
biote-2016 - Read the Docs
biote-2016
Release
O. Maya
September 02, 2016
Antes de empezar
1
Índice
1.1 Linux . . . . . . . . . .
1.2 MacOS . . . . . . . . .
1.3 Windows . . . . . . . .
1.4 GNU/Linux . . . . . . .
1.5 Terminal . . . . . . . .
1.6 Entorno . . . . . . . . .
1.7 Preprocesamiento . . .
1.8 Asignación taxonómica
1.9 Asignación funcional . .
1.10 Install . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
3
.
3
. 49
. 52
. 53
. 56
. 76
. 80
. 93
. 98
. 100
i
ii
biote-2016, Release
Este es un curso que me hubiera gustado recibir cuando empecé a usar GNU/Linux en el ámbito bioinformático,
especialmente en el análisis de datos “metagenómicos”.
Antes de empezar
1
biote-2016, Release
2
Antes de empezar
CHAPTER 1
Índice
El curso está organizado en las siguientes secciones:
• Antes de empezar
• Sistema UNIX
• Análisis 16S
1.1 Linux
Un sistema Linux requiere de al menos tres particiones:
• Una partición primaria para el directorio raíz: /
• Una partición lógica para el área de intercambio: swap
• Y una partición lógica para los archivos: /home
1.1.1 Tipos de particiones
Primaria Es la partición necesaria que es necesaria para almacenar y bootear el sistema operativo.
Extendida Partición que puede ser subdividida en unidades lógicas y es vista como un contenedor de ellas. No es
booteable. Puede ser dividida hasta en 24 particiones lógicas.
Lógica Las particiones lógicas son aquellas creadas en la partición extendida.
3
biote-2016, Release
Ejemplo Servidor Debian “Amidala”:
1.1.2 Instalación de Ubuntu
Bipartición
Pasos para instalar Ubuntu en un sistema con Windows:
4
Chapter 1. Índice
biote-2016, Release
1.1. Linux
5
biote-2016, Release
6
Chapter 1. Índice
biote-2016, Release
1.1. Linux
7
biote-2016, Release
8
Chapter 1. Índice
biote-2016, Release
1.1. Linux
9
biote-2016, Release
10
Chapter 1. Índice
biote-2016, Release
1.1. Linux
11
biote-2016, Release
12
Chapter 1. Índice
biote-2016, Release
1.1. Linux
13
biote-2016, Release
14
Chapter 1. Índice
biote-2016, Release
1.1. Linux
15
biote-2016, Release
16
Chapter 1. Índice
biote-2016, Release
1.1. Linux
17
biote-2016, Release
18
Chapter 1. Índice
biote-2016, Release
1.1. Linux
19
biote-2016, Release
20
Chapter 1. Índice
biote-2016, Release
1.1. Linux
21
biote-2016, Release
22
Chapter 1. Índice
biote-2016, Release
1.1. Linux
23
biote-2016, Release
Nativa
Pasos para instalar Ubuntu de manera exclusiva:
24
Chapter 1. Índice
biote-2016, Release
1.1. Linux
25
biote-2016, Release
26
Chapter 1. Índice
biote-2016, Release
1.1. Linux
27
biote-2016, Release
28
Chapter 1. Índice
biote-2016, Release
1.1. Linux
29
biote-2016, Release
30
Chapter 1. Índice
biote-2016, Release
1.1. Linux
31
biote-2016, Release
32
Chapter 1. Índice
biote-2016, Release
1.1. Linux
33
biote-2016, Release
34
Chapter 1. Índice
biote-2016, Release
1.1. Linux
35
biote-2016, Release
36
Chapter 1. Índice
biote-2016, Release
1.1. Linux
37
biote-2016, Release
38
Chapter 1. Índice
biote-2016, Release
1.1. Linux
39
biote-2016, Release
40
Chapter 1. Índice
biote-2016, Release
1.1. Linux
41
biote-2016, Release
42
Chapter 1. Índice
biote-2016, Release
1.1. Linux
43
biote-2016, Release
44
Chapter 1. Índice
biote-2016, Release
1.1. Linux
45
biote-2016, Release
46
Chapter 1. Índice
biote-2016, Release
1.1. Linux
47
biote-2016, Release
1.1.3 Configuración de Ubuntu
Descargar y ejecutar el siguiente script:
wget --trust-server-names https://goo.gl/cEtm0J
chmod +x post_install_ubuntu1604.sh
sudo ./post_install_ubuntu1604.sh
Aparecerá el siguiente mensaje:
# -------------------------------------------------------------#
#
#
# Ubuntu 16.04 LTS Post-installation script - CINVESTAV 2016 #
#
#
# -------------------------------------------------------------#
Finishing the installation may take several minutes
Do you want to continue (y/n)?
Al finalizar tendremos nuestro sistema listo para utilizar.
48
Chapter 1. Índice
biote-2016, Release
1.2 MacOS
En Mac, el shell está disponible a través de la Terminal en Applications -> Utilities -> Terminal. Arrástrala a tu Dock
para fácil acceso.
1.2.1 XCode
En la terminal copia y pega el siguiente comando:
xcode-select --install
Te aparecerá el siguiente mensaje en tu pantalla. Click Install cuando aparezca.
“Lee” y acepta el License Agreement ;)
1.2. MacOS
49
biote-2016, Release
Una vez que el software esté instalado, Done. ¡Listo!
50
Chapter 1. Índice
biote-2016, Release
1.2.2 Homebrew
Homebrew es un gestor de paquetes para OS X. Para instalar escribe en una terminal:
/usr/bin/ruby -e "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/master/install)"
Puedes instalar algunos de los comandos que utilizaremos en este curso:
brew install wget htop vim git python
XQuartz
QIIME para Mac utiliza librerías de XQuartz X11, por lo que es necesario instalarlas. Descarga el paquete XQuartz
X11 e instálalo. Dichas librerías proporcionan una versión de X.org X Window System para MacOS.
Dependencias de R
Descarga e instala el PKG de R. Posteriormente, en Terminal, iniciar R e instalar las dependencias necesarias para
MacQIIME.
