehension in a health care context rather than as a test of the broader concept of health literacy.25 Specifically, the S-TOFHLA might not evaluate aspects of health literacy other than reading, such as oral health literacy, navigation, and culture. The limitations of the instrument as a measure of health literacy could attenuate its association with some health outcomes. A third potential limitation that might be more general to health literacy research is the challenge of including sufficiently large numbers of participants with marginal/inadequate levels of literacy to detect the effects of health literacy levels. As noted, our goal was a sample of approximately 50% marginal/inadequate health literacy, but our actual sample had 30% marginal/inadequate health literacy. We targeted patients for inclusion in the health literacy substudy using formal education as a proxy, and our results reinforced the conclusion of prior research that formal education and health literacy, while related, are distinct.13 Several previous studies of health literacy using the S-TOFHLA (not all on cancer) obtained samples with even lower percentages of marginal/ inadequate levels of health literacy.15,16,26 The lower-than-expected numbers of participants with marginal/inadequate levels of proficiency in studies of health literacy suggest that selection bias might influence which patients enter these studies. To be eligible for inclusion in our sample, participants had to enroll in CanCORS, complete a baseline survey, and be administered the S-TOFHLA. Patients who died before completing any of these steps could 100 Cancer Control not have participated in the sample. For included patients, the mean number of days from cancer diagnosis to CanCORS enrollment and baseline survey was 150 days and from diagnosis to S-TOFHLA administration was 640 days. It is possible that, if the level of health literacy was associated with rates of survival, then patients with CRC who died before enrolling in CanCORS, or before they could complete the baseline survey or the S-TOFHLA, might have had disproportionately low levels of health literacy. Poor health and difficulty completing surveys among those with low levels of health literacy might systematically limit these patients’ participation in studies of health literacy. Future studies of health literacy should be designed to account for this possibility. Conclusions In the context of previous research, patients with CRC could benefit from health care professionals’ sensitivity toward, and adjustment to, different levels of patient health literacy. In addition, health care professionals could consider that any of their patients might have difficulty understanding and making decisions about health care. Therefore, clear communication is likely to help both lower and higher health literacy patients. Among patients with stage 3/4 disease, those with lower levels of health literacy were less likely to receive chemotherapy compared with patients with higher levels of health literacy. To provide high-quality, patient-centered care, health care professionals should consider strategies of clear communication and patient engagement, recognizing that health literacy might affect physician–patient interactions and choices in medical care. References 1. Ad Hoc Committee on Health Literacy for the Council on Scientific Affairs, American Medical Association. Health literacy: report of the Council on Scientific Affairs. JAMA. 1999;281(6):552-557. 2. Smith SG, Curtis LM, Wardle J, et al. Skill set or mind set? Associations between health literacy, patient activation and health. PLoS One. 2013;8(9):e74373. 3. Baker DW, Parker RM, Williams MV, et al. Health literacy and the risk of hospital admission. J Gen Intern Med. 1998;13(12):791-798. 4. Gazmararian JA, Baker DW, Williams MV, et al. Health literacy among Medicare enrollees in a managed care organization. JAMA. 1999;281(6):545-551. 5. Kalichman SC, Ramachandran B, Catz S. Adherence to combination antiretroviral therapies in HIV patients of low health literacy. J Gen Intern Med. 1999;14(5):267-273. 6. von Wagner C, Semmler C, Good A, et al. Health literacy and self-efficacy for participating in colorectal cancer screening: the role of information processing. Patient Educ Couns. 2009;75(3):352-357. 7. American Cancer Society. Cancer Facts & Figures 2014. Atlanta, GA: American Cancer Society; 2014. 8. Ayanian JZ, Chrischilles EA, Fletcher RH, et al. Understanding cancer treatment and outcomes: the Cancer Care Outcomes Research and Surveillance Consortium. J Clin Oncol. 2004;22(15):2992-2996. 9. Malin JL, Ko C, Ayanian JZ, et al. Understanding cancer patients’ experience and outcomes: development and pilot study of the Cancer Care Outcomes Research and Surveillance patient survey. Support Care Cancer. 2006;14(8):837-848. 10. Baker DW, Williams MV, Parker RM, et al. Development of a brief test to measure functional health literacy. Patient Educ Couns. 1999;38(1):33-42. 11. Nurss J, Parker RM, Williams MV, et al. Test of Functional Health January 2015, Vol. 22, No. 1 Literacy in Adults. Snow Camp, NC: Peppercorn Books & Press, 2001. 12. National Cancer Institute. Colon cancer treatment (PDQ). http://www. cancer.gov/cancertopics/pdq/treatment/colon/Patient/page5. Accessed October 13, 2014. 13. Baker DW. Reading between the lines: deciphering the connections between literacy and health. J Gen Intern Med. 1999;14(5):315-317. 14. Jolly BT, Scott JL, Feied CF, et al. Functional illiteracy among emergency department patients: a preliminary study. Ann Emerg Med. 1993;22(3):573-578. 15. Koay K, Schofield P, Gough K, et al. Suboptimal health literacy in patients with lung cancer or head and neck cancer. Support Care Cancer. 2013;21(8):2237-2245. 16. von Wagner C, Knight K, Steptoe A, et al. Functional health literacy and health-promoting behaviour in a national sample of British adults. J Epidemiol Community Health. 2007;61(12):1086-1090. 17. Williams MV, Parker RM, Baker DW, et al. Inadequate functional health literacy among patients at two public hospitals. JAMA. 1995;274(21):1677-1682. 18. Baker DW, Gazmararian JA, Sudano J, et al. The association between age and health literacy among elderly persons. J Gerontol B Psychol Sci Soc Sci. 2000;55(6):S368-S374. 19. Weiss BD, Hart G, McGee DL, et al. Health status of illiterate adults: relation between literacy and health status among persons with low literacy skills. J Am Board Fam Pract. 1992;5(3):257-264. 20. Gazmararian J, Baker D, Parker R, et al. A multivariate analysis of factors associated with depression: evaluating the role of health literacy as a potential contributor. Arch Intern Med. 2000;160(21):3307-3314. 21. Friedman DB, Hoffman-Goetz L. A systematic review of readability and comprehension instruments used for print and web-based cancer information. Health Educ Behav. 2006;33(3):352-373. 22. Lee PP. Why literacy matters. Links between reading ability and health. Arch Ophthalmol. 1999;117(1):100-103. 23. Howard-Pitney B, Winkleby MA, Albright CL, et al. The Stanford Nutrition Action Program: a dietary fat intervention for low-literacy adults. Am J Public Health. 1997;87(12):1971-1976. 24. Schillinger D, Piette J, Grumbach K, et al. Closing the loop: physician communication with diabetic patients who have low health literacy. Arch Intern Med. 2003;163(1):83-90. 25. DeWalt DA, Pignone MP. Reading is fundamental: the relationship between literacy and health. Arch Intern Med. 2005;165(17):1943-1944. 26. Rogers ES, Wallace LS, Weiss BD. Misperceptions of medical understanding in low-literacy patients: implications for cancer prevention. Cancer Control. 2006;13(3):225-229. January 2015, Vol. 22, No. 1 Cancer Control 101 Pathology Report Familial Gastrointestinal Stromal Tumor Syndrome: Report of 2 Cases With KIT Exon 11 Mutation Derek H. Jones, MD, Jamie T. Caracciolo, MD, Pamela J. Hodul, MD, Jonathan R. Strosberg, MD, Domenico Coppola, MD, and Marilyn M. Bui, MD, PhD Background: As with cases of sporadic gastrointestinal stromal tumor (GIST), familial GIST syndrome arises from mutations in KIT or PDGFRA. Only a few dozen such families have been described in the literature. Methods: Cases of 2 individuals from 2 different newly described kindreds with familial GIST syndrome were retrospectively reviewed. Pertinent immunohistochemical stains, including CD117, CD34, DOG1, desmin, and S100, were performed. Samples from each case were sent to outside facilities for molecular analysis. A review of the relevant literature was performed and the number of familial GIST syndrome cases reported was updated through July 2014. Results: In case 1, a woman 40 years of age with a family history of GIST presented with abdominal pain and gastrointestinal bleeding. Biopsy of a gastric mass revealed spindle-cell type GIST. Molecular analysis revealed a heterozygous mutation of p.Asp579del in exon 11 of KIT. The patient was placed on imatinib therapy and an initial positive response was demonstrated by imaging. Disease regression was seen on computed tomography, and several GIST tumors were surgically resected. The patient has had stable disease since surgery. In case 2, an asymptomatic woman 29 years of age presented for screening due to a family history of GIST. One small nodule was noted in her stomach and another was noted in the duodenum; both were surgically resected. The patient recovered well following surgery. The GIST in this patient was noted to have similar histological, immunohistochemical, and molecular findings as case 1. Conclusions: Imatinib has often been shown to be an effective therapy in both the familial and sporadic forms of GIST. There is no standard protocol for addressing the surveillance of patients with spindle-cell type GIST seen in the setting of familial GIST syndrome and with a p.Asp579del mutation of exon 11 on KIT. Introduction Although gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumor of the gastrointestinal tract, the incidence of familial GIST syndrome is rare, representing fewer than 5% of cases.1 Familial GIST syndrome is an autosomal dominant disease caused by germline mutations in KIT or PDGFRA, both of which are also involved in the initiation of most sporadic GIST cases. Other hereditable forms of GIST include deficiencies in the succinate dehydrogenase complex, Carney–Stratakis syndrome, and type 1 neurofibromatosis.1-3 From the University of South Florida Morsani College of Medicine (DHJ) and the Departments of Diagnostic Imaging and Interventional Radiology ( JTC), Gastrointestinal Oncology (PJH, JRS, DC), Anatomic Pathology (DC, MMB), and Sarcoma (MMB), H. Lee Moffitt Cancer Center & Research Institute, Tampa, Florida. Submitted August 17, 2014; accepted August 29, 2014. Address correspondence to Marilyn M. Bui, MD, PhD, Moffitt Cancer Center, 12902 Magnolia Drive, Tampa, FL 33612. E-mail: [email protected] No significant relationships exist between the authors and the companies/organizations whose products or services may be referenced in this article. 102 Cancer Control In 2013, Neuhann et al4 summarized 24 cases of familial GIST syndrome arising from KIT. In addition to this publication, 5 other cases of familial GIST syndrome have been described that involve KIT5-9 and 3 reported cases of familial GIST syndrome arising from PDGFRA.10-12 The 2 additional kindreds described in this report raise the total number of reported familial GIST syndrome cases to 34. Here we present clinical, gross, histological, immunological, and molecular findings of 2 cases of familial GIST involving the same mutation in exon 11 of KIT. We also provide an update of the literature on familial GIST syndrome since the work of Neuhann et al4 and review its molecular findings and clinical characteristics. Knowledge of this entity is important for the appropriate management of this condition among these patients. Materials and Methods Clinical, radiological, and pathological data from the 2 patients were retrospectively reviewed following the research guidelines of the University of South Florida and the Moffitt Cancer Center in Tampa, Florida. The tissues were processed, sectioned, January 2015, Vol. 22, No. 1 and stained according to the guidelines from the College of American Pathologists. The hematoxylin and eosin stain and immunohistochemical studies were performed at the histology laboratory of the Moffitt Cancer Center. The immunohistochemical staining was carried out using the Discovery XT System (Ventana Medical Systems, Tucson, Arizona) per the manufacturer’s protocol. Samples from each case were subjected to genetic analysis. Case 1 was performed with Sanger sequencing by ARUP Laboratories (Salt Lake City, Utah), and case 2 was performed with Sanger sequencing by Knight Diagnostic Laboratories (Portland, Oregon). Clinical and Radiological Information Case 1 A 40-year-old woman presented with epigastric pain and recurrent gastrointestinal bleeding. Her family history was significant for GIST in her aunt and grandmother. The patient history included anemia and a small, spontaneously healing gastrointestinal perforation. Laboratory values, including liver aminotransferases and bilirubin, were within normal limits. Contrast-enhanced abdominal computed tomography (CT) with intravenous contrast demonstrated numerous hypervascular, exophytic, mural-based, soft-tissue masses arising from the stomach, duodenum, and jejunum (Fig 1A). No evidence of hepatic or intraperitoneal metastatic disease was seen. Biopsies of an exophytic gastric mass and a peripancreatic duodenal mass were performed with the guidance of endoscopic ultrasonography. Both tumors were diagnosed as GIST. Molecular analysis revealed a mutation of exon 11 on KIT, and imatinib therapy was initiated. The patient underwent follow-up CT with oral and intravenous contrast 6 months following the initiation of imatinib therapy that demonstrated the decreased size of disease sites consistent with a partial response to therapy (Fig 1B). However, within 9 months of initiating imatinib, the patient presented with increased abdominal pain. Abdominal CT was repeated and demonstrated an increase in size of several lesions, including the duodenal mass (Fig 1C). Given the relatively limited extent of her multifocal disease and the absence of metastatic disease, surgical debulking was recommended. She underwent partial gastrectomy, pancreas-sparing duodenectomy, proximal jejunal resection, and cholecystectomy. The patient recovered well from surgery and reports that she is feeling better. CT was obtained 4 months following surgery, and the imaging demonstrated no evidence of recurrence or distant metastatic disease. Case 2 A woman 29 years of age who was asymptomatic presented for screening secondary to a family history of January 2015, Vol. 22, No. 1 familial GIST in her mother and grandmother. After an initial evaluation, the patient exhibited mild epigastric discomfort and subsequently presented with a mildly elevated level of alanine transaminase, possibly due to hepatic steatosis. The patient did not exhibit anemia and other laboratory values were not abnormal. Endoscopic ultrasonography performed with esophagogastroduodenoscopy had demonstrated subcentimeter, mural-based, hypoechoic, gastric, and duodenal soft-tissue nodules (Fig 2). These small, submucosal, intramural nodules were isodense and isoenhancing to the gastric and duodenal walls; therefore, they were not readily evident with CT imaging. Abdominopelvic CT did not demonstrate evidence of hepatic metastatic disease or peritoneal carcinomatosis. Both lesions A B C Fig 1A–C. — Serial CT scans at a similar level over time from case 1. (A) Axial contrast-enhanced abdominal CT demonstrates a 3.9-cm exophytic, mural-based, hypervascular duodenal mass (white arrow) and a similarly appearing jejunal mass (yellow arrow). The red asterisk denotes the duodenum. (B) Six months after CT shown in Fig 1A, both masses demonstrated decreased size on imatinib therapy consistent with tumor response. The duodenal mass measured 2.9 cm. No new lesions or interval development of metastatic disease was seen. (C) Six months after CT shown in Fig 1B, the duodenal mass increased in size and measured 3.3 cm, which was consistent with resistance to imatinib therapy. CT = computed tomography. Cancer Control 103 Fig 2. — Endoscopic ultrasonography from case 2 demonstrates a submucosal, intramural, hypoechoic, soft-tissue nodule of 6 × 5 mm in size within the posterior wall of the second portion of the duodenum. CBD = common bile duct, GIST, gastrointestinal stromal tumor, HOP = head of pancreas, POSTR = posterior. Fig 3. — Gross findings for case 1. Duodenal and proximal jejunal resection with an expanded cross-section of tumor demonstrates 6 nodules with diameters up to 4.0 cm on the serosal aspect of the resection. Nodules are pink-tan to tan-white in appearance. were subsequently surgically resected. The patient is not on imatinib therapy and was recovering well 1 month following surgery. observed; some extended to the serosa. Histologically, the resected tumor specimens were of similar morphology as the gastric and duodenal biopsies (Fig 4A–C). The mitotic rate was 3 per 5 mm2. Minimal tumor necrosis was observed (approximately 5% of total tumor volume), corresponding to a minimal treatment effect. The viability rate of the tumor cells observed was 90% to 95%. Tumors were diagnosed as spindle-cell type GIST and staged as T2M0N0. Gross, Histological, and Immunohistochemical Findings Case 1 Histologically, cells from both the gastric and duodenal mass biopsies were of spindle-cell morphology and largely uniform in appearance. The cells had cigar-shaped nuclei and pale eosinophilic cytoplasm. Skeinoid fibers were present in the interstitium. Necrosis was not identified. Mitotic rate could not be accurately determined due to the small sample size. The tumor was immunohistochemically positive for CD117 and DOG1, while negative for CD34, desmin, actin, S-100, and pan-keratin (AE1/AE3/CAM 5.2). Findings were consistent with the diagnosis of spindle-cell type GIST. The surgical resection specimens consisted of a 3.0 × 1.3 × 0.8-cm section of the lesser gastric curvature, a 2.8 × 1.0 × 0.5-cm section of the posterior stomach wall, a 5.4 × 2.4 × 1.8-cm wedge of an anterior gastric wall body mass, a 3.8 × 1.0 × 0.8-cm wedge of a posterior gastric wall mass, and a section of duodenum and proximal jejunum approximately 44.0 cm in length with attached adipose tissue yellow-tan in color on the serosal aspect (Fig 3). In the gastric sections, various nodules up to 1.0 cm extending to the serosa that ranged from tan-pink, tan-white, to white in color were observed. In the duodenal and proximal jejunal resections, approximately 6 tan-white to pinktan nodules ranging from 0.6 to 4.0 cm in size were 104 Cancer Control Case 2 Macroscopically, a 1.5 × 0.6 × 0.4-cm section of the anterior gastric mucosa was resected. It contained a circumscribed nodule that was tan-white in color and measured 1.1 × 0.9 × 0.6 cm in size. In addition, a 0.9 × 0.6 × 0.5-cm section of duodenum containing a small nodule was resected. It consisted of cauterized fibrotic tissue brown-tan in color. Histologically, findings were also consistent with spindle-cell type GIST. The mitotic rate was 0 per 5 mm2. The tumor was immunohistochemically positive for CD117 (Fig 5A), strongly positive for DOG1 (Fig 5B), and negative for S100, actin, and desmin. The Ki-67 labeling index was between 1% and 2%. Cell necrosis was not identified. Molecular Analysis Case 1 The core biopsy of the gastric mass guided by endoscopic ultrasonography was found to have a mutation on exon 11 of KIT. A deletion of GAT was present in positions 1735 to 1737 (c.1735_1737delGAT), which January 2015, Vol. 22, No. 1 A A B B C Fig 4A–C. — Nodules from jejunal and duodenal resection of case 1. (A) Histology from a 4.0-cm nodule. Longitudinal positioning of the cells is observed. H & E, × 60. (B) Histology from a 0.6-cm nodule. Cigar-shaped nuclei with a uniform appearance and distinct, spindle-cell morphology. H & E, × 20. (C) High power image of the nodule from Fig 4B. Skeinoid fibers are visible in the interstitium. H & E, × 60. H & E = hematoxylin and eosin. resulted in the loss of aspartic acid at position 579 (p.Asp579del). The mutation was heterozygous. Exon 9 from KIT was intact. Case 2 Molecular studies were performed from a peripheral blood sample. Similar to the results from case 1, a heterozygous p.Asp579del mutation was detected in exon 11 of KIT. Discussion Mutations in familial GIST syndrome involve KIT and PDGFRA, which are the same genes mutated in 80% January 2015, Vol. 22, No. 1 Fig 5A–B. Histological and immunohistochemical images from stomach tumor in case 2. (A) CD117 was diffusely positive in the cell cytoplasm. CD117, × 40. (B) DOG1 was strongly positive in the cell membranes and cytoplasm. DOG1, × 40. to 88% of cases of sporadic GIST.13 Wild-type GIST arising from other molecular pathways may comprise up 15% of GIST cases,13 and they are typically negative for KIT and PDGFRA mutations but positive for mutations of BRAF V600E, the RAS family, and the succinate dehydrogenase complex.1 However, it is possible that cases of wild-type GIST might be less common than previously thought. For example, one study suggests that 1% to 2% of GIST previously regarded as the wild-type form may actually harbor mutations in exon 8 of KIT.14 The Table demonstrates similarities and differences between the molecular basis of familial and sporadic GIST caused by KIT mutations.15,16 In familial GIST syndrome, KIT mutations in exon 9 have not been reported as they have with sporadic GIST. The reasons for a lack of exon 9 mutations in familial GIST syndrome remain unclear, but reasons may become elucidated as more information is gained on familial GIST. Six cases have been reported of familial GIST syndrome due to mutations in KIT exon 13 and 4 cases have been reported due to mutations in exon 17.4,6,8,9 By comparison, exon 9 mutations account for 15% of KIT mutations in the setting of sporadic GIST, whereas exons 13 and 17 account for 2% and 1%, respectively.15 Although pure insertions involving KIT are documented in sporadic GIST, none have been documented in Cancer Control 105 Table. — KIT Findings for GIST Genetic Finding Sporadic GIST Familial GIST Syndrome Involved KIT exons 8, 9, 11, 13, 17 8, 11, 13, 17 Type of mutation Substitution Duplication Deletion Insertion Deletion–insertion Substitution Duplication Deletion Deletion–insertion KIT mutations involving exon 11, % 70–9015,26 65 GIST = gastrointestinal stromal tumor. familial GIST syndrome (see Table).15,16 Our literature review demonstrates that, as a whole, mutations of exon 11 of KIT are more common among sporadic GIST than familial GIST syndrome. However, given the small number of familial GIST syndrome cases to compare with the much larger number of sporadic GIST cases, these differences must be thoroughly examined. In the future, reports of additional cases of familial GIST will help achieve a better analysis of these underlying similarities and differences. Twenty families with familial GIST have arisen from exon 11 of KIT as described by Neuhann et al,4 Nakai et al,5 Adela et al,7 and the cases presented here. KIT and PDGFRA are members of the family of class 3 tyrosine kinase receptors. The juxtamembrane domain of this family is highly conserved and has been demonstrated to have an inhibitory effect on the kinase domain of the tyrosine kinase receptor.17 The juxtamembrane domain of KIT is encoded by exon 11.13 A mutation in this region has been molecularly modeled to disrupt the usual autoinhibitory state of KIT, leading an activated gain-of-function state.18 Because of this gain-of-function type mutation, individuals are typically heterozygous for these mutations. The current report describes 2 additional cases of a mutation of p.Asp579del in exon 11 of KIT, adding 2 kindreds to the 3 previously reported families with familial GIST syndrome demonstrating this mutation.18-20 Mutations causing familial GIST syndrome have no clear differentiating factors from mutations causing sporadic GIST, and the mutation of p.Asp579del has also been documented in sporadic GIST.21 The p.Asp579del mutation is not within the region of high-frequency mutations noted at positions 556 to 560 in exon 11 of KIT in patients with GIST.18 However, 11 of the 20 cases of familial GIST syndrome with a mutation in exon 11 of KIT are located in this high-frequency region. Of these 11 cases, 7 are from the most common mutation in familial GIST syndrome, p.Val559Ala. The KIT exon 11 mutation p.Val559Ala is also a common missense mutation in GIST.22 A large study found that patients with deletions in exon 11 of KIT from positions 562 to 579 have significantly higher risk for meta106 Cancer Control static disease than patients with mutations at positions 550 and 561, suggesting that patients with the mutation of p.Asp579del, such as the patients in this report, may be at high risk for metastatic disease.23 These 2 cases also contribute to the body of evidence suggesting that phenotypic features cannot reliably indicate the presence of familial GIST syndrome. As with sporadic GIST, nonspecific signs and symptoms in familial GIST syndrome include gastrointestinal bleeding, abdominal pain, ulcer-type symptoms, and a variety of other gastrointestinal complaints. No specific serum markers are currently used to routinely screen or diagnose GIST. Familial GIST syndrome has classically been associated with hyperpigmentation, urticaria pigmentosa, and dysphagia. However, a growing number of cases do not demonstrate these symptoms, including our 2 patients. Notably, patient age can be a guiding feature as patients with familial GIST syndrome typically present at least 10 years prior to patients with sporadic GIST, presenting at a median age of 60 to 65 years.1,2 Patients with familial GIST syndrome or other GIST-related syndromes, such as type 1 neurofibromatosis, typically present with multifocal disease, whereas most patients with sporadic GIST usually present with solitary primary tumors.24 Sporadic GIST may present with multiple gastrointestinal masses in the setting of metastatic disease or from independent mutations. Metastatic disease may be diagnosed in the setting of peritoneal sarcomatosis, the second most common pattern of metastatic disease following hepatic metastatic disease. Approximately 11% of patients presenting with GIST have metastatic disease identified at the time of initial diagnosis.25 Several studies indicate that a substantial portion of patients with multifocal disease may arise from independent mutational events. Two large studies, one by Agaimy et al26 and the other by Gasparotto et al,27 indicate the presence of multiple molecular origins in multifocal GIST in 7 of 11 and 6 of 10 cases, respectively. The presence of multiple mutations could be explained by premutational epigenetic changes.26 Current guidelines from the National Comprehensive Cancer Network recommend mutational testing for KIT and PDGFRA in the primary evaluation of GIST, but they make no recommendations for further analysis in the case of multifocal disease.28 Due to the variety of possible etiologies in patients presenting with multifocal disease, genetic screening for familial GIST syndrome may be a prudent step in the initial evaluation of multifocal disease. In addition, genetic counseling for individuals with familial GIST syndrome should be considered to identify other family members at risk. In general, histological and immunohistochemical features do not differentiate cases of familial GIST syndrome from cases of sporadic GIST. Both feature January 2015, Vol. 22, No. 1 spindle cell or, less frequently, epithelioid histologies. Both are usually positive for CD117 and DOG1 and negative for desmin, keratin, and S100. Similar to sporadic GIST, CD34 variability has also been noted in familial GIST syndrome. A high rate of response to imatinib has been well documented in cases of sporadic GIST, and familial GIST may also be sensitive to imatinib treatment. Typically, patients with mutations of exon 11 of KIT have an especially strong response to imatinib.28 Tarn et al18 reported that the mutations of exon 11 on KIT may not affect the nucleotide-binding site, thereby lowering the probability of imatinib resistance. However, recent data demonstrate that changes to the juxtamembrane domain can affect the structure of the kinase domain, thus resulting in imatinib resistance, such as the case with p.Val559Ile.29 Kleinbaum et al20 noted a strong response to imatinib in patients with familial GIST syndrome who had a KIT exon 11 p.Asp579del mutation. Nine of the 11 family members who did not receive imatinib eventually died from metastatic GIST, whereas all 4 patients receiving imatinib achieved stable disease for more than 4 years.20 Typically, sunitinib is a second-line therapy following treatment failure with imatinib. Currently, several reports have focused on the general topic of familial GIST syndrome. Burgoyne et al1 provided a review of nonsporadic GIST, and Corless et al13 provided a comprehensive review of the molecular basis of GIST. No guidelines currently exist for screening patients with familial GIST, nor do any clear indications exist for the role or extent of surgery in the setting of multifocal disease. The patient in case 1 had symptomatic multifocal tumors and achieved palliation with neoadjuvant imatinib followed by surgical resection, whereas the patient in case 2 was asymptomatic and chose to undergo surgery for her identifiable sites of disease. It is unclear whether the removal of small, low-grade tumors will ultimately improve her long-term prognosis, particularly because additional tumors are likely to develop over her lifetime. The role of imatinib as a chemopreventive agent in familial GIST is also unclear. Although it is possible that imatinib can prevent or delay growth of multifocal GIST in this population, it is uncertain to what extent imatinib therapy improves long-term prognoses among patients with nonmetastatic, low-grade tumors. Conclusions Familial gastrointestinal stromal tumor syndrome is a rare disease, but the number of associated families identified continues to expand. The 2 cases identified in this report add to the growing body of evidence that will better help researchers and clinicians understand the epidemiology, characteristics, and optimal treatment of January 2015, Vol. 22, No. 1 familial gastrointestinal stromal tumor syndrome. References 1. Burgoyne AM, Somaiah N, Sicklick JK. Gastrointestinal stromal tumors in the setting of multiple tumor syndromes. Curr Opin Oncol. 