R #inicia la terminal de R
install.packages(c('randomForest', 'ape', 'vegan', 'optparse', 'gtools', 'klaR', 'RColorBrewer', 'bio
source("http://bioconductor.org/biocLite.R") #define el repositorio de Bioconductor
biocLite(c('metagenomeSeq', 'DESeq2')) #instala paquetes de Bioconductor
QIIME
Para instalar MacQIIME, la versión QIIME para MacOS, es necesario seguir los siguientes pasos:
cd $HOME
wget ftp://ftp.microbio.me/pub/macqiime-releases/MacQIIME_1.9.1-20150604_OS10.7.tgz
tar -xvf MacQIIME_1.9.1-20150604_OS10.7.tgz
cd MacQIIME_1.9.1-20150604_OS10.7/
./install.s && cd .. && rm -r MacQIIME_1.9.1-20150604_OS10.7/
1.2. MacOS
51
biote-2016, Release
1.3 Windows
En Windows, utilizaremos MobaXTerm para explorar la parte correspondiente a Shell. La mayoría de las herramientas
utilizadas para el manejo de datos de NGS están optimizadas para sistemas operativos UNIX.
1.3.1 MobaXterm
Descargar e instalar MobaXterm.
52
Chapter 1. Índice
biote-2016, Release
1.4 GNU/Linux
1.4.1 Qué es GNU/Linux
En términos simples, Linux es un sistema operativo. En la siguiente figura se muestra un diagrama general de cómo
está constituido un sistema GNU/Linux.
Kernel
El kernel es principalmente responsable de cuatro principales funciones:
• Manejo del sistema de memoria
• Manejo de programas de software
• Manejo de hardware
• Manejo de sistema de archivos
1.4. GNU/Linux
53
biote-2016, Release
Sistema de ficheros
El sistema de ficheros está constituido principalmente por los siguientes directorios:
Directorio
/
/bin
/boot
/dev
/etc
/home
/lib
/media
/mnt
/opt
/proc
/root
/sbin
/run
/sys
/tmp
/usr
/var
Uso
raiz del directorio virtual
directorio de binarios, donde se almacenan la mayor parte de los programas de GNU/Linux
directorio boot, donde están los archivos de arranque
directorio de dispositivos, donde Linux crea los nodos de dispositivos
configuración del sistema de directorio de archivos
directorio home, donde Linux crea los directorios de usuarios
directorio de librerías, donde se almacenan librerías del sistema y de aplicaciones
directorio de media, un lugar comun para puntos de montaje de dispositivos extraibles
directorio de montaje, otro lugar común para puntos de montaj de dispositivos extraibles
directorio opcional, frecuentemente utilizado para software de terceros
directorio de procesos, donde se almacena información actual de hardware y de procesos
directorio home del superusuario root
directorio de sistema binario, ahí se almacenan utilidades GNU con nivel de administrador
directorio de corrida, ahí se almacenan datos de corrida durante procesos del sistema
directorio de sistema, donde se almacena información del sistema de hardware
directorio temporal, donde se crean y destruyen archivos temporales de trabajo
directorio binario de usuario, dondese guardan los programas ejecutable de usuario
directorio variable, para archivos que cambian frecuentemente, como archivos de registro
Antes de los sistemas gráficos, la única manera de interactuar con los sistemas Linux era a través de texto en una
interfaz de linea de comandos (CLI) provista por el shell.
1.4.2 Distribuciones GNU/Linux
Una distribución Linux (coloquialmente llamada distro) es una distribución de software basada en el
núcleo Linux que incluye determinados paquetes de software para satisfacer las necesidades de un grupo
específico de usuarios, dando así origen a ediciones domésticas, empresariales y para servidores.
54
Chapter 1. Índice
biote-2016, Release
1.4.3 Shell
El shell es un programa que toma los comandos del teclado y los comunica al sistema operativo para que se lleven a
cabo. La mayoría de las distribuciones Linux proporcionan un programa shell del GNU Project llamado bash.
A continuación se muestra un directorio parcial con dos usuarios en /home. Por ejemplo, la ruta para el usuario Dan
sería /home/dan/
NOTA: es importante distinguir entre /home/dan, /Home/Dan/, /home/Dan/. Linux es sensible a mayúsculas y minúsculas.
Los archivos personales de usuario a menudo se encuentran en /home/my-user-name. Existen muchas maneras de
1.4. GNU/Linux
55
biote-2016, Release
localizarlos o de referir al directorio de home.
cd Sin argumentos, el comando cd nos lleva directamente al directorio home.
Variable HOME La variable de entorno HOME contiene el hombre de nuestro directorio home.
$ echo $HOME
/home/otoniel
El comando `echo` imprime el argumento de la variable `$HOME`
~ Cuando se usa en lugar de directorio, la tilde es interpretada por el shell como el nombre del directorio home.
$ echo
/home/otoniel
Otra manera de localizar home es con el comando pwd
$ pwd
/home/otoniel
“print working directory”
Emuladores de terminal
Cuando se utiliza una interfaz gráfica se necesita un programa llamado emulador de terminal para interactuar con el
shell. Los más populares son gnome-terminal, konsole, terminator.
1.5 Terminal
Abrimos el emulador de terminal de nuestra preferencia. Deberíamos ver algo como esto:
[zorbax@linux ~]$
Esto se llama el prompt del shell y aparece dondequiera que el shell esté listo para aceptar un input. Algunas veces
puede variar la apariencia, dependiendo de la distribución, pero usualmente incluirá username@machinename, seguido
del directorio actual de trabajo (lo veremos más adelante) y un signo de dolar. Si el útimo caracter del prompt es un hash
(#) en lugar de iun signo de dolar, la terminal tiene privilegios de superusuario. Probemos la terminal, introduzcamos
algunas letras sin sentido:
[zorbax@linux ~]$ gfhbfhbn3tu
Como no tienen ningún sentido, el shell nos los hará saber y nos dará otra oportunidad.
bash: gfhbfhbn3tu: command not found
[zorbax@linux ~]$
1.5.1 Navegación
Ahora, lo primero que necesitamos aprender es cómo navegar en el sistema de archivos Linux:
• pwd — Imprime el directorio actual de trabajo.
• cd — Cambia de directorio.
56
Chapter 1. Índice
biote-2016, Release
• ls — Lista los archivos de directorios.
Como Windows, un sistema operativo Unix-like, como Ubuntu, orgnaiza sus archivos en lo que se llama estructura
jerárquica de directorios. Esto significa que están organizados en un sistema ramificado de directorios ( o folders),
los cuales pueden contener archivos u otros directorios.El primer directorio en el sistema de archivos es llamado el
directorio raiz (root).
pwd
Cuando entramos al sistema, o cuando abrimos un emulador de terminal el directorio de trabajo actual será siempre el
directorio Home. Para obtener la ruta completa del lugar donde nos encontremos utilizaremos el comando pwd (print
working directory):
[zorbax@linux ~]$ pwd
/home/zorbax
Para listar los archivos y directorios en el directorio de trabajo actual usamos el comando ls:
[zorbax@linux ~]$ ls
bin Desktop Documents
Downloads
Images
Music
Videos
cd
Para cambiar el directorio de trabajo se utiliza el comando cd seguido de la ruta (pathname) de directorio de trabajo
deseado. Una ruta es la dirección completa que tomaremos entre las ramas del directorio para llegar al directorio
deseado. Las rutas pueden ser absolutas o relativas.