2014;26(4):408-414. 2. Fletcher BJ, Hogendoorn PCW, Mertens F. WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyon: IARC Press; 2013: 164-167. 3. Postow MA, Robson ME. Inherited gastrointestinal stromal tumor syndromes: mutations, clinical features, and therapeutic implications. Clin Sarcoma Res. 2012;2(1):16. 4. Neuhann TM, Mansmann V, Merkelbach-Bruse S, et al. A novel germline KIT mutation (p.L576P) in a family presenting with juvenile onset of multiple gastrointestinal stromal tumors, skin hyperpigmentations, and esophageal stenosis. Am J Surg Pathol. 2013;37(6):898-905. 5. Nakai M, Hashikura Y, Ohkouchi M, et al. Characterization of novel germline c-kit gene mutation, KIT-Tyr553Cys, observed in a family with multiple gastrointestinal stromal tumors. Lab Invest. 2012;92(3):451-457. 6. Peña-Irún A, Villa-Puente M, García-Espinosa R, et al. Familial gastrointestinal stroma tumor due to mutation in exon 13 (K642E) of the KIT gene [In Spanish]. Med Clin (Barc). 2012;139(11):512-513. 7. Adela Avila S, Peñaloza J, González F, et al. Dysphagia, melanosis, gastrointestinal stromal tumors and a germinal mutation of the KIT gene in an Argentine family. Acta Gastroenterol Latinoam. 2014;44(1):9-15. 8. Yamanoi K, Higuchi K, Kishimoto H, et al. Multiple gastrointestinal stromal tumors with novel germline c-kit gene mutation, K642T, at exon 13. Hum Pathol. 2014;45(4):884-888. 9. Wadt K, Andersen MK, Hansen TV, et al. A new genetic diagnosis of familiar gastrointestinal stromal tumour [In Danish]. Ugeskr Laeger. 2012;174(21):1462-1464. 10. Chompret A, Kannengiesser C, Barrois M, et al. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology. 2004;126(1):318-321. 11. de Raedt T, Cools J, Debiec-Rychter M, et al. Intestinal neurofibromatosis is a subtype of familial GIST and results from a dominant activating mutation in PDGFRA. Gastroenterology. 2006;131(6):1907-1912. 12. Pasini B, Matyakhina L, Bei T, et al. Multiple gastrointestinal stromal and other tumors caused by platelet-derived growth factor receptor alpha gene mutations: a case associated with a germline V561D defect. J Clin Endocrinol Metab. 2007;92(9):3728-3732. 13. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878. 14. Huss S, Künstlinger H, Wardelmann E, et al. A subset of gastrointestinal stromal tumors previously regarded as wild-type tumors carries somatic activating mutations in KIT exon 8 (p.D419del). Mod Pathol. 2013;26(7):1004-1012. 15. Gounder MM, Maki RG. Molecular basis for primary and secondary tyrosine kinase inhibitor resistance in gastrointestinal stromal tumor. Cancer Chemother Pharmacol. 2011;67(suppl 1):S25-S43. 16. Miettinen M, Lasota J. Gastrointestinal stromal tumors. Gastroenterol Clin North Am. 2013;42(2):399-415. 17. Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5(6):464-471. 18. Tarn C, Merkel E, Canutescu AA, et al. Analysis of KIT mutations in sporadic and familial gastrointestinal stromal tumors: therapeutic implications through protein modeling. Clin Cancer Res. 2005;11(10):3668-3677. 19. Lasota J, Miettinen M. A new familial GIST identified. Am J Surg Pathol. 2006;30(10):1342. 20. Kleinbaum EP, Lazar AJ, Tamborini E, et al. Clinical, histopathologic, molecular and therapeutic findings in a large kindred with gastrointestinal stromal tumor. Int J Cancer. 2008;122(3):711-718. 21. Nakahara M, Isozaki K, Hirota S, et al. A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology. 1998;115(5):1090-1095. 22. Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology. 2008;53(3):245-266. 23. Emile JF, Théou N, Tabone S, et al. Clinicopathologic, phenotypic, and genotypic characteristics of gastrointestinal mesenchymal tumors. Clin Gastroenterol Hepatol. 2004;2(7):597-605. 24. Haller F, Schulten HJ, Armbrust T, et al. Multicentric sporadic gastrointestinal stromal tumors (GISTs) of the stomach with distinct clonal origin: differential diagnosis to familial and syndromal GIST variants and peritoneal metastasis. Am J Surg Pathol. 2007;31(6):933-937. 25. Nilsson B, Bumming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era--a population-based study in western Sweden. Cancer. 2005;103(4):821-829. 26. Agaimy A, Dirnhofer S, Wünsch PH, et al. Multiple sporadic gastrointestinal stromal tumors (GISTs) of the proximal stomach are caused by different somatic KIT mutations suggesting a field effect. Am J Surg Pathol. 2008;32(10):1553-1559. 27. Gasparotto D, Rossi S, Bearzi I, et al. Multiple primary sporadic gastrointestinal stromal tumors in the adult: an underestimated entity. Clin Cancer Cancer Control 107 Res. 2008;14(18):5715-5721. 28. von Mehren M, Randall RL, Benjamin RS, et al. Gastrointestinal stromal tumors, version 2.2014. J Natl Compr Canc Netw. 2014;12(6):853-862. 29. Nakagomi N, Hirota S. Juxtamembrane-type c-kit gene mutation found in aggressive systemic mastocytosis induces imatinib-resistant constitutive KIT activation. Lab Invest. 2007;87(4):365-371. 108 Cancer Control January 2015, Vol. 22, No. 1 Tumor Biology Current and Emerging Therapies for Bone Metastatic Castration-Resistant Prostate Cancer Jeremy S. Frieling, David Basanta, PhD, and Conor C. Lynch, PhD Background: A paucity of therapeutic options is available to treat men with metastatic castration-resistant prostate cancer (mCRPC). However, recent developments in our understanding of the disease have resulted in several new therapies that show promise in improving overall survival rates in this patient population. Methods: Agents approved for use in the United States and those undergoing clinical trials for the treatment of mCRPC are reviewed. Recent contributions to the understanding of prostate biology and bone metastasis are discussed as well as how the underlying mechanisms may represent opportunities for therapeutic intervention. New challenges to delivering effective mCRPC treatment will also be examined. Results: New and emerging treatments that target androgen synthesis and utilization or the microenvironment may improve overall survival rates for men diagnosed with mCRPC. Determining how factors derived from the primary tumor can promote the development of premetastatic niches and how prostate cancer cells parasitize niches in the bone microenvironment, thus remaining dormant and protected from systemic therapy, could yield new therapies to treat mCRPC. Challenges such as intratumoral heterogeneity and patient selection can potentially be circumvented via computational biology approaches. Conclusions: The emergence of novel treatments for mCRPC, combined with improved patient stratification and optimized therapy sequencing, suggests that significant gains may be made in terms of overall survival rates for men diagnosed with this form of cancer. Introduction Prostate cancer is the second most common cancer in American men with approximately 233,000 newly diagnosed cases in 2014.1 With an aging population, the incidence of prostate cancer is likely to continue to increase. Patients whose disease is detected at an early stage benefit from a range of treatment strategies, including radiotherapy and prostatectomy, with survival rates near 100%.2 However, the clinical reality is that many men present with advanced stages of the disease. Currently, the main treatment option for men with advanced cancer is hormone therapy. Historic contributions from Huggins and Hodges3 in 1941 revealed that removing androgens could inhibit the progression of prostate cancer. These earFrom the Departments of Tumor Biology (JSF, CCL) and Integrated Mathematical Oncology (DB), H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida. Submitted August 6, 2014; accepted September 11, 2014. Address correspondence to Conor C. Lynch, PhD, Department of Tumor Biology, Moffitt Cancer Center, 12902 Magnolia Drive, SRB-3, Tampa, FL 33612. E-mail: [email protected] No significant relationships exist between the authors and the companies/organizations whose products or services may be referenced in this article. This work was supported in part by funding support that Dr Lynch received from the National Cancer Institute (RO1CA143094). January 2015, Vol. 22, No. 1 ly observations paved the way for the development of androgen-deprivation therapy — either surgically or chemically — which has remained the standard treatment for men with advanced disease for the last 70 years. Despite the initial response to androgen deprivation for most men, the disease typically progresses to a castration-resistant state within 18 to 24 months.4 Castration-resistant prostate cancer (CRPC) is defined by disease progression that, despite chemical castration, is often indicated by rising levels of prostate-specific antigen (PSA).5 The development of resistance to hormonal intervention and why the disease progresses is not fully understood, although some mechanisms have been demonstrated, with the majority focusing on the continued androgen receptor (AR) activity in addition to TMPRSS2/ERG fusion, PTEN, Nkx3.1, and EGR1. As the disease progresses, the CRPC ultimately metastasizes (mCRPC). Patients with mCRPC have a poor prognosis and a predicted survival rate of fewer than 2 years from the initial time of progression, comprising a large portion of the 30,000 prostate cancer-related deaths per year.6,7 Currently, mCRPC is an incurable disease and represents a major clinical hurdle. Prostate cancer preferentially metastasizes to bone.8 As the disease transitions from castration senCancer Control 109 Fig 1. — Approved and developing mCRPC therapies and their targets. mCRPC has experienced a rapid expansion of treatment options over the last decade. Better understanding of mechanisms of progression has allowed for the improvement of broad-acting options such as chemotherapy and hormonal therapy as well as the development of novel targeted therapies to modulate the immune system and microenvironment. mCRPC = metastatic castration-resistant prostate cancer. sitive to castration resistant, the incidence of bone metastasis increases, with more than 90% of patients with mCRPC developing bone metastases.9,10 Patients with mCRPC who are symptomatic are at a high risk for skeletal-related events (SREs), including spontaneous fracture and spinal cord compression, that are a source of significant pain and decreased quality of life.11 Pain from the metastases is a major component of the disease and is an important aspect to be considered regarding a patient’s treatment regimen. Depending on the level of pain, medications ranging from ibuprofen to morphine are prescribed.12 Because prostate to bone metastases are primarily bone-forming sclerotic lesions, bone scanning using technetium-99m is often preferred for diagnosis due to the incorporation of the radionuclide tracer into regions of new bone formation by osteoblasts.13 Magnetic resonance imaging (MRI) and positron emission tomography (PET)/computed tomography 110 Cancer Control (CT) are also used for detection. A trial comparing 18F–sodium fluoride PET/CT, 18F-fluorodeoxyglucose PET/CT, MRI, and technetium-99m identified strengths for each modality.14 However, the ability to detect occult or micrometastases less than 5 mm remains a current limitation for each imaging technique. Approved Therapeutic Options Currently, mCRPC remains incurable, and many treatment options are palliative in nature. However, the treatment landscape of mCRPC is expanding both in broad-spectrum and targeted therapies that are likely to positively impact overall survival rates within the next decade. This expansion began with docetaxel, which, in 2004, was the first therapy to provide improved survival rates to patients with mCRPC. However, many patients develop resistance.15 To combat this issue, 5 new agents have received approval by the US Food and Drug Administration (FDA) to treat January 2015, Vol. 22, No. 1 mCRPC since 2010 (abiraterone acetate, enzalutamide, cabazitaxel, radium-223, and sipuleucel-T).16 Some of these agents may be administered in combination with steroids, such as prednisone, which has been shown to decrease testosterone levels and reduce tumor growth as well as counteract adverse events (eg, nausea, allergic reactions, inflammation, pain).17,18 Recently FDA-approved agents that target the cancer and host compartments are discussed below and are also illustrated in Fig 1. Targeting Metastatic Castration-Resistant Prostate Cancer Cells One of the defining measures of mCRPC is resistance to androgen deprivation. The mechanism of castration resistance is not fully understood but inroads have been made. For example, prostate cancer cells circumvent castration by overexpressing and increasing the sensitivity of the AR to residual androgens, acquiring AR gene mutations that lead to functional gain or promiscuous ligand interactions, splice variants resulting in constitutive AR activation, and post-translational modifications affecting the stability, localization, and activity of the receptor.19 Alternative methods utilized by prostate cancer cells to synthesize dihydrotestosterone (DHT) have also been shown to circumvent androgen deprivation methods.20-22 Efforts to target DHT synthesis have resulted in FDA-approved androgen deprivation therapy (ADT) options. Abiraterone acetate is one such option that works by inhibiting the activity of the CYP17A1 enzyme, thereby preventing androgen synthesis. Abiraterone has improved the overall survival and radiographic progression-free survival rates of men with mCRPC.23,24 Another therapeutic strategy for