Una ruta absoluta comienza con el directorio root y sigue el arbol de directorio hasta la ruta del directorio deseado.
Por ejemplo, la ruta /usr/bin, donde la mayoría de los progrmas del sistema son instalados, indica que en el
directorio raiz (root, representado por un / ), hay un directorio llamado usr que contiene un directorio llamado bin.
[zorbax@linux ~]$ cd /usr/bin
[zorbax@linux bin]$ pwd
/usr/bin
[zorbax@linux bin]$ ls
... ...
Hemos cambiado del directorio actual a /usr/bin. Cuando cambiamos de directorio, el prompt cambió automáticamente
y muestra el nombre del directorio actual.
Una ruta relativa empieza desde el directorio actual hacia el directorio de destino. Para lograrlo utiliza unos símbolos
especiales que representan la posición relativa en el árbol del sistema de archivos. Estos símbolos son . (punto) y el
.. (punto punto).
El símbolo . refiere al directori de trabajo actual y .. refiere al directorio de trabajo parental. Funciona de la siguiente
manera, cambiemos nuevamente a /usr/bin:
[zorbax@linux ~]$ cd /usr/bin
[zorbax@linux bin]$ pwd
/usr/bin
Si deseamos cambiar al directorio parental de /usr/bin, el cual es /usr, podemos hacerlo de dos maneras: usando
una ruta absoluta:
[zorbax@linux bin]$ cd /usr
[zorbax@linux usr]$ pwd
/usr
1.5. Terminal
57
biote-2016, Release
o con una ruta relativa:
[zorbax@linux bin]$ cd ..
[zorbax@linux usr]$ pwd
/usr
Los dos métodos producen resultados idénticos. ¿Cuál usar? Aquel que requiera teclear menos. De la misma manera
podemos cambiar de directorio de trabajo de /usr a /usr/bin de dos maneras diferentes, usando una ruta absoluta:
[zorbax@linux usr]$ cd /usr/bin
[zorbax@linux bin]$ pwd
/usr/bin
O una ruta relativa:
[zorbax@linux usr]$ cd ./bin
[zorbax@linux bin]$ pwd
/usr/bin
Algo importante que debo aclarar es que en la mayoría de los casos se omite ./ porque está implícito. Escribiendo en
la termina:
[zorbax@linux usr]$ cd bin
se obtiene el mismo resultado. En general, si no se especifica la ruta a algo se asume que se trata del directorio actual.
Algunos atajos útiles:
• cd Cambia del directorio de trabajo actual al directorio home
• cd - Cambia del directorio de trabajo actual al directorio de trabajo anterior.
• cd ~ Cambia del directorio de trabajo actual al directorio home del usuario actual, cd ~username cambia
al home de cualquier otro usuario.
Nix:
# Filenames
# Hiden filenames
# Extensions
1.5.2 Exploración del sistema
Ahora que nos sabemos mover en la terminar aprenderemos otros comandos básicos:
• ls — Listar el contenido de directorios.
• file — Determinar el tipo de archivo.
• less — Ver el contenido de un archivo.
ls
El comando más usado es ls y por buenas razones. Gracias a ls podemos ver el contenido de un directorio y
determinar una variedad de atributos importantes de archivos y directorios.
[zorbax@linux ~]$ ls
bin Desktop Documents
Downloads
Images
Music
Videos
Además del directorio actual de trabajo, podemos especificar el directorio a listar:
58
Chapter 1. Índice
biote-2016, Release
[zorbax@linux ~]$ ls /usr
bin etc games include lib
lib32
local
sbin
share
src
O incluso podemos listar varios directorios. En este caso listamos el directoro home de nuestro usuario ( representado
por el caracter ~) y el directorio /usr.
[zorbax@linux ~]$ ls ~ /usr
/home/zorbax:
bin Desktop Documents Downloads
/usr:
bin etc
games
include
lib
Images
lib32
local
Music
sbin
Videos
share
src
También podemos cambiar el formato de salida que releve un poco más de detalle:
[zorbax@linux~]$ ls -l
total 84
drwxr-xr-x 57 zorbax 4096
drwxr-xr-x 2 zorbax 16384
drwxr-xr-x 6 zorbax 12288
drwxr-xr-x 6 zorbax 32768
drwxr-xr-x 2 zorbax 4096
drwxr-xr-x 10 zorbax 4096
drwxr-xr-x 2 zorbax 4096
ago
ago
ago
ago
ago
ago
ago
18
20
17
22
18
18
19
17:43
20:57
12:35
09:33
11:17
11:28
23:12
bin
Desktop
Documents
Downloads
Images
Music
Videos
Agregando la opción -l al domando cambiamos el formato a large.
Opciones y argumentos
Algo importante a considerar acerca de cómo funcionan la mayoría de los comandos es que a menudo son seguidos de
una o más opciones que modifican su comportamiento, y además, uno o más argumentos, los ítems sobre los cuales
los comandos actuan. Así, la mayoría de los comandos lucen de la siguiente manera:
command -options arguments
La mayoría de los comandos usan opciones que corresponden a un solo caracter precedido por un guión, por ejemplo
-l. Aunque los comandos del GNU Project también aceptan opciones en versión larga, que consiste en una palabra
precedidad por dos guiones. Por ejemplo, en el comando ls -lt, -l produce una salida en formato detallado y -t
ordena los resultados por fecha de la última modificación.
[zorbax@linux~]$ ls -lt
total 84
drwxr-xr-x 6 zorbax 32768
drwxr-xr-x 2 zorbax 16384
drwxr-xr-x 2 zorbax 4096
drwxr-xr-x 57 zorbax 4096
drwxr-xr-x 10 zorbax 4096
drwxr-xr-x 2 zorbax 4096
drwxr-xr-x 6 zorbax 12288
ago
ago
ago
ago
ago
ago
ago
22
20
19
18
18
18
17
09:33
20:57
23:12
17:43
11:28
11:17
12:35
Downloads
Desktop
Videos
bin
Music
Images
Documents
Agregamos la opción --reverse para ordenar de forma inversa el orden listado a detalle (–reverse es equivalente a
-r en la versión corta).
[zorbax@linux~]$ ls -lt --reverse
total 84
drwxr-xr-x 6 zorbax 12288 ago 17
drwxr-xr-x 2 zorbax 4096 ago 18
drwxr-xr-x 10 zorbax 4096 ago 18
drwxr-xr-x 57 zorbax 4096 ago 18
drwxr-xr-x 2 zorbax 4096 ago 19
1.5. Terminal
12:35
11:17
11:28
17:43
23:12
Documents
Images
Music
bin
Videos
59
biote-2016, Release
drwxr-xr-x
drwxr-xr-x
2 zorbax 16384 ago 20 20:57 Desktop
6 zorbax 32768 ago 22 09:33 Downloads
Personalmente las opciones que más utilizo son: ls -ltrh y ls -ltrhR.
file
Mientras exploramos el sistema, es útil saber qué tipo de archivo contiene. Para realizarlo utilizaremos el comando
file. El nombre del archivo no requiere reflejar su contenido, así puede haber un archivo txt y lo puedo nombrar
como mp3.
[zorbax@linux~]$ echo "Esto es un simulacro" > the_next_friday.mp3
[zorbax@linux~]$ file the_next_friday.mp3
the_next_friday.mp3: ASCII text
[zorbax@linux~]$ file song.mp3
song.mp3: Audio file with ID3 version 2.4.0, contains: MPEG ADTS, layer III, v1,
64 kbps, 48 kHz, St
[zorbax@linux~]$ file bin/fastqc
bin/fastqc: symbolic link to /home/zorbax/bin/FastQC/fastqc
[zorbax@linux~]$ file seqtk
seqtk: ELF 64-bit LSB executable, x86-64, version 1 (SYSV), dynamically linked, interpreter /lib64/ld
Hay muchos tipos de archivos. De hecho, una de las ideas de los sistemas operativos tipo Unix es que “todo es un
archivo”.
less
El comando less es un programa para visualizar texto. En el sistema Linux hay muchos archivos que contienen texto
legible. El uso del comando es:
less filename
Una vez iniciado, el programa permitirá ver todo el texto de principio a fin desplazándonos con up-down. Para salir
hay que presionar la tecla Q.
less /etc/passwd
1.5.3 Manipulación de archivos
Hasta este punto estamos listos para algo de trabajo en el MundoReal™. A continuación veremos los siguientes
comandos:
• mkdir — Crear directorios.
• cp — Copiar archibos y directorios.
• mv — Mover/renombrar archivos y directorios.
• rm — Eliminar archivos y directorios.
• ln — Crear vínculos simbólicos.
Estos cinco comandos son los comandos más frecuentes. Se utilizan para manejar archivos y directorios. Para ser
honesto, algunas tareas realizadas por estos comandos son más fáciles de realizar con un gestor gráfico de archivos.
Con el gestor de archivos sólo arrastramos y soltamos en un directorio, copiamos, pegamos, borramos archivos y así.
60
Chapter 1. Índice
biote-2016, Release
Sin embargo, el poder y flexibilidad que dan estos comandos es la razón principal por la que se siguen usando. Es fácil
realizar acciones sencillas en el gestor de archivos, pero tareas más complicadas pueden realizarse de manera más fácil
con programas de la linea de comandos.
Comodines
Antes de empezar a usar los comandos es necesario hablar de aquellas características que hacen que los comandos
sean poderosos. La shell usa nombres de archivos para actuar, y provee además caracteres especiales para ayudar a
especificar rápidamente grupos específicos de Estos caracteres especiales son llamados comodines. Usando dichos
comodines (también llamados globbing) permite seleccionar nombres de archivo basadas en patrones de caracteres.
Los dos comodines más comunes son: *, cualquier caracter y ?, cualquier caracter único. Por ejempo:
• * -> Todos los archivos
• g* -> Todos los archivos que comiencen con g
• *fa -> Todos los archivos que terminen con fa
• b*.txt -> Cualquier archivo que comience con b seguido de cuakquier caracter y termina con .txt
mkdir
El comando mkdir sirve para crear directorios. Funciona de la siguiente manera:
mkdir directory...
Cuando escribo puntos suspensivos después de un argumentos en la descripción del comando, como arriba, quiere
decir que el argumento puede ser repetido, en este caso:
mkdir dir1
creará un directorio llamado dir1, mientras que:
mkdir dir1 dir2 dir3
creará tres directorios llamados, dir1, dir2, dir3, respectivamente.
Además se puede crear directorios anidados con la opción -p:
mkdir -p this/is/a/nested/folder
mkdir -p /this/is/a/nested/folder #Este comando dará error ¿por qué?
También se pueden crear directorios con base en un número consecutivo:
mkdir sample{01..10}
Creará diez folders, desde el folder sample01 hasta sample10.
cp
El comando cp copia archivos y directorios. Se puede utilizar de dos maneras diferentes. Para copiar un solo archivo
o directorio item1 al archivo o directorio item2 y:
cp item1 item2
para copiar multiples ítems (archivos o directorios) a un directorio.
cp item... directory
Las opciones más comunes son:
1.5. Terminal
61
biote-2016, Release
• -i, Antes de sobreescribir un archivo existente, pregunta al usuario por confirmación. Si no se especifica esta
opción cp sobreescribirá silenciosamente los archivos.
• -r, copia recursiva de directorios y su contenido. Opción requerida para copiar directorios.
• -u, cuando se copian archivos de un directorio a otro, copia sólo los archivos que, o no existan o sean más
nuevos que los archivos existentes correspondientes en el directorio de destino.
• -v, Despliega información de proceso de copiado.
Ejemplos:
Copiar el archivo file1 al archivo file2. Si el archivo file2 extiste, sobreescribirá su contenido con el contenido del
archivo file1. Si el archivo file2 no existe, se creará:
cp file1 file2
Lo mismo de arriba, excepto que si el archivo file2 existe, el usuario es notificado antes de sobreescribir el archivo:
cp -i file1 file2
Copiar archivo file1 y el archivo file2 en el directorio dir1. Es necesario que el directorio dir1 exista:
cp file1 file2 dir1
Usando un comodín, todos los archivos del directorio dir1 son copiados en dir2. El directorio dir2 debe de existir:
cp dir1/* dir2
Copia el directorio dir1 (y su contenido) al directorio dir2. Si el directorio dir2 no existe, se creará y tendrá el mismo
contenido que el directorio dir1:
cp -r dir1 dir2
mv
El comando mv renombra y mueve archivos, dependiendo de cómo es utilizado. Em ambos casos el archivo original
dejará de existir después de ejecutarlo. Se puede utilizar en la misma manera que cp, para mover o renombrar archivos
o directorio item1 a item2:
mv item1 item2
o mover uno o más ítems de un directorio a otro.
mv item... directory
Algunas opciones de mv las comparte con cp:
• -i, Antes de mover un archivo existente, pregunta al usuario por confirmación. Si no se especifica esta opción
mv sobreescribirá silenciosamente los archivos.