preventing androgen utilization by mCRPC cells is to directly target the AR with reagents such as flutamide, nilutamide, and bicalutamide. Enzalutamide was recently approved for the treatment of mCRPC in a postdocetaxel setting without the administration of corticosteroids.25,26 Enzalutamide has a superior affinity to the AR compared with other AR antagonists and works by preventing nuclear translocation of the receptor, DNA binding, and recruitment of coactivators of the AR to increase overall survival rates and delay the onset of SREs.27-29 Results of a phase 3 trial demonstrated enzalutamide activity in patients naive to chemotherapy, and FDA approval of enzalutamide as a first-line therapeutic option for mCRPC may be on the horizon.30 A list of approved therapies for the treatment of mCRPC appears in Table 1.15,23,24,27,28,31-36 In addition to ADT strategies, taxane-derived chemotherapies are commonly used to treat mCRPC. Docetaxel was the first therapy to demonstrate a beneficial effect on overall survival rates accompanied by improved quality of life for men with mCRPC, January 2015, Vol. 22, No. 1 Table 1. — Approved Therapies for the Treatment of Metastatic Castration-Resistant Prostate Cancer Drug Target Effect CYP17A1 Reduces circulating testosterone levels23,24 Microtubules Microtubule stabilization, interrupts cell cycle31 RANKL Decreases bone resorption34 Microtubules Microtubule stabilization, interrupts cell cycle15,36 AR AR antagonism, prevents signaling27,28 Radium-223 Bone Localized radiation35 Sipuleucel-T Ex vivo activation of PBMCs via GM-CSF and PAP T-cell activation32 Zoledronic acid Osteoclasts Decreases bone resorption33 Abiraterone acetate Cabazitaxel Denosumab Docetaxel Enzalutamide AR = androgen receptor, GM-CSF = granulocyte-macrophage colony-stimulating factor, PAP = prostatic acid phosphatase, PBMC = peripheral blood mononucleated cell, RANKL = receptor activator of nuclear κB ligand. and it has since become the standard therapy for mCRPC.15,36 Cabazitaxel is a more recent derivative of the taxoids that has shown increases in overall survival rates, improvements in progression-free survival rates, and improved PSA response rates in men with mCRPC.31,37 Cabazitaxel-associated toxicities were minor, leading to the FDA approval of the therapy for the treatment of patients with mCRPC after treatment with docetaxel.38 Targeting the Microenvironment Given the heterogeneity of mCRPCs and the likelihood of ADT/chemotherapy resistance, targeting the genetically stable host microenvironment supporting the mCRPC represents an attractive treatment approach. Immune evasion is a hallmark of cancer progression, and the goal of sipuleucel-T is to make mCRPC more visible to cytotoxic T cells.32,39 Sipuleucel-T is an autologous immunotherapy approved for the treatment of asymptomatic or minimally symptomatic mCRPC.40 Sipuleucel-T harnesses the properties of the patient’s immune system by collecting peripheral blood mononuclear cells and activating them ex vivo by exposing them to a fusion protein consisting of prostatic acid phosphatase (PAP; commonly expressed by prostate cancer cells) and granulocyte-macrophage colony-stimulating factor. Patients receive 3 separate infusions of the activated cells at 2-week intervals to generate PAP-expressing dendritic cells that activate T cells to recognize and eliminate PAP-expressing prostate cancer cells.32 Most mCRPCs arise in the bone matrix where they induce extensive bone remodeling by stimulating osteoblasts and osteoclasts. The process promotes the growth of the mCRPCs via the solubilization of bone matrix–sequestered growth factors, causing Cancer Control 111 Fig 2A–C. — Dormancy and the “vicious cycle” in bone marrow niches. (A) Disseminated tumor cells can home to the vascular niche and cluster on stable endothelium. Decreased expression of thrombospondin 1 combined with activation of transforming growth factor β and periostin in areas of “sprouting” vasculature can result in the outgrowth of tumor cells. (B) Cancer cells may also home to the endosteal niche via mechanisms such as chemokine motif 12/ chemokine receptor 4 where they compete with quiescent hematopoietic stem cells for osteoblast interaction. Subsequently, the cancer cells can be maintained in a dormant state via interactions with GAS6- and ANXA2-expressing niche osteoblasts or proliferate into metastases. (C) A “vicious cycle” occurs between tumor cells and other cells of the bone microenvironment. Factors secreted by the tumor cells act on osteoblasts, leading to the increased production of RANKL. RANKL subsequently promotes the differentiation of osteoclast precursors into mature, bone-resorbing osteoclasts that degrade the bone and release additional factors into the microenvironment, providing positive feedback to the cancer cells. Matrix metalloproteinases 2, 7, and 9 contribute to the vicious cycle by regulating factors such as vascular endothelial growth factor A, RANKL, and transforming growth factor β, whereas myeloid-derived suppressor cells contribute by releasing protumorigenic factors, suppressing T cells, and differentiating into osteoclasts. RANKL = receptor activator of nuclear κB ligand. pain and SREs (eg, pathological fractures). Therefore, preventing the interaction of cancer and bone has been a major focus of treatment for several decades. Bisphosphonates, such as zoledronic acid, are reagents that can “stick” to bones undergoing remodeling; upon resorption by osteoclasts, they can induce apoptosis and limit the amount of cancer-induced bone disease.41 In the clinical setting, zoledronic acid has demonstrated a benefit for patients with mCRPC by delaying the time to SRE incidence.33 However, no increase in overall survival rates has been demonstrated. Receptor activator of nuclear κB ligand (RANKL) is a molecule critical for the maturation and activation of bone-resorbing osteoclasts. Denosumab is a fully humanized monoclonal antibody that prevents RANKL interaction with the RANK receptor.42 For patients with bone mCRPC, 112 Cancer Control a significant delay has been demonstrated in the time to first SRE compared with zoledronic acid.34 Evidence suggests that denosumab may have direct effects on tumor burden, particularly tumor cells expressing RANK.43,44 Furthermore, preclinical in vivo animal studies have highlighted the efficacy of docetaxel/ denosumab treatment in increasing median survival rates, suggesting that combination approaches with denosumab could enhance the overall survival rates of men with mCRPC.45 At the time of publication, the most recent agent to receive FDA approval for mCRPC is radium-223.46 The bone-seeking properties of radium-223 (and other similar radiopharmaceuticals) make it useful for the treatment of bone metastases. Although most radiopharmaceuticals emit ß particles, radium-223 emits α particles to deliver more localized radiation January 2015, Vol. 22, No. 1 (< 100 µm distance) to induce cell death via DNA damage.47 In a study of men with mCRPC previously treated with radiotherapy, radium-223 showed improved rates of overall survival, time to PSA progression, and reduced alkaline phosphatase levels (a measure of bone remodeling).35 In addition, radium-223 delays the time to first SRE.35 Previous radiopharmaceuticals used to treat mCRPC were effective at reducing pain alone. Therefore, radium-223 represents an important step forward for the field.46 stable disease in 51 patients.59 Decreases in circulating tumor cells were also observed, thus serving as a further indication of efficacy. Based on these positive data, phase 3 trials were initiated; however, the results of one of those phase 3 trials indicated that orteronel administered in combination with prednisone failed to significantly impact overall survival rates compared with placebo but did provide a benefit in radiographic progression free survival rates in both chemotherapy naive and postchemotherapy mCRPC.48,49 Emerging Therapeutic Options Targeting the Microenvironment Tasquinimod: In addition to the approval of some small molecule inhibitors, several novel inhibitors are, at the time of publication, in various phases of clinical trials for mCRPC. Tasquinimod, a quinoline-3-carboxamide derivative, is being investigated in men with mCRPC (NCT01234311, NCT00560482). Tasquinimod provides an antiangiogenic effect by upregulating thrombospondin-1 (TSP-1) and downregulating the gene expression of vascular endothelial growth factor (VEGF), the C-X-C chemokine receptor (CXCR) 4, and lysyl oxidase.60 It has also been shown to reduce the expression levels of C-X-C chemokine motif (CXCL) 12 and inhibit S100A9, both of which are important molecules implicated in tumorigenesis and angiogeneOrteronel sis.50,60-63 The results of a phase 2 trial in patients naive Similar to abiraterone acetate, orteronel inhibits to chemotherapy showed improved rates of median CYP17A1 to reduce circulating levels of testosterprogression-free survival (7.6 months vs 3.3 months).57 one. However, orteronel possesses specificity toward In addition, the study showed bone alkaline phoslyase activity, leaving the synthesis of adrenal cortiphatase levels, a correlate of bone turnover, were sol unaltered.17,20 Therefore, orteronel is less likely stabilized in patients receiving tasquinimod. Following than abiraterone acetate to require the concomitant the favorable outcome of the phase 2 trial, a phase 3 administration of corticosteroids.25,58 Phase 2 trials trial comparing tasquinimod to placebo was initiated demonstrated a significant reduction in serum levels in patients with mCRPC naive to chemotherapy.50 of PSA that led to 10 partial responses and 22 cases of Cabozantinib: Cabozantinib is a tyrosine kinase inhibitor that blocks c-MET and VEGF recepTable 2. — Experimental Therapies for the Treatment of Metastatic Castration-Resistant tor 2 and is already apProstate Cancer proved for the treatment of medullary thyroid Drug Target Effect Study Results cancer. This fact, comCabozantinib c-MET Inhibits tyrosine kinase Partial resolution of bone lesions, bined with its oral adactivity decreases number of CTCs, VEGF-R2 decreases pain51 ministration, makes it a favorable candidate for Custirsen Clusterin Improves response Extended median survival, extends to docetaxel PFS, improves PSA declines52 further investigation and Ipilimumab CTLA-4 T-cell activation Ongoing54,55 development in mCRPC. Phase 2 clinical trials have Nivolumab PD-1 T-cell activation Ongoing53 shown that cabozantinib Orteronel CYP17A1 Reduces circulating Decreases number of CTCs, results in partial resolu(17,20 lyase activity) testosterone levels improves radiographic PFS48,49 tion of bone lesions in Prostvac-VF Delivery of PSA transgene T-cell activation Improves median survival32,56 56% of patients and proTasquinimod Thrombospondin Antiangiogenic, reduces Improves median PFS, stable bone vided complete resolution S100A9 MDSC recruitment alkaline phosphatase levels50,57 in 19%.51 A total of 64% CTC = circulating tumor cell, CTLA = cytotoxic T-lymphocyte antigen 4, MDSC = myeloid-derived suppressor cell, PD-1 = programmed cell death 1, PFS = progression-free survival, PSA = prostate-specific antigen, VEGF-R2 = vascular had an improvement in endothelial growth factor receptor 2. pain and 46% were able Despite the growing number of FDA-approved agents to treat mCRPC, room remains to improve upon the therapeutic options available to patients and clinicians. For example, although approximately 50% of patients with mCRPC will respond to docetaxel, most patients develop resistance and disease progression within 1 year of beginning treatment.36 However, some treatments that target cancer and support the microenvironment are currently in clinical trials that have the potential to provide health care professionals with new therapeutic options to treat men diagnosed with mCRPC (see Fig 1). A list of these experimental therapies appears in Table 2.32,48-57 January 2015, Vol. 22, No. 1 Cancer Control 113 to decrease or discontinue narcotics.51 An additional exploratory analysis updated the results of this phase 2 trial and indicated a reduction of more than 30% in the bone scan lesion area and also indicated a reduction in circulating tumor cells.64 Multiple phase 3 trials focused on the treatment of mCRPC with cabozantinib are either ongoing or in the recruiting stages (NCT01428219, NCT01703065, NCT01995058, NCT01605227, NCT01834651, NCT01599793, NCT01522443, NCT01683994). At the time of publication, NCT01605227 failed to reach efficacy in men with mCRPC. Custirsen: Custirsen is an antisense oligonucleotide that targets clusterin, a chaperone induced by stress and detected at elevated levels in several tumor types, including prostate cancer.65 Studies of clusterin have demonstrated its antiapoptotic and prosurvival activities in prostate cancer that are believed to be associated with docetaxel resistance.66 As such, inhibiting clusterin concomitantly with docetaxel may increase the time until docetaxel resistance in mCRPC. Phase 2 trials of weekly intravenous custirsen plus docetaxel extended median survival rates from 16.9 months to 23.8 months compared with single-agent docetaxel.67,68 Subsequent to treatment, significant decreases in clusterin levels were noted in patients treated with custirsen.67,68 A second phase 2 trial evaluating custirsen plus prednisone compared with mitoxantrone plus prednisone in patients with mCRPC who previously failed first-line docetaxel showed an increase of 4.3 months in median overall survival and a 3.8-month increase in progression-free survival as well as improved declines in PSA.52 Phase 3 trials of custirsen are ongoing (NCT01578655), although its benefits may be limited to patients expressing high levels of clusterin.69 Prostvac-VF: The use of cancer vaccines aims to generate an immune response to specific tumor antigens. The Prostvac vaccine uses a fowlpox and vaccinia platform to deliver the PSA transgene to antigen-presenting cells, which, in turn, express and present the antigen to T cells and T-cell activation.70 In addition to PSA, the vaccine has been engineered to include B7-1, ICAM-1, and LFA-3 antigen-presenting cell costimulatory molecules.71 Phase 2 trials in patients with mCRPC have shown improvements of 8 to 9 months in median survival rates.56,72 The results of these trials suggest that Prostvac offers an improvement compared with sipuleucel-T and have resulted in the initiation of a phase 3 trial (NCT01322490). Nivolumab: Blocking the programmed cell death 1 (PD-1)/programmed death ligand 1 (PD-L1) immunosuppressive axis has received much attention in recent years. Nivolumab is a monoclonal antibody that inhibits the interaction between PD-L1 and T-cell expressed PD-1, preventing tumor-induced loss of 114 Cancer Control T-cell effector function.73 In trials of melanoma, 80% of patients responded to nivolumab therapy.74 However, limited studies in CRPC have not been as promising; phase 1 studies have failed to reach objective responses and others have shown limited or lack of PD-L1 expression by CRPCs or the immune infiltrates.53,73 However, it is possible that prospective, individual patients with mCRPC with high levels of PD-L1 could benefit from nivolumab. Ipilimumab: As cancer progresses, it can express inhibitory ligands such as B7-1, B7-2, and PD-L1 to suppress the immune system. Ipilimumab is a monoclonal antibody that inhibits T-cell–expressed cytotoxic T-lymphocyte antigen 4 from interacting with antigen-presenting cell B7-1 and B7-2 ligands but not those on tumor cells, allowing for the continued immune-mediated destruction of tumor cells. Ipilimumab has been studied in melanoma and is the only FDA-approved immune checkpoint inhibitor on the market.40 Despite encouraging results in early clinical trials, the results of a phase 3 trial of patients with mCRPC receiving bone-directed radiotherapy prior to 10 mg/kg ipilimumab or placebo revealed no significant improvement in overall survival rates.54,55 However, individual analysis of patient subsets indicated that ipilimumab may benefit men with low disease burden, thus emphasizing the importance of appropriate patient selection.16,55 Therapeutic Opportunities on the Horizon Treatment options to extend the overall survival of patients diagnosed with mCRPC remains a major clinical challenge. Therefore, understanding the factors that drive the process of metastasis, the homing of the metastasis to organs (eg, bone), and how prostate cancer cells form life-threatening active metastases once in the bone warrants extensive research to generate new therapies to cure the disease. Although metastasis is classically thought of as a linear sequence of events beginning with the dissemination and invasion of tumor cells from the primary site and ending with proliferation at the metastatic site, recent evidence suggests that the first steps of metastasis can occur before a patient’s tumor is diagnosed (Fig 2).75 This “step 0” of the metastatic cascade results in the nonrandom priming of future sites of metastasis, a concept known as the “premetastatic niche.” Premetastatic Niche Primary tumor-derived factors have been implicated in the development of premetastatic niches in distant organs.76 Through a series of in vivo experiments, it was illustrated that conditioned media derived from highly metastatic cancer cells lines, such as the B-16 melanoma cell line, could stimulate the mobilization of bone marrow–derived VEGF receptor 1+ VLA4+ Id3+ January 2015, Vol. 22, No. 1 hematopoietic precursor cells to develop premetastatic niche sites, including the lungs, liver, spleen, kidney, and testes.76 Cancer-derived exosomes have been implicated as the mechanism for facilitating long distance, tumor–stroma interactions and initiating the premetastatic niche.77 Exosomes are microvesicles measuring 30 nm to 100 nm that contain a variety of functional proteins and messenger/micro RNAs.78 In the context of premetastatic niche formation, B16-F10–derived exosomes have been labeled and shown to “home” to common sites of melanoma metastasis.75 Furthermore, in the premetastatic niche, exosomes can educate bone marrow–derived cells to support metastatic tumor growth via the horizontal transfer of the c-MET protein.75 c-MET inhibitors, such as cabozantinib, could be used to prevent the development of premetastatic niches and, thus, mitigate the ability of cancers to metastasize to new sites. Exosome shedding has also been demonstrated in prostate cancer, and studies have shown the presence of microvesicles termed oncosomes (0.5–5 µm) in prostate cancer–conditioned media. Oncosomes contain a variety of signal transduction proteins, including Akt and Src, and can interact with tumor and stromal cells to elicit disease-promoting responses.79 In addition, a correlation exists between a Gleason score higher than 7 and the number of oncosomes present in patient plasma.80 Based on these findings, it is plausible that prostate cancer–derived exosomes can play a role in the formation of premetastatic niches in the bone microenvironment. Emerging evidence also suggests that prostate cancer cells homing to the bone microenvironment can occupy the endosteal niche, the vascular niche, or both.81 Defining Factors Controlling the Homing of Bone Metastatic Castration-Resistant Prostate Cancer An unsolved question regarding metastasis is why prostate cancer has such a predilection for the bone microenvironment. More than a century ago, Paget82 formulated the “seed and soil” hypothesis to address this question. His hypothesis suggested that metastasis is a challenging process that requires “fertile soil” for outgrowth but begins long before the “seed” meets the “soil.”82 Ewing83 challenged Paget’s hypothesis in the 1920s, proposing that metastasis was instead dependent on anatomy, vasculature, and lymphatics. Metastasis by anatomy would become the accepted model until the 1970s when modern experiments rekindled interest in the “seed and soil” hypothesis, notably observing that circulating tumor cells reach the vasculature of all organs, but only certain organs are receptive for metastasis.84,85 In reality, prostate to bone metastasis occurs by a blend of both hypotheses: It metastasizes first to the pelvic lymph node and then to sites in the bone, including iliac crests, January 2015, Vol. 22, No. 1 sacrum wings, L1 to L5 vertebrae, T8 to T12 vertebrae, ribs, manubrium, humeral heads, and femoral necks.86 Although 15% to 30% of prostate to bone metastases are due to cells traveling through the Batson plexus to the lumbar spine, it is clear that molecular factors, such as chemokines and integrins, underpin the propensity for prostate cancer cells to metastasize to the skeleton.11 Elucidating those factors could help identify new therapies to prevent bone metastatic CRPC. Bone is the home of regulatory sites for hematopoietic stem cells (HSCs), which are cells localized to the vascular and endosteal niches where they either await hematopoietic demand or reside in a quiescent state.81 One well-defined signaling axis implicated in metastasis is that between stromal cell–derived factor 1/CXCL12 and its receptor CXCR4, a system normally utilized by HSCs homing to the niche.87 CXCL12 expression is increased in the premetastatic niche, and studies in prostate cancer have demonstrated that tumor cells with high bone-homing capacity express CXCR4 and CXCR7 to parasitize the HSC niche.76,88,89 Furthermore, CXCR4 expression correlates with poor prognosis.90 Additional axes, including MCP-1/CCR2 and CXCL16/CXCR6, have also been found to contribute to the progression of prostate cancer through increases in proliferation, migration, and invasion.91,92 Disseminated Tumor Cells and Dormancy Evidence suggests that tumor cells disseminated from the prostate localize to the bone marrow niche, displace HSCs, and either proliferate to form a metastatic mass or enter a state of dormancy.93 Dissemination from the primary site to reside in distant environments is an early event seen in prostate cancer, as patients who undergo prostatectomy may present with metastases many years later.94,95 Disseminated tumor cells (DTCs) reside in the bone marrow niche where they can remain dormant and resistant to chemotherapy for long periods of time (> 10 years) before emerging to form metastatic outgrowths.94 Although most patients with prostate cancer harbor DTCs, not all will develop metastases, suggesting that mechanisms exist to maintain DTC dormancy as well as to promote awakening.95 Several bone marrow–dependent mechanisms have been identified as modulators of prostate cancer DTC dormancy. In the endosteal niche, the osteoblast expression of Anxa2 combined with the expression of the Anxa2 receptor (Anxa2R) by HSCs is important in regulating HSC homing to the niche. Anxa2R expression is elevated in metastatic prostate tumor cells and, as such, the Anxa2/Anxa2R axis can be hijacked to promote the homing of prostate tumor cells to the niche. Interrupting the interaction between Anxa2 and Anxa2R is sufficient to reduce tumor burden in the niche.96 Evidence has revealed that the ligation of Anxa2 with Anxa2R stimulates the expression of Cancer Control 115 the Axl receptor tyrosine kinase.97 Axl, along with Tyro3 and Mer, are receptors for osteoblast-expressed growth arrest-specific 6 (GAS6).98 As was the case with Anxa2/Anxa2R, the GAS6/Axl interaction typically occurs between HSCs and osteoblasts and is one mechanism of controlling HSC dormancy.98 Engaging osteoblast-expressed GAS6 and tumor cell–expressed Axl yields a similar result that includes growth arrest and enhanced drug resistance in prostate cancer cells.97 Following-up on these observations, data show that these activities may be specific to the Axl receptor compared with other GAS6 receptors.98 A high ratio of Axl to Tyro3 expression encourages maintenance of a dormant state, whereas reducing the expression of Axl and increasing the expression of Tyro3 has been shown to promote outgrowth.98 Interactions between osteoblasts and tumor cells may be important to DTC dormancy. Prostate cancer cells that bind with osteoblasts also upregulate the expression of TANK-binding kinase 1 (TBK1). In vitro and in vivo knockdown of TBK1 resulted in decreased drug resistance, suggesting that TBK1 may also play a role in dormancy and drug resistance.100 A high p38:ERK ratio has been shown to maintain dormancy of squamous carcinoma cells, whereas interactions with the microenvironment can stimulate a switch to high ERK:p38 and reverse dormancy.101 Bone marrow–derived transforming growth factor (TGF) β2 has been implicated in maintaining the dormancy of DTCs by p38 activation, and inhibiting either the TGF-β receptor 1 or p38 leads to the proliferation and metastasis of DTCs.102 Similarly, bone morphogenetic protein 7 triggers prostate cancer DTC dormancy in part by activating p38.103 Although much focus has been on the endosteal niche, the vascular niche also has implications for DTC dormancy. Through the use of advanced imaging techniques, dormant DTCs have been shown to home to perivascular niches in the bone marrow and the lungs.104 These niches promote dormancy through the expression of TSP-1; however, dormancy is lost in regions of sprouting vasculature due to a loss of TSP-1 and the activation of TGF-β and periostin.104 In vivo experiments in mice receiving bone marrow transplantation revealed that fewer HSCs successfully engraft in tumor-bearing mice, suggesting that the tumor cells occupying the niche outcompete HSCs for residence.105 In addition, expanding the endosteal osteoblast niche with parathyroid hormone (PTH) promoted metastasis, whereas decreasing the size of the niche using conditional osteoblast knockout models reduced dissemination.105 Tumor cells can also be forced out of the niche using methods to mobilize HSCs, perhaps offering an opportunity for therapeutic intervention.105 Filgrastim is an agent that mobilizes HSCs out of the niche, and plerixafor blocks the interaction with stromal cell–derived factor 1 by acting as 116 Cancer Control a CXCR4 antagonist to mobilize HSCs.106 Both agents have been approved by the FDA and may serve as a method of awakening and forcing the DTCs into circulation where they would become vulnerable to chemotherapy. A small molecule inhibitor specific to CXCR6 but not other chemokine receptors was developed for investigating the CXCL16/CXCR6 axis.107 Although the clinical utility of such an inhibitor must be investigated, the selectivity of small molecule antagonists could aid in the targeting of dormant tumor cells. Therapeutic Opportunities for “Active” mCRPC Although therapies to prevent the homing and establishment of mCRPC in the bone microenvironment are important clinical tactics, many patients in the clinical setting present with “active” bone metastases that cause extensive bone remodeling. Defining the mechanisms that control cell–cell communication between the metastases and the microenvironment are also likely to reveal important therapeutic targets. Osteomimicry: A recurring theme in bone metastasis is the hijacking of normal bone mechanisms by tumor cells. The concept of osteomimicry is that bone metastatic prostate cells acquire the ability to produce proteins typically restricted to bone cells, such as osteoblasts, to survive and proliferate in the otherwise restrictive bone microenvironment.108 Select genes normally expressed in bone have been detected in prostate cells, including osteocalcin, osteopontin, bone sialoprotein, osteonectin, RANK, RANKL, and PTH-related protein.108-111 The expression of these genes appears to be associated with the metastatic capacity of the cells. Studies in both the PC3 and LNCaP cell lines have shown that the expression of osteonectin is highest in the more invasive and metastatic sublines, including the LNCaP metastatic variant C4-2B.109 Analysis of patient samples support these findings, showing that osteonectin staining in prostate to bone metastases was more intense than from soft-tissue metastases.109 In addition to changes in gene expression, prostate tumor cells may adopt biological activities usually specific to bone cells. In vitro studies indicate that human C4-2B prostate tumor cells are capable of depositing hydroxyapatite and contributing to mineralization, a common feature of the sclerotic lesions observed in vivo.110 Due to the shared expression of specific bone genes between tumor and stroma cells, these common proteins could be used to simultaneously target both compartments. Understanding that soluble factors like bone morphogenetic protein 2, RANKL, TGF-β, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor are partially responsible for inducing osteomimetic genes may also provide options to specifically target osteomimicry and establish bone outgrowths.111 It has January 2015, Vol. 22, No. 1 been suggested that promoters for the common genes between the tumor and stroma cells could be utilized to drive the expression of therapeutic genes, thus targeting both the stroma and tumor cells.108 Halting the Vicious Cycle of Bone Metastases: Once the DTCs awaken and establish micrometastases, continued outgrowth arises through the interaction with multiple stromal cell types, growth factors, and enzymes in a process known as the vicious cycle model.112 Prostate to bone metastases are characterized by areas of mixed osteogenesis and osteolysis