• -u, cuando se mueven archivos de un directorio a otro, mueve sólo los archivos que, o no existan o sean más
nuevos que los archivos existentes correspondientes en el directorio de destino.
• -v, Despliega información de proceso de mv.
Ejemplos:
Mover archivo file1 al archivo file2. Si el archivo file2 existe, se sobreescribirá con el contenido del archivo file1. Si el
archivo file2 no existe se creará. En ambos casos el archivo file1 deja de existir.
62
Chapter 1. Índice
biote-2016, Release
mv file1 file2
Al igual que el ejemplo anterior, excepto que si el archivo file2 existe, el usuario es notificado antes de sobreescribir el
archivo:
mv -i file1 file2
Mover los archivos file1 y file2 al directorio dir1. El directorio dir1 debe de existir.
mv file1 file2 dir1
Mueve el directorio dir1 ( y su contenido) al directorio dir2. Si el directorio dir2 no existe, crea el directorio dir2,
mueve el contenido del directorio dir1 al directorio dir2, y borra el directorio dir2.
mv dir1 dir2
rm
El comando rm se utiliza para borrar archivos y directorios. Como esta linea, donde item es el nombre de uno o más
archivos o directorios:
rm item...
¡¡¡PRECAUCIÓN CON rm!!!
En los sistemas operativos tipo Unix, como el GNU/Linux, no existe el comando undo, o ctrl + Z, por lo menos en la
terminal. Una vez que se borra algo con rm es irrecuperable. Linux asume que eres inteligente y sabes lo que estás
haciendo (sic). Tengan cuidado con los comodines. Esto le sucedió al primo de un amigo: digamos que se requiere
borrar los archivos FASTA de nuestro directorio, basta con:
rm *.fasta
lo cual es correcto, pero si accidentalmente se coloca un espacio entre * y .fasta
rm * .fasta
el comando rm borraré todos los archivos del directorio y dara error debido a que no encuentra archivos con la
extensión FASTA.
TIP: siempre que utilicen comodines con rm (además de revisar cuidadosamente mientras se escribe), prueben el
comodín con ls. Esto les mostrará los archivos a borrar. Luego presionar la flecha up para llamar nuevamente el
comando y reemplazar ls con rm.
Las opciones más comunes son:
• -i, antes de borrar un archivo pregunta al usuario por confirmación. Si no se especifica se borrarán los archivos
de manera silenciosa.
• -r, borra recursivamente los directorios. Esto significa que si el directorio tiene subdirectorios, éstos serán
borrados también. Para borrar un directorio esta opción debe de especificarse.
• -f, Ignora los archivos no existentes y no pregunta nada.
• -v, Despliega mensajes informativos conforme se lleva acabo el proceso de borrado.
Manipulating Files and Directories
Ejemplos:
Borrar el archivo file1 silenciosamente.
1.5. Terminal
63
biote-2016, Release
rm file1
Antes de borrar el archivo file1 notifica al usuario por confirmación.
rm -i file1
Borra el archivo file1 y el directorio dir1 y su contenido.
rm -r file1 dir1
Lo mismo que el anterior, excepto que si tanto el archivo file1 o el directorio dir1 no existe, rm continuará silenciosamente.
rm -rf file1 dir1
¿Cuál sería el inconveniente de ejecutar rm -rf /*? ¿Por qué?
ln
Quizá cuando estemos navegando en los directorios encontraremos una entrada como esta, por ejemplo en
$HOME/bin:
lrwxrwxrwx 1 zorbax 30 oct 28
2014 fastqc -> /home/zorbax/bin/FastQC/fastqc
La primera letra de los atributos es l, y pareciera ser que el archivo tiene dos nombres, uno en $HOME/bin y otro en
$HOME/bin/FastQC/fastqc. Este tipo de archivos es llamdo enlaces simbólicos ( soft link o también symlink).
En la mayoría de los sistemas Unix es posible tener un archivo referenciado con múltiples nombres. Ésta es una
característica muy útil ya que permite tener el archivo disponible sin necesidad de copiar nuevamente el archivo.
Suponiendo que tenemos archivos fastq provenientes de secuenciación masiva. Los enlaces simbólicos nos permitirá
ir renombrando los archivos sin necesidad de modificar los datos crudos. Además, nos permite ‘copiar’, como enlaces
simbólicos, en diferentes carpetas sin necesidad de duplicar archivos y sobre todo sin necesidad de generar más espacio
en el disco duro.
[zorbax@linux raw_data]$ pwd
/home/zorbax/Documents/raw_data
[zorbax@linux data]$ ls
... ... *fastq files ... ...
Supongamos que quiero trabajar en el directorio analysis y necesito que los archivos fastq estén en
/home/zorbax/Documents/Dropbox/analysis/qiime/input:
cd /home/zorbax/Documents/Dropbox/analysis/qiime/input
ln -s /home/zorbax/Documents/raw_data/*fastq .
La estructura del comando es:
ln -s /path/to/file /path/to/symlink
Los enlaces simbólicos se borrar con rm y al hacerlo unicamente de borra el enlace, no el archivo.
1.5.4 Redireccionamiento I/O
Una de las mejores características de la linea de comandos es la redirección I/O (input-output). Esta características
implica que se puede redireccionar el input y el output de comandos hacia y desde harchivos, así como conectar multiples comandos para generar útiles lineas de procesos - *pipelines - de comandos. Para descubrir dicha característica
debemos de conocer los siguientes comandos:
64
Chapter 1. Índice
biote-2016, Release
• cat — Concatena archivos.
• sort — Ordena lineas de texto.
• uniq — Reporta u omite lineas repetidas.
• wc — Imprime la cuenta de salto de linea, palabra y byte de cada linea.
• grep — Imprime linead que coninciden con un patrón.
• head — Imprime en pantalla la salida de la primera parte de un archivo.
• tail — Imprime la última parte de un archivo.
• tee — Lee de un input estándar y escribe a un output estándar y a archivos.
Entrada, salida y error estándar
Muchos de los programas que utilizamos producen una salida-output de alguna clase. Esta salida-output frecuentemente consiste en dos tipos. Primero, tenemos el resultado del programa, el cual es, los datos que el programa está
diseñado a producir. Segundo, tenemos mensajes de estatus y de error que nos dice cómo está llendo el programa.
Si observamos el comando ls, vemos que muestra su resultado y su mensaje de error en la pantalla. Manteniendo
en mente lo que escribí hace un momento de que “todo es un archivo”, el programa ls envía su resultado hacia
un archivo especial llamado salida estándar (standar output, frecuentemente stdout) y su mensaje de estatus a otro
archivo especial llamado standar error (sterr)
Por default, tanto el error y la salida estándar están vinculados a la pantalla y no se guardan en un archivo en el disco.
Además, muchos programas toman la entrada de algo llamado entrada standar (stdin), el cual está por default, anexo
al teclado. La redirección I/O nos permite cambiar a dónde va la salida y de dónde viene la entrada. Normalmente, la
salida va a la pantalla y la entrada viene del teclado, pero con la redirección I/O podemos cambier ese orden.
Redirección a salida estándar
Para redireccionar la salida estándar a otro archivo en lugar de la pantalla, utilizamos el operador de redirección >
seguido del nombre del archivo. ¿Por qué habríamos de hacer eso? A menudo es útil almacenar en un archivo la salida
de un comando. Por ejemplo, podríamos mandar la salida del comando ls a un al archivo ls_output.txt en lugar de la
pantalla.
ls -l /usr/bin > ls_output.txt
La finalidad del comando ls es obtener una lista de /usr/bin y enviar el resultado al archivo ls_output.txt.
Si examinamos un poco más el archivo de salida, notaremos que es un archivo de texto algo extenso.
ls -l ls_output.txt
> -rw-r--r-- 1 zorbax 198059 ago 20 05:26 ls_output.txt
Si vemos el contenido del archivo con less observaremos que de hecho contiene la salida del comando ls.
less ls_output.txt
Repitamos la redirección pero ahora démosle un pequeño giro. Cambiaremos el nombre del directorio a uno que no
existe.
ls -l /bin/usr > ls_output.txt
> ls: cannot access /bin/usr: No such file or directory
Obtuvimos un mensaje de error. Esto tiene sentido pues especificamos un directorio que no existe /bin/usr, pero
¿por qué el mensaje de error de desplegó en la pantalla en lugar de redireccionarse al archivo ls_output.txt. La
respuesta es que el programa ls no envió el mensaje de error a la salida estándar, lo envió al error estándar por lo
1.5. Terminal
65
biote-2016, Release
que el mensaje fue enviado a la pantalla. Si revisamos nuevamente nuestro archivo nos daremos cuenta de que está
vacío.
ls -l ls_output.txt
> -rw-r--r-- 1 zorbax 0 ago 20 05:28 ls_output.txt
Podemos redireccionar la salida al archivo pero en lugar de sustituirlo podemos reescribir el contenido con el operador
>> :
ls -l /usr/bin >> ls-output.txt
Usando este operador el resultado se agregará al archivo de salida, en lugar de sobreescribirlo. Si el archivo ni existe
lo creará, al igual que >.
/usr/bin >> ls_output.txt
/usr/bin >> ls_output.txt
/usr/bin >> ls_output.txt
ls -l ls_output.txt
> -rw-r--r-- 1 zorbax 594177 ago 20 05:38 ls_output.txt
Repetimos el comando tres veces, así que obtendremos un archivo de salida tres veces más grande que el original, ya
que la salida del comando se va añadiendo al archivo ls_output.txt.
Redireccionamiento a error estándar
Para redireccionar el error estándar no existe un operador asignado, sim embargo, podemos referirlo a su descriptor
de archivo.
Un programa puede generar muchos tipos de archivo, pero siempre referirá los primeros tres a la entrada, salida y error
estándar. El shell los refiere con los descriptores de archivos 0, 1 , y 2 , respectivamente. Ya que el error estándar es
igual al descriptor de archivo 2 podemos redireccionar el error estándar con esta notación:
ls -l /bin/usr 2> ls_error.txt
EL descriptor 2 está colocado antes del operador de redirección para realizar la redirección del error estándar al archivo
ls_error.txt
Redireccionamiento de salida y error estándar a un archivo
Hay casos en los que se desea capturar todo la salida de un comando a un archivo. Para hacerlo, redireccionaremos
tanto la salida estándar y el error estándar al mismo tiempo. Hay dos formas de hacerlo, la forma tradicional, que
funciona con versiones antiguas de shell.
66
Chapter 1. Índice
biote-2016, Release
ls -l /bin/usr > ls_output.txt 2>&1
Usando este método, se realizan dos redirecciones.
Primero redirigimos la salida estándar al archivo
ls_output.txt, y luego redireccionamos el descriptor de archivo 2 (error estándar) al descriptor de archivo 1
(salida estándar) usando la notación 2>&1
El orden de la redirección es importante. La redirección del error estándar siempre debe de ocurrir después de la
redirección a la salida estándar o no funcionará. En el ejemplo anterior, > ls_output.txt 2>&1 redirecciona
el error estándar al archivo ls_output.txt , pero si se cambia el orden a 2>&1 > ls-output.txt el error
estándar es redireccionado a la pantalla.
Otra manera de combinar la redirección:
ls -l /bin/usr &> ls_output.txt
En este ejemplo, se utiliza la notación &> para redireccionar tanto la salida estándar y el error estándar al archivo
ls_output.txt.
Silenciando salida no deseada
En otras ocasiones no deseamos guardar la salida de un comando, principalmente para los mensajes de error y de
estado. El sistea provee una manera de redireccionar la salida a un archivo especial llamado /dev/null, el cual es
en pocas palabras una papelera de bits. que acepta entrada datos pero no hace nada con ellos. Para suprimir mensajes
de error de un comando:
ls -l /bin/usr 2> /dev/null
Redireccionamiento de entrada estándar
cat El comando cat lee uno mas archivos y los copia a la salida estándar:
cat file...
También se usa para mostrar el contenido de archivos de texto sin paginar el contenido.
cat ls_output.txt
Linea de procesos
Hemos visto que es importante tener control acerca de la salida de un comando, de dónde viene y hacia dónde va (vía
redirección), pero también es posible que la salida de un comando pase a través de un comando a la entrada de otro
comando (vía pipe). Este proceso es conocido como pipeline o linea de procesos. Cada proceso se separa con un pipe
( | ).
command1 | command2
Para demostrar una linea de procesos necesitamos aprender algunos comandos. Hemos visto un comando que acepta
la entrada estándar, less. Podemos utilizar dicho comando para imprimir en pantalla, página por página, la salida de
cualquier comanto que envíe su resultado a la salida estándar:
ls -l /usr/bin | less
Podemos examinar la salida de cualquier comando que produzca salida estándar.