that give rise to painful lesions.113 A number of tumor-derived factors, including PTH-related protein, interleukin (IL) 1, IL-6, and IL-11, have been shown to interact with osteoblasts and stimulate the production of RANKL.114 RANKL is a crucial molecule for osteoclast differentiation; therefore, it contributes to the extensive bone remodeling seen in bone metastasis. In addition to bone destruction, osteoclast-mediated bone resorption also releases a multitude of bone-derived factors such as TGF-β, insulin growth factor, platelet-derived growth factor, and fibroblast growth factor. These factors provide positive feedback via interaction with their respective receptors on the surface of tumor cells, thus promoting the proliferation and continued production of tumor-derived factors.114 The vicious cycle is continually evolving to include other cell types, cytokines, proteases, and therapeutics.115-118 Several studies have shown contributory roles for highly expressed host matrix metalloproteinases (MMPs) in the vicious cycle, including the regulation of latent TGF-β and VEGF-A bioavailability by MMP-2 and MMP-9, and the generation of a soluble form of RANKL by MMP-7, which promotes osteoclastogenesis and mammary tumor–induced osteolysis in vivo.119-121 In recent years, the interactions with immune cells have become an integral part of the vicious cycle. For example, T cells stimulate and inhibit the formation of osteoclasts, and the recruitment of regulatory T cells to bone marrow may inhibit osteoclastogenesis. Myeloid-derived suppressor cells suppress T cells and release angiogenic, tumor-promoting factors. Recruited myeloid-derived suppressor cells have also been shown to differentiate into osteoclasts.118 Although the need for therapies aimed at the early stages of metastasis has been emphasized, patients will still present in the later stages of the disease; therefore, improving therapies for these patients must still remain a priority. The interactions between tumor and stromal cells in the vicious cycle model offer many opportunities to intervene. Therapies such as zoledronic acid and denosumab interfere with the osteolytic component of the vicious cycle; however, therapies to inhibit the unique osteosclerotic component of prostate to bone metastases are lacking. Many roles for specific MMPs have been elucidated January 2015, Vol. 22, No. 1 in the vicious cycle,115,120,121 and the development of MMP inhibitors with improved specificity is perhaps a promising method to modulate the vicious cycle.122 From these discoveries, it is becoming evident that the metastasis of prostate cancer is not a linear, stepwise procedure. Defining the mechanisms that control CRPC metastasis may help elucidate new therapeutic targets that directly impact the cancer cells and the processes that facilitate the formation of a premetastatic niche, niche seeding, dormancy, and the vicious cycle.123 Such new discoveries are highly likely to impact the clinical treatment of patients with mCRPC. Upcoming Challenges Our knowledge of the mechanisms driving the progression of prostate cancer is growing. Although several new therapies that target both the cancer cells and the supporting microenvironment and are likely to increase overall survival rates for men with mCRPC, new challenges are also emerging, particularly within the context of tumor heterogeneity. Heterogeneity is a key aspect of cancer evolution and is a clinical reality in many cancers, including prostate cancer.124-126 Greater heterogeneity facilitates the evolution of the treatment resistance of cancer but also gives the cancer a number of phenotypic strategies that allow for growth in select microenvironments (eg, bone). Emerging studies suggest that most patients would be best served by therapies tailored toward cancer cells harboring common aberrations as well as by therapies geared toward smaller subpopulations who could potentially become the dominant-resistant population.127 The therapies described herein constitute new ways in which to expand the number of potential options for the treatment of heterogeneous bone metastatic CRPCs. However, a challenge emerging with the advent of these therapies is how to rationally design a treatment strategy for individual patients. Current guidelines from the National Comprehensive Cancer Network provide recommendations for applying the sequence of existing therapies to patients with mCRPC based on individual patient parameters. However, some studies suggest that altering the sequence or the combination of existing therapies can have a profound impact on overall survival rates.128 To circumvent costly and time-consuming clinical trials assessing the combination and sequence alterations of a new line of targeted therapies currently in clinical trials, alternative approaches are required. In this regard, integrating computational models and genetic algorithms with individual patient-derived biological data might lead to the rapid optimization of therapy choice and sequence. In the preclinical setting, the power of this integrated approach has been demonstrated. Recent studies have discovered how appropriate drug combinations guided by comCancer Control 117 putational models could minimize prostate cancer progression in vivo.129 Therefore, the refinement and validation of these approaches may assist in overcoming the challenges posed by cancer heterogeneity. Conclusions Metastatic castration-resistant prostate cancer is an incurable disease, but the advent of new therapies, combined with an enhanced understanding of the underlying biology, suggests that significant improvement in overall survival is within reach. An increase in the number of available treatment options will be challenging from a clinical perspective with regard to patient stratification and in selecting the optimal therapy sequence, combination, or both. However, integrating computational models and genetic algorithms based on individual patient data may help overcome this challenge and allow for the delivery of individualized treatment for patients with this disease. References 1. American Cancer Society. Cancer Facts & Figures 2014. Atlanta, GA: American Cancer Society; 2014. 2. American Cancer Society. Survival rates for prostate cancer. http:// www.cancer.org/cancer/prostatecancer/detailedguide/prostate-cancer-survival-rates. Accessed October 15, 2014. 3. Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1:293-297. 4. Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011;8(1):12-23. 5. Cookson MS, Roth BJ, Dahm P, et al; American Urological Association (AUA). Castration-Resistant Prostate Cancer. Linthicum, MD: AUA; 2014. https://www.auanet.org/common/pdf/education/clinical-guidance/Castration-Resistant-Prostate-Cancer.pdf. Accessed October 15, 2014. 6. Huang X, Chau CH, Figg WD. Challenges to improved therapeutics for metastatic castrate resistant prostate cancer: from recent successes and failures. J Hematol Oncol. 2012;5:35. 7. American Cancer Society. What are the key statistics about prostate cancer? http://www.cancer.org/cancer/prostatecancer/detailedguide/prostate-cancer-key-statistics. Accessed October 15, 2014. 8. Keller ET, Brown J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem. 2004;91(4):718-729. 9. Bubendorf L, Schöpfer A, Wagner U, et al. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol. 2000;31(5):578-583. 10. Crawford ED, Petrylak D. Castration-resistant prostate cancer: descriptive yet pejorative? J Clin Oncol. 2010;28(23):e408. 11. Benjamin R. Neurologic complications of prostate cancer. Am Fam Physician. 2002;65(9):1834-1840. 12. American Cancer Society. Preventing and treating prostate cancer spread to bone. http://www.cancer.org/cancer/prostatecancer/detailedguide/ prostate-cancer-treating-treating-pain. Accessed October 15, 2014. 13. Tomblyn M. The role of bone-seeking radionuclides in the palliative treatment of patients with painful osteoblastic skeletal metastases. Cancer Control. 2012;19(2):137-144. 14. Iagaru A, Young P, Mittra E, et al. Pilot prospective evaluation of 99mTcMDP scintigraphy, 18F NaF PET/CT, 18F FDG PET/CT and whole-body MRI for detection of skeletal metastases. Clin Nucl Med. 2013;38(7):e290-296. 15. Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351(15):1513-1520. 16. Agarwal N, Di Lorenzo G, Sonpavde G, et al. New agents for prostate cancer. Ann Oncol. 2014;25(9):1700-1709. 17. American Cancer Society. Prednisone. http://www.cancer.org/treatment/ treatmentsandsideeffects/guidetocancerdrugs/prednisone. Accessed October 15, 2014. 18. National Comprehensive Cancer Network. NCCN clinical practice guidelines in prostate cancer. Version 2.2014. http://www.nccn.org. Accessed October 15, 2014. 19. Egan A, Dong Y, Zhang H, et al. Castration-resistant prostate cancer: adaptive responses in the androgen axis. Cancer Treat Rev. 2014;40(3):426-433. 20. Chang KH, Li R, Papari-Zareei M, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl 118 Cancer Control Acad Sci U S A. 2011;108(33):13728-13733. 21. Ishizaki F, Nishiyama T, Kawasaki T, et al. Androgen deprivation promotes intratumoral synthesis of dihydrotestosterone from androgen metabolites in prostate cancer. Sci Rep. 2013;3:1528. 22. Chang KH, Li R, Kuri B, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154(5):1074-1084. 23. de Bono JS, Logothetis CJ, Molina A, et al; COU-AA-302 Investigators. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995-2005. 24. Ryan CJ, Smith MR, de Bono JS, et al; COU-AA-302 Investigators. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368(2):138-148. 25. Pinto Á. Beyond abiraterone: new hormonal therapies for metastatic castration-resistant prostate cancer. Cancer Biol Ther. 2014;15(2):149-155. 26. US Food and Drug Administration. Enzalutamide (XTANDI capsules). http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm317997.htm. Accessed October 15, 2014. 27. Scher HI, Fizazi K, Saad F, et al; AFFIRM Investigators. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187-1197. 28. Fizazi K, Albiges L, Massard C, et al. Novel and bone-targeted agents for CRPC. Ann Oncol. 2012;23(suppl 10):x264-x267. 29. Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787-790. 30. Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate vancer before chemotherapy. N Engl J Med. 2014371(5):424-433. 31. de Bono JS, Oudard S, Ozguroglu M, et al; TROPIC Investigators. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147-1154. 32. Kantoff PW, Higano CS, Shore ND, et al; IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411-422. 33. Saad F, Gleason DM, Murray R, et al; Zoledronic Acid Prostate Cancer Study Group. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J Natl Cancer Inst. 2004;96(11):879-882. 34. Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet. 2011;377(9768):813-822. 35. Parker C, Nilsson S, Heinrich D, et al; ALSYMPCA Investigators. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213-223. 36. Tannock IF, de Wit R, Berry WR, et al; TAX 327 Investigators. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502-1512. 37. Mita AC, Denis LJ, Rowinsky EK, et al. Phase I and pharmacokinetic study of XRP6258 (RPR 116258A), a novel taxane, administered as a 1-hour infusion every 3 weeks in patients with advanced solid tumors. Clin Cancer Res. 2009;15(2):723-730. 38. National Cancer Institute. FDA approval for cabazitaxel. National Cancer Institute. 2013. http://www.cancer.gov/cancertopics/druginfo/fda-cabazitaxel. Accessed October 15, 2014. 39. National Cancer Institute. FDA approval for sipuleucel-T. http://www. cancer.gov/cancertopics/druginfo/fda-sipuleucel-T. Accessed October 15, 2014. 40. Schweizer MT, Drake CG. Immunotherapy for prostate cancer: recent developments and future challenges. Cancer Metastasis Rev. 2014;33(23):641-655. 41. Rogers MJ, Watts DJ, Russell RG. Overview of bisphosphonates. Cancer. 1997;80(8 suppl):1652-1660. 42. National Cancer Institute. FDA approval for denosumab. http://www. cancer.gov/cancertopics/druginfo/fda-denosumab. Accessed October 15, 2014. 43. Helo S, Manger JP, Krupski TL. Role of denosumab in prostate cancer. Prostate Cancer Prostatic Dis. 2012;15(3):231-236. 44. Armstrong AP, Miller RE, Jones JC, et al. RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate. 2008;68(1):92-104. 45. Miller RE, Roudier M, Jones J, et al. RANK ligand inhibition plus docetaxel improves survival and reduces tumor burden in a murine model of prostate cancer bone metastasis. Mol Cancer Ther. 2008;7(7):2160-2169. 46. National Cancer Institute. FDA approval for radium 223 dichloride. http:// www.cancer.gov/cancertopics/druginfo/fda-radium-223-dichloride. Accessed October 15, 2014. 47. Cheetham PJ, Petrylak DP. Alpha particles as radiopharmaceuticals in the treatment of bone metastases: mechanism of action of radium-223 chloride (Alpharadin) and radiation protection. Oncology (Williston Park). 2012;26(4):330-337, 341. 48. De Wit R FK, Jinga V, Efstathiou E, et al. Phase 3, randomized, placebo-controlled trial of orteronel (TAK-700) plus prednisone in patients with chemotherapy-naive metastatic castration-resistant prostate cancer (mCRPC) (ELM-PC4 trial). J Clin Oncol. 2014;32(suppl 5):5008. 49. Dreicer R JR, Oudard S, Efstathiou E, et al. Results from a phase 3, randomized, double-blind, multicenter, placebo-controlled trial of orteronel January 2015, Vol. 22, No. 1 (TAK-700) plus prednisone in patients with metastatic castration-resistant prostate cancer (mCRPC) that has progressed during or following docetaxelbased therapy (ELM-PC5 trial). J Clin Oncol. 2014;32(suppl 4):7. 50. Osanto S, van Poppel H, Burggraaf J. Tasquinimod: a novel drug in advanced prostate cancer. Future Oncol. 2013;9(9):1271-1281. 51. Smith DC, Smith MR, Sweeney C, et al. Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol. 2013;31(4):412-419. 52. Saad F, Hotte S, North S, et al; Canadian Uro-Oncology Group. Randomized phase II trial of Custirsen (OGX-011) in combination with docetaxel or mitoxantrone as second-line therapy in patients with metastatic castrate-resistant prostate cancer progressing after first-line docetaxel: CUOG trial P-06c. Clin Cancer Res. 2011;17(17):5765-5773. 53. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443-2454. 54. Slovin SF, Higano CS, Hamid O, et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol. 2013;24(7):1813-1821. 55. Gerritsen WR, Sharma P. Current and emerging treatment options for castration-resistant prostate cancer: a focus on immunotherapy. J Clin Immunol. 2012;32(1):25-35. 56. Gulley JL, Arlen PM, Madan RA, et al. Immunologic and prognostic factors associated with overall survival employing a poxviral-based PSA vaccine in metastatic castrate-resistant prostate cancer. Cancer Immunol Immunother. 