1.5. Terminal
67
biote-2016, Release
Filtros
Las lineas de procesos son útiles para realizar operaciones complejas con datos.
Frecuentemente se usa de una manera en que se refiere a ellos como filtros. Los filtros pueden tomar la entrada,
cambiar algo y enviarlo a la salida estándar.
sort Si deseamos combinar una lista de todos los programas ejecutable en /bin y /usr/bin, ordenarlos y visualizar la lista. Las opcines más comunes son -n para ordenar los valores numéricos y -r, para obtener la lista en
orden inverso.
ls /bin /usr/bin | sort | less
uniq Este comando nos permite omitir lineas repetidas. Se usa a menudo junto con sort. Utiliza como entrada
estándar una lista de datos ordenados y por defecto elimina las lineas repetidas. Con la opción -c podemos obtener
68
Chapter 1. Índice
biote-2016, Release
cuántas veces está repetida la linea:
ls /bin /usr/bin | sort | uniq | less
Si queremos ver las lineas duplicadas, utilizamos la versión -d
ls /bin /usr/bin | sort | uniq -d | less
wc El comando wc (word count) es utilizado para mostrar el número de lineas, palabras y bytes que contiene un
archivo.
wc ls_output.txt
3303 28338 198698 ls_output.txt
Los tres números corresponden a lineas, palabras y bytes, respectivamente, contenidas en el archivo
ls_output.txt. Al agregarlo al pipeline es una manera fácil de contar elementos, en este caso, nos interesa
el número de lineas.
ls /bin /usr/bin | sort | uniq | wc -l
3452
grep El comando grep imprime lineas que coinciden con un patrón de texto.
grep pattern [file...]
El ‘patrón’ que grep reconozca puede ser muy complejos ( grep -v "^#\|^’\|^\/\/" file.txt ), pero
para fines prácticos del curso nos enfocaremos en coindicencias con texto.
Digamos que queremos encontrar aquellos archivos de nuestra lista de programas que tengan la palabra zip en el
nombre:
ls /bin /usr/bin | sort | uniq | grep zip
bbunzip2
bzip2
bzip2recover
funzip
gpg-zip
gunzip
gzip
lzip
lzip.lzip
mzip
p7zip
preunzip
prezip
prezip-bin
unzip
unzipsfx
zip
zipcloak
zipdetails
zipgrep
zipinfo
zipnote
zipsplit
1.5. Terminal
69
biote-2016, Release
Algunas de las opciones más útiles de grep: -i - Hace que el comando ignore mayúsculas y minúsculas. Las
búsquedas en Unix son sensible a dichas diferencias. -v - Le dice al programa que imprima aquellas que no
coincidan con el patrón. -c - Le dice al program que cuente cuántas veces se encuentra el patrón en la lista.
head/tail A menudo no queremos todo la salida de un comando. Estos comandos nos ayudan a obtener la primera
parte y la última parte de un archivo, las primeras y las últimas 10 lineas, respectivamente. El número de lineas puede
ser modificado con la opción -n.
head -n 5 ls_output.txt
total 547960
-rwxr-xr-x 1 root
39464
lrwxrwxrwx 1 root
8
-rwxr-xr-x 1 root
106
-rwxr-xr-x 1 root
96
mar 14
mar 16
ene 26
mar 1
2015
2015
2013
2015
[
2to3 -> 2to3-2.7
2to3-2.6
2to3-2.7
Estos comandos también funcionan con la salida de otros comandos:
ls /usr/bin | tail -n 5
zipinfo
zipnote
zipsplit
zjsdecode
zlib-flate
El comando tail tiene una opción que nos permite monitorear archivos en tiempo real. En el siguiente ejemplo
veremos mensajes del archivo /var/log/messages. Para ello se requiere permisos de superusuario, más adelante
explicaremos en qué consiste dichos privilegios.
tail -f /var/log/messages
Aug 24 22:21:38 amidala kernel:
Aug 24 22:21:38 amidala kernel:
Aug 24 22:21:38 amidala kernel:
Aug 24 22:21:38 amidala kernel:
Aug 24 22:21:38 amidala kernel:
Aug 24 22:21:38 amidala kernel:
Aug 24 22:21:38 amidala kernel:
Aug 24 22:21:38 amidala kernel:
70
[2371005.944927]
[2371005.944927]
[2371005.944943]
[2371005.944944]
[2371005.944944]
[2371005.944946]
[2371005.944947]
[2371005.944948]
CPU3:
CPU7:
CPU4:
CPU0:
CPU1:
CPU2:
CPU5:
CPU6:
Package
Package
Package
Package
Package
Package
Package
Package
temperature/speed
temperature/speed
temperature/speed
temperature/speed
temperature/speed
temperature/speed
temperature/speed
temperature/speed
normal
normal
normal
normal
normal
normal
normal
normal
Chapter 1. Índice
biote-2016, Release
tee
Tomando una analogía de la plomería, Unix provve un comanto llamado tee el cual crea una “T” en nuestra linea de
procesos. Este comando lee la entrada estándar y lo copia a la salida estándar (permitiendo que los datos sigan en la
linea de procesos) y a uno o más archivos. Este comando es útil para capturar el contenido de una linea de procesos
en una etapa intermediaria o final de procesamiento.
ls /usr/bin | tee ls.txt | grep zip
funzip gpg-zip lzip lzip.lzip mzip p7zip preunzip prezip prezip-bin unzip unzipsfx zip zipcloak zipdetails
zipgrep zipinfo zipnote zipsplit
Ejemplo
A continuación les dejo un ejemplo muy rebuscado para terminar un proceso. En el apartado *Tareas veremos una
manera más sencilla hacerlo. El comando ps aux obtendrá una lista de los procesos que están corriendo además de
alguna información adicional. La segunda columna es el número identificador de cada proceso (PID).
ps aux
USER
root
root
zorbax
zorbax
zorbax
zorbax
zorbax
zorbax
zorbax
zorbax
zorbax
zorbax
zorbax
PID %CPU %MEM
VSZ
RSS TTY
715 0.0 0.2 431276 14280 ?
853 0.0 0.1 278484 6088 ?
1942 0.0 0.2 366064 13884 ?
1985 0.1 0.6 359468 39172 ?
1990 0.0 0.6 584088 36148 ?
2008 0.1 1.0 1238336 62616 ?