2010;59(5):663-674. 57. Pili R, Häggman M, Stadler WM, et al. Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J Clin Oncol. 2011;29(30):4022-4028. 58. Yamaoka M, Hara T, Hitaka T, et al. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J Steroid Biochem Mol Biol. 2012;129(3-5):115-128. 59. Agus DB SW, Shevrin DH, Hart L, et al. Safety, efficacy, and pharmacodynamics of the investigational agent orteronel (TAK-700) in metastatic castration-resistant prostate cancer (mCRPC): updated data from a phase I/ II study. J Clin Oncol. 2012;30(suppl 5):98. 60. Jennbacken K, Welén K, Olsson A, et al. Inhibition of metastasis in a castration resistant prostate cancer model by the quinoline-3-carboxamide tasquinimod (ABR-215050). Prostate. 2012;72(8):913-924. 61. Hiratsuka S, Watanabe A, Aburatani H, et al. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8(12):1369-1375. 62. Hiratsuka S, Watanabe A, Sakurai Y, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10(11):1349-1355. 63. Olsson A, Björk A, Vallon-Christersson J, et al. Tasquinimod (ABR215050), a quinoline-3-carboxamide anti-angiogenic agent, modulates the expression of thrombospondin-1 in human prostate tumors. Mol Cancer. 2010;9:107. 64. Scher HI, Smith MR, Sweeney C, et al. An exploratory analysis of bone scan lesion area (BSLA), circulating tumor cell (CTC) change, pain reduction, and overall survival (OS) in patients with castration-resistant prostate cancer (CRPC) treated with cabozantinib (cabo): updated results of a phase II nonrandomized expansion (NRE) cohort. J Clin Oncol. 2013;31(suppl):5026. 65. Cochrane DR, Wang Z, Muramaki M, et al. Differential regulation of clusterin and its isoforms by androgens in prostate cells. J Biol Chem. 2007;282(4):2278-2287. 66. Zellweger T, Chi K, Miyake H, et al. Enhanced radiation sensitivity in prostate cancer by inhibition of the cell survival protein clusterin. Clin Cancer Res. 2002;8(10):3276-3284. 67. Chi KN, Bjartell A, Dearnaley D, et al. Castration-resistant prostate cancer: from new pathophysiology to new treatment targets. Eur Urol. 2009;56(4):594-605. 68. Chi KN, Hotte SJ, Yu EY, et al. Randomized phase II study of docetaxel and prednisone with or without OGX-011 in patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(27):4247-4254. 69. Higano CS. Potential use of custirsen to treat prostate cancer. Onco Targets Ther. 2013;6:785-797. 70. Madan RA, Arlen PM, Mohebtash M, et al. Prostvac-VF: a vector-based vaccine targeting PSA in prostate cancer. Expert Opin Investig Drugs. 2009;18(7):1001-1011. 71. DiPaola RS, Plante M, Kaufman H, et al. A phase I trial of pox PSA vaccines (PROSTVAC-VF) with B7-1, ICAM-1, and LFA-3 co-stimulatory molecules (TRICOM) in patients with prostate cancer. J Transl Med. 2006;4:1. 72. Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(7):1099-1105. 73. Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064-5074. 74. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122-133. January 2015, Vol. 22, No. 1 75. Peinado H, Alečković M, Lavotshkin S, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18(6):883-891. 76. Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820-827. 77. Webber J, Steadman R, Mason MD, et al. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010;70(23):9621-9630. 78. Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569-579. 79. Di Vizio D, Kim J, Hager MH, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;69(13):5601-5609. 80. Di Vizio D, Morello M, Dudley AC, et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol. 2012;181(5):1573-1584. 81. Taichman RS. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood. 2005;105(7):2631-2639. 82. Paget G. Remarks on a case of alternate partial anaesthesia. Br Med J. 1889;1(1462):1-3. 83. Ewing J. Neoplastic Diseases. 6th ed. Philadelphia: W.B. Saunders; 1928. 84. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453-458. 85. Poste G, Fidler IJ. The pathogenesis of cancer metastasis. Nature. 1980;283(5743):139-146. 86. Roudier MP, Vesselle H, True LD, et al. Bone histology at autopsy and matched bone scintigraphy findings in patients with hormone refractory prostate cancer: the effect of bisphosphonate therapy on bone scintigraphy results. Clin Exp Metastasis. 2003;20(2):171-180. 87. Lapidot T, Kollet O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2m(null) mice. Leukemia. 2002;16(10):1992-2003. 88. Wang J, Loberg R, Taichman RS. The pivotal role of CXCL12 (SDF-1)/ CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 2006;25(4):573-587. 89. Singh RK, Lokeshwar BL. The IL-8-regulated chemokine receptor CXCR7 stimulates EGFR signaling to promote prostate cancer growth. Cancer Res. 2011;71(9):3268-3277. 90. Akashi T, Koizumi K, Tsuneyama K, et al. Chemokine receptor CXCR4 expression and prognosis in patients with metastatic prostate cancer. Cancer Sci. 2008;99(3):539-542. 91. Lu Y, Cai Z, Galson DL, et al. Monocyte chemotactic protein-1 (MCP1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate. 2006;66(12):1311-1318. 92. Lu Y, Wang J, Xu Y, et al. CXCL16 functions as a novel chemotactic factor for prostate cancer cells in vitro. Mol Cancer Res. 2008;6(4):546-554. 93. Shiozawa Y, Havens AM, Pienta KJ, et al. The bone marrow niche: habitat to hematopoietic and mesenchymal stem cells, and unwitting host to molecular parasites. Leukemia. 2008;22(5):941-950. 94. Vessella RL, Pantel K, Mohla S. Tumor cell dormancy: an NCI workshop report. Cancer Bio Ther. 2007;6(9):1496-1504. 95. Lam HM, Vessella RL, Morrissey C. The role of the microenvironment-dormant prostate disseminated tumor cells in the bone marrow. Drug Discov Today Technol. 2014;11:41-47. 96. Shiozawa Y, Havens AM, Jung Y, et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J Cell Biochem. 2008;105(2):370-380. 97. Shiozawa Y, Pedersen EA, Patel LR, et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia. 2010;12(2):116-127. 98. Taichman RS, Patel LR, Bedenis R, et al. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS One. 2013;8(4):e61873. 99. Dormady SP, Zhang XM, Basch RS. Hematopoietic progenitor cells grow on 3T3 fibroblast monolayers that overexpress growth arrest-specific gene-6 (GAS6). Proc Natl Acad Sci U S A. 2000;97(22):12260-12265. 100. Kim JK, Jung Y, Wang J, et al. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia. 2013;15(9):1064-1074. 101. Bragado P, Sosa MS, Keely P, et al. Microenvironments dictating tumor cell dormancy. Recent Results Cancer Res. 2012;195:25-39. 102. Bragado P, Estrada Y, Parikh F, et al. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38β/β signalling. Nat Cell Biol. 2013;15(11):1351-1361. 103. Kobayashi A, Okuda H, Xing F, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med. 2011;208(13):2641-2655. 104. Ghajar CM, Peinado H, Mori H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15(7):807-817. 105. Shiozawa Y, Pedersen EA, Havens AM, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121(4):1298-1312. 106. Pedersen EA, Shiozawa Y, Pienta KJ, et al. The prostate cancer bone marrow niche: more than just ‘fertile soil’. Asian J Androl. 2012;14(3):423-427. 107. Hershberger PM, Peddibhotla S, Sugarman E, et al. Probing the Cancer Control 119 CXCR6/CXCL16 Axis: targeting prevention of prostate cancer metastasis. Probe Reports from the NIH Molecular Libraries Program. Bethesda, MD: National Center for Biotechnology Information; 2012. 108. Koeneman KS, Yeung F, Chung LW. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate. 1999;39(4):246-261. 109. Thomas R, True LD, Bassuk JA, et al. Differential expression of osteonectin/SPARC during human prostate cancer progression. Clin Cancer Res. 2000;6(3):1140-1149. 110. Lin DL, Tarnowski CP, Zhang J, et al. Bone metastatic LNCaP-derivative C4-2B prostate cancer cell line mineralizes in vitro. Prostate. 2001;47(3):212-221. 111. Chu GC, Chung LW. RANK-mediated signaling network and cancer metastasis. Cancer Metastasis Rev. 2014;33(2-3):497-509. 112. Mundy GR. Mechanisms of bone metastasis. Cancer. 1997;80(8 suppl):1546-1556. 113. Roudier MP, Morrissey C, True LD, et al. Histopathological assessment of prostate cancer bone osteoblastic metastases. J Urol. 2008;180(3):1154-1160. 114. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2(8):584-593. 115. Lynch CC. Matrix metalloproteinases as master regulators of the vicious cycle of bone metastasis. Bone. 2011;48(1):44-53. 116. Faccio R. Immune regulation of the tumor/bone vicious cycle. Ann N Y Acad Sci. 2011;1237:71-78. 117. Casimiro S, Guise TA, Chirgwin J. The critical role of the bone microenvironment in cancer metastases. Mol Cell Endocrinol. 2009;310(1-2):71-81. 118. Cook LM, Shay G, Aruajo A, et al. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metastasis Rev. 2014;33(2-3):511-525. 119. Lynch CC, Vargo-Gogola T, Martin MD, et al. Matrix metalloproteinase 7 mediates mammary epithelial cell tumorigenesis through the ErbB4 receptor. Cancer Res. 2007;67(14):6760-6767. 120. Thiolloy S, Edwards JR, Fingleton B, et al. An osteoblast-derived proteinase controls tumor cell survival via TGF-beta activation in the bone microenvironment. PLoS One. 2012;7(1):e29862. 121. Bruni-Cardoso A, Johnson LC, Vessella RL, et al. Osteoclast-derived matrix metalloproteinase-9 directly affects angiogenesis in the prostate tumor-bone microenvironment. Mol Cancer Res. 2010;8(4):459-470. 122. Tauro M, McGuire J, Lynch CC. New approaches to selectively target cancer associated matrix metalloproteinase activity. Cancer Metastasis Rev. 2014. [In press]. 123. Esposito M, Kang Y. Targeting tumor-stromal interactions in bone metastasis. Pharmacol Ther. 2014;141(2):222-233. 124. Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing [Erratum appears in N Engl J Med. 2012;367(10):976]. N Engl J Med. 2012;366(10):883-892. 125. Gerlinger M, Catto JW, Orntoft TF, et al. Intratumour heterogeneity in urologic cancers: from molecular evidence to clinical implications. Eur Urol. 2014. [Epub ahead of print]. 126. Drake JM, Graham NA, Lee JK, et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci U S A. 2013;110(49):E4762-E4769. 127. Gallaher J, Cook LM, Gupta S, et al. Improving treatment strategies for patients with metastatic castrate resistant prostate cancer through personalized computational modeling. Clin Exp Metastasis. 2014. [Epub ahead of print]. 128. Sweeney C, Chen YH, Carduccie MA, et al. Impact on overall survival (OS) with chemohormonal therapy versus hormonal therapy for hormone-sensitive newly metastatic prostate cancer (mPrCa): an ECOG-led phase 3 randomized trial. J Clin Oncol. 2014;32(5 suppl):LBA2. 129. Zhao B, Pritchard JR, Lauffenburger DA, et al. Addressing genetic tumor heterogeneity through computationally predictive combination therapy. Cancer Discov. 2014;4(2):166-174. 120 Cancer Control January 2015, Vol. 22, No. 1 Ten Best Readings Relating To Transfusion Medicine Glynn SA, Busch MP, Dodd RY, et al. Emerging infectious agents and the nation’s blood supply: responding to potential threats in the 21st century. Transfusion. 2013;53(2):438-454. This review includes a history and current status of transfusion-transmissible risks of HIV, hepatitis C virus, West Nile virus, Trypanosoma cruzi, Babesia, dengue viruses, and variant Creutzfeldt–Jakob disease prion. It addresses the challenges noted and measures taken by the field of transfusion medicine to minimize the risk of emerging infectious disease agents. Mitigation strategies such as specific donor deferral criteria, screening assays, discontinuation of blood collection in specific geographic areas, and pathogen reduction technologies are discussed. Price TH, McCullough J, Ness P, et al. A randomized controlled trial on the efficacy of high-dose granulocyte transfusion therapy in neutropenic patients with infection. Paper presented at: 56th ASH Annual Meeting and Exposition; San Francisco, CA; December 6–9, 2014. https://ash.confex. com/ash/2014/webprogram/Paper68775.html. Accessed November 20, 2014. The authors report on the results of the Resolving Infection in Neutropenia With Granulocytes study. The recently completed randomized controlled trial studied the efficacy of high-dose granulocyte transfusion therapy. provide clinical recommendations on the appropriate use of platelet transfusion in adults. The strongest recommendation was for the use of prophylactic transfusion to reduce the risk of spontaneous bleeding in hospitalized adult patients with therapy-induced hypoproliferative thrombocytopenia. For such patients, a threshold of 10,000/µL or less should be used, and doses up to a single apheresis unit or equivalent are sufficient; greater doses are not more effective, and lower doses are equally effective. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med. 2013;368(1):11-21. As compared with a liberal transfusion strategy, a restrictive strategy significantly improved outcomes in patients with acute upper gastrointestinal bleeding. Stanworth SJ, Estcourt LJ, Powter G, et al; TOPPS Investigators. A no-prophylaxis platelet-transfusion strategy for hematologic cancers. N Engl J Med. 2013;368(19):1771-1780. The results of this study support the need for the continued use of prophylaxis with platelet transfusion and show the benefit of such prophylaxis for reducing bleeding compared with no prophylaxis. Carson JL, Grossman BJ, Kleinman S, et al; Clinical Transfusion Medicine Committee of the AABB. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Intern Med. 2012;157(1):49-58. The AABB (formerly American Association of Blood Banks) developed this practice guideline to provide clinical recommendations for hemoglobin concentration thresholds and other clinical variables that trigger red blood cell (RBC) transfusions in hemodynamically stable adults and children. The strongest recommendation was for adhering to a restrictive transfusion strategy (7–8 g/dL) in stable patients in the hospital. Wandt H, Schaefer-Eckart K, Wendelin K, et al; Study Alliance Leukemia. Therapeutic platelet transfusion versus routine prophylactic transfusion in patients with haematological malignancies: an open-label, multicentre, randomised study. Lancet. 2012;380(9850):1309-1316. According to results from this study, therapeutic platelet transfusion could become the new standard of care after autologous stem cell transplantation; however, prophylactic platelet transfusion should remain the standard for patients with acute myeloid leukemia. This new strategy should be used by select hematology centers if their health care team is well educated and experienced in the new approach and can react in a timely way to the first signs of central nervous system bleeding. Kaufman RM, Djulbegovic B, Gernsheimer T, et al. Platelet transfusion: a clinical practice guideline from the AABB. Ann Intern Med. 2014. http://annals.org/article.aspx?articleid=1930861. Accessed November 20, 2014. The AABB developed this practice guideline to Steiner ME, Triulzi DJ, Assmann SF, et al. Randomized trial results: red cell storage age is not associated with a significant difference in multiple organ dysfunction score or mortality in transfused cardiac surgery patients. Paper presented at: AABB Annual Meeting; Philadelphia, PA; October 25–28, January 2015, Vol. 22, No. 1 Cancer Control 121 2014. http://onlinelibrary.wiley.com/doi/10.1111/ trf.12845/full. Accessed November 20, 2014. Cardiac patients often require multiple RBC units, so they may be exposed to units that have been stored the longest. This study aimed to determine whether a difference in patient outcomes occurred after the transfusion of units stored for 10 days or less versus units stored 21 days or longer. Holst LB, Haase N, Wetterslev J, et al; TRISS Trial Group; Scandinavian Critical Care Trials Group. Lower versus higher hemoglobin threshold for transfusion in septic shock. N Engl J Med. 2014;371(15):1381-1391. Among patients with septic shock, the rates of 90-day mortality and ischemic events as well as use of life support were similar among those assigned to blood transfusion at a higher hemoglobin threshold and those assigned to blood transfusion at a lower threshold; the latter group received fewer transfusions. Schwartz J, Winters JL, Padmanabhan A, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145-284. This Sixth Edition of the American Society for Apheresis (ASFA) Special Issue has further improved the process of using evidence-based medicine in the recommendations by consistently applying the category and GRADE system definitions and eliminate level of evidence criteria. This article consists of 78 fact sheets for therapeutic indications in ASFA categories I to IV and includes multiple clinical presentations and scenarios that are individually graded and categorized. 122 Cancer Control January 2015, Vol. 22, No. 1 Contact and General Information Cancer Control: Journal of the Moffitt Cancer Center is published by H. Lee Moffitt Cancer Center & Research Institute and is included in Index Medicus®/ MEDLINE® and EMBASE®/Excerpta Medica, Thomson Reuters Science Citation Index Expanded (SciSearch®) and Journal Citation Reports/Science Edition. Cancer Control currently has an impact factor of approximately 2.655. This peer-reviewed journal contains articles on the spectrum of actions and approaches needed to reduce the impact of human malignancy. Cancer Control is sent at no charge to approximately 15,000* medical professionals, including oncologists in all subspecialties, selected primary care physicians, medical researchers who specialize in oncology, and others who have a professional interest in cancer control. To Cancel a Subscription: Please send your cancellation request, including your name and address, to the Editorial Coordinator, as listed to the left. To Order an Individual, Institutional, or International Subscription: For information on subscription rates, please contact the Editorial Coordinator. To Request Permission to Reprint Copyrighted Materials: To request permission to reprint tables, figures, or materials copyrighted by Cancer Control, please contact the Editorial Coordinator. * This includes approximately 1,000 oncologists from over 85 countries. To Access the Journal Online: Most issues and supplements of Cancer Control are available at cancercontroljournal.org. To Change Your Mailing Address: Please provide your old address and your new address to: Veronica Nemeth, Editorial Coordinator Cancer Control Journal Moffitt Cancer Center MBC-JRNL 12902 Magnolia Drive Tampa, FL 33612 E-mail:[email protected] Fax: 813-449-8680 Phone:813-745-1348 To Inquire About Reprints: For information on obtaining reprints and pricing information, please contact the Editorial Coordinator. To Submit an Article for Publication: The editor welcomes submission of manuscripts pertaining to all phases of oncology care for possible publication in Cancer Control. Articles are subject to editorial evaluation and peer review. Author guidelines are available online at cancercontroljournal.org. Follow us on Twitter @MoffittResearch #MoffittCCJ Celebrating Over 20 Years of Publishing YOUR OPINION COUNTS! Access our readership survey at cancercontroljournal.org January 2015, Vol. 22, No. 1 Cancer Control 123 FACULTY POSITION: CUTANEOUS MEDICAL ONCOLOGIST Moffitt Cancer Center, an NCI-designated Comprehensive Cancer Center, is seeking a Medical Oncologist for its Cutaneous Oncology Program. A competitive salary package with excellent benefits, a high level of clinical resources, and outstanding infrastructural research support are available, including protected time for research endeavors. The prospective candidate will be appointed at the Assistant Member level or higher if warranted. Extensive Cancer Center Core facilities for translational research are available, and their use by the candidate for innovative clinical trials will be encouraged. The patient population at Moffitt Cancer Center is a diverse and outstanding resource for the conduct of clinical trials. At Moffitt, significant growth in clinical and translational research, laboratory space resources, and faculty recruitment will be a high priority in the next decade. In 2007, Moffitt established the Comprehensive Melanoma Research Center made possible by a generous philanthropic gift of $20.4 million from Donald A. Adam. This Center conducts research in melanoma and translates it into cutting-edge patient treatment. Moffitt was also recently awarded an NIH Specialized Program in Research Excellence (SPORE) grant for melanoma, and the prospective candidate will be expected to have a substantive role in the clinical and translational research activities of the SPORE and will have access to the career development and other developmental resources provided by the SPORE. Applicants must have a Florida medical license or be eligible for one, an MD, or MD/PhD and be board certified or eligible in internal medicine and board eligible/certified or equivalent in medical oncology. The applicant should be familiar with a multidisciplinary academic clinical practice setting. The successful candidate must have clinical expertise in melanoma and a desire to participate in and design clinical trials, including those involving drug development. Familiarity with other cutaneous malignancies besides melanoma is a plus. Background in clinical and/or translational research is essential, as is an interest in education and teaching. Knowledge of scientific research methods, knowledge of federal guidelines related to conducting clinical trials, knowledge of quality assurance, and excellent spoken and written communication skills are required. An opportunity exists to participate in the clinical activities of other Moffitt clinical programs as well. For inquiries about the position, contact Vernon K. Sondak, MD, Chair, Cutaneous Oncology Department, at [email protected] or 813-745-8788. To apply, visit our Web page at MOFFITT.org/careers. The H. Lee Moffitt Cancer Center & Research Institute, a rapidly growing NCI-designated Comprehensive Cancer Center, is committed to education through a wide range of residency and fellowship programs. The Cancer Center is composed of a large ambulatory care facility, a 206-bed hospital, with a 36-bed blood and marrow transplant program, 15 state-of-the-art operating suites, a 30-bed intensive care unit, a high-volume screening program, and a basic science research facility. The Moffitt Research Institute is composed of approximately 150 principal investigators, 58 laboratories, and 306,000 square feet of research space. The Moffitt Cancer Center is affiliated with the University of South Florida. Primary and secondary university appointments are available as applicable. Academic rank is commensurate with qualifications and experience. 124 Cancer Control January 2015, Vol. 22, No. 1 FACULTY POSITION: NEUROLOGIST Moffitt Cancer Center’s Neuro-Oncology Department is seeking a neurologist. The Neuro-Oncology Program employs an interdisciplinary approach, offering comprehensive therapy for patients with primary and metastatic tumors of the brain and spinal cord, as well as neurological complications of cancer and its treatments. In addition to focusing on the neurological complications of cancer and its treatment, the neurologist would provide in-house consultation for general neurological problems to the Cancer Center. The successful candidate will develop a strong, clinical, or translational program in general neurology in cancer. The ideal candidate will have significant expertise in general neurology in a cancer setting with an emphasis in neurology. Clinical research and the ability to work closely with an interdisciplinary team of experts, including neurosurgical oncology, neuropathology, neuroradiology, neuropsychology, and laboratory scientists, are required. Moffitt Cancer Center has strong preclinical programs in immunotherapy, drug discovery, genomics, cell-based therapies, and bioinformatics. There is also an extraordinary effort in personalized medicine partnering with the biotechnology/pharmaceutical industry. The Neuro-Oncology Program at Moffitt is a high-volume program, with approximately 500 new patients with brain tumors every year, and is active in the initiation and completion of numerous clinical trials with a well-developed clinical and translational research infrastructure. The Neuro-Oncology Program is an active participant in the National Comprehensive Cancer Network. Successful candidates must have a Florida medical license or be eligible for one, an MD, be board certified/ eligible in neurology, and fellowship trained in neurology. Experience in a clinical, multidisciplinary academic setting is preferred. A commitment to develop clinical research studies is required. The candidate should be experienced in performing and interpreting electroencephalography and electromyography. With a very active Cancer Spine Program and Neurosurgical Division, experience in physical and rehabilitation medicine would be desirable. For inquiries about the position, contact Peter Forsyth, MD, Chair, Department of Neuro-Oncology, at [email protected] or 813-745-3063. To apply, visit our Web page at MOFFITT.org/careers. Moffitt Cancer Center provides a tobacco-free work environment. It is an equal opportunity, affirmative action employer and a drug-free workplace. January 2015, Vol. 22, No. 1 Cancer Control 125 LEADING TREATMENT FOR WOMEN WITH CANCER At Moffitt, we offer a patient-centric approach coupled with the latest advances in breast and gynecologic oncology, taking into account fertility-sparing therapies and sexual health counseling. Our team utilizes comprehensive treatment plans and recovery programs to provide women with the best quality of life before, during and after cancer treatment. TO REFER A PATIENT, CALL 1-888-MOFFITT OR VISIT REFER2MOFFITT.com MOFFITT CANCER CENTER 12902 MAGNOLIA DRIVE, TAMPA, FL MOFFITT CANCER CENTER AT INTERNATIONAL PLAZA 4101 JIM WALTER BOULEVARD, TAMPA, FL 126 Cancer Control January 2015, Vol. 22, No. 1 REFER2MOFFITT.COM ACCESS REAL-TIME RECORDS AND PATIENT UPDATES AN ALL-IN-ONE RESOURCE SITE NOW AVAILABLE FOR PHYSICIAN PRACTICES As Florida’s largest multispecialty cancer center, Moffitt Medical Group welcomes you and your staff to our physician portal resource site. The site is a valuable tool for following your patients’ progress as well as accessing Moffitt’s clinical services. • Real-time shared patient record information • Search the physician directory • Process easy online referrals • View treatment & procedure information • Access open clinical trials • Find lectures & CME schedules TO REFER A PATIENT, CALL 1-888-MOFFITT OR VISIT REFER2MOFFITT.com MOFFITT CANCER CENTER 12902 MAGNOLIA DRIVE, TAMPA, FL MOFFITT CANCER CENTER AT INTERNATIONAL PLAZA 4101 JIM WALTER BOULEVARD, TAMPA, FL MOFFITT CANCER CENTER, TAMPA, FL 1-888-MOFFITT | MOFFITT.org January 2015, Vol. 22, No. 1 Cancer Control 127 advances in the management of multiple Save the Date! presents Advances in the Management of Multiple Myeloma March 6—7, 2015 Loews Don Cesar Hotel St. Petersburg Beach, FL Course Directors: Melissa Alsina, MD, Rachid Baz, MD, and Kenneth H. Shain, MD, PhD Moffitt Cancer Center, Tampa, FL Conference Overview: The Advances in the Management of Multiple Myeloma conference is designed to foster the exchange of the most recent advances in the biology and treatment of multiple myeloma. National and international leading experts in the field will present in a format that promotes discussion and interaction with participants. Target Audience: This educational program is directed toward hematologists, medical and surgical oncologists, and BMT physicians who diagnose, treat, and manage multiple myeloma. Other health care professionals interested in the diagnosis, treatment, and care of patients with multiple myeloma are also invited to attend. To be added to the conference mailing list, contact: Moffitt Cancer Center | Marsha Moyer, MBA | 813-745-2286 | [email protected] 128 Cancer Control January 2015, Vol. 22, No. 1 3 RD A N N U A L S TAT E - O F -T H E - A R T Neuro-Oncology CONFERENCE SAVE THE DATE March 19—20,2015 Sheraton Sand Key Clearwater Beach, FL CONFERENCE HIGHLIGHTS • Renownedfaculty representingthecountry’s leadinginstitutions •Updatesonbrainand spinetumors •Preconferencedinner presentationonMarch19 •Interactivecasepresentations withdiscussion • Nursetracksession • Callforabstracts COURSE DIRECTORS Peter Forsyth, MD • Frank Vrionis, MD, MPH, PhD CONFERENCE CONTACT [email protected] • MOFFITT.org/NeuroOncology2015 PROVIDED BY January 2015, Vol. 22, No. 1 Cancer Control 129 Save the Date 9TH ANNUAL Business of Biotech Conference Collaborate to Innovate Friday, April 17, 2015 7:45 AM — 4:00 PM Vincent A. Stabile Research Building Moffitt Cancer Center | Tampa, FL MAKING THE VISION OF CURING CANCER A REALITY As the only NCI-designated Comprehensive Cancer Center based in Florida, Moffitt is a national innovator in research and is at the forefront of transforming cancer care for better patient outcomes, prevention and cures. KEYNOTE SPEAKER: Henri A. Termeer Former CEO, Chairman and President of Genzyme Corporation. He retired following the acquisition of Genzyme by Sanofi in a transaction valued at more than $20 billion. Register today for the Business of Biotech 2015 conference by using the QR code. TO LEARN MORE ABOUT OTMC ALLIANCES, EMAIL [email protected] 130 Cancer Control January 2015, Vol. 22, No. 1


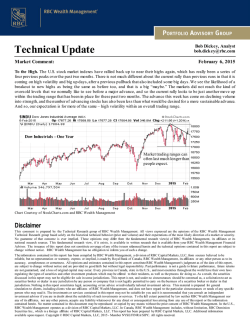



© Copyright 2026