2026 0.0 0.9 511608 54480 ?
2028 0.1 3.3 2686964 198416 ?
2418 17.6 30.6 3080640 1811536 ?
2562 0.0 0.9 502464 57128 ?
2571 0.0 0.1 29496 11464 pts/0
2598 0.0 0.1 29500 11436 pts/1
2605 0.0 0.1 29496 11420 pts/2
1.5. Terminal
STAT
Ssl
SLsl
Ssl
Sl
Sl
Sl
Sl
Sl
Sl
Sl
Ss
Ss
Ss+
START
TIME COMMAND
ago23
0:09 /usr/sbin/NetworkManager --no-daemon
ago23
0:00 /usr/sbin/lightdm
ago23
0:00 mate-session
ago23
1:41 marco
ago23
0:05 mate-panel
ago23
1:31 caja
ago23
0:07 /home/zorbax/bin/cloud
ago23
1:31 /usr/bin/python /usr/local/bin/turpi
ago23 201:07 firefox-esr
ago23
0:07 /usr/bin/python /usr/bin/terminator
ago23
0:00 /bin/bash
ago23
0:00 /bin/bash
ago23
0:00 /bin/bash
71
biote-2016, Release
zorbax
zorbax
zorbax
zorbax
2908
5646
11221
11260
0.0
0.0
0.0
0.0
0.0 13260 1648 ?
0.2 47704 12756 pts/0
0.5 438880 32236 ?
0.0 19104 2552 pts/1
S
S+
Sl
R+
ago23
13:07
18:21
18:24
0:00
0:02
0:00
0:00
/bin/bash /usr/bin/atom
python -m SimpleHTTPServer
pluma
ps aux
Sólo estoy intersado en un proceso pluma, así que utilizo grep para obtener la lines que me interesa.
ps aux | grep pluma
zorbax
zorbax
11221
11400
0.0
0.0
0.5 438880 32236 ?
0.0 12752 2232 pts/1
Sl
S+
18:21
18:31
0:00 pluma
0:00 grep --color=auto pluma
Obtengo una lista breve de los procesos que estoy corriendo, ps aux para obtener la lista completa y grep para
filtrar con base en el patrón que deseo. Ahora, utilizo la opción -v en grep para que excluya las lineas que contengan
cierto patrón, en este caso grep
ps aux | grep pluma | grep -v grep zorbax 11221 0.0 0.5 438880 32236 ? Sl 18:21 0:00 pluma
Ahora que tengo aislada la linea de mi proceso deseo obtener su PID para poder terminarlo. Para ello deseo imprimir
sólo la segunda palabra de la linea obetenida. Afortunadamente la mayoría de versiones Unix tienen awk, un lenguaje
de programación que se especializa en datos tabulares. Una característica muy útil de este lenguaje es que se puede
usar en una linea de procesos.
ps aux | grep pluma | grep -v grep | awk ‘{print $2}’ 11221
Una vez obtenido el PID es necesario ‘pasar’ el número al comando kill, el cual finalizará el proceso. Para ello
necesitaremos xargs.
ps aux | grep pluma | grep -v grep | awk '{print $2}' | xargs kill
Misión cumplida :D
1.5.5 Permisos
Los sistemas Unix son multitarea y multiusuarios. Esto significa que más de una persona puede usar la computadora
al mismo tiempo. Aunque sólo tenga un teclado y un monitor, aún puede ser utilizada por más de un usuario. Si la
computadora está conectada a una red o a internet, los usuarios se pueden conectar de manera remota vía ssh (Tareas)
En esta parte veremos algunos comandos encargados de proteger los archivos de acciones de otro usuario:
• chmod — Cambia el modo de un archivo.
• su — Ejecuta la shell como otro usuario.
• sudo — Ejecuta comandos como otro usuario.
• chown — Cambia el propietario de un archivo.
• passwd — Cambia la contraseña de un usuario.
Propietario, miembros de grupo y otros
En algún momento de los ejercicios anteriores, cuando estabamos viendo lo de error y la salida estándar nos encontramos con el siguiente problema. Intentaremos con otro archivo:
file /etc/shadow
/etc/shadow: regular file, no read permission
72
Chapter 1. Índice
biote-2016, Release
less /etc/shadow
/etc/shadow: Permission denied
La razón del ‘error’ es que los usuarios regulares no tienen permiso de leer ese archivo. En Unix todos los usuarios
tienen sus propios archivos y directorios. Cuando un usuario es propietario de un archivo o directorio, el usuario tiene
control sobre su acceso, así que sólo él decide qué grupo puede acceder o si bien lo decide que sea accesible a todo el
mundo. El usuario puede cambiar la propiedad, el modo y el acceso a los archivos y directorios.
Lectura, escritura y ejecución
Los derechos de acceso a los archivos y directorios son definidos en términos de acceso de lectura, acceso de escritura
y acceso de ejecución. Con el comando ls -l nos podemos dar cuenta de cómo son implementados:
ls -l
drwx-----6 4.0K
drwxr-xr-x
9 16K
-rwxr-xr-x
1 194K
lrwxrwxrwx
1
28
-rwxr-xr-x
1 3.0K
-rwxr-xr-x
1 1.3M
drwxr-xr-x 13 4.0K
drwxr-xr-x
5 4.0K
drwxr-xr-x 313 12K
drwxr-xr-x
3 4.0K
may
jul
jul
jul
jul
jul
ago
ago
ago
ago
13
14
16
25
25
28
3
4
10
17
11:55
17:34
21:25
00:07
10:34
00:06
19:53
16:40
07:15
12:22
BRIG-0.95-dist/
pandaseq/
seqtk*
thunderbird -> /opt/thunderbird/thunderbird*
monitor*
usearch61*
Phinch/
lefse/
R/
python/
Ejemplos
1.5. Terminal
73
biote-2016, Release
• -rwx------ — Un archivo regular es legible, grabable y executable por el propietario del archivo. Nadie más
tiene acceso.
• -rw------- — Un archivo regular que es legible y grabable por el propietario del archivo. Nadie más tiene
acceso.
• -rw-r--r-- — Un archivo regular que es legible y grabable por el propietario del archivo. Los mientros del
grupo del propietario pueden leer el archivo. Todos los demás también pueden leer el archivo.
• --rwxr-xr-x — Un archivo regular que es legible, grabable y ejecutable por el propietario del archivo. El
archivo también puede ser leído y ejecutado por todos los demás.
• -rw-rw---- — Un archivo regular que sólo es legible y grabable por el propietario del archivo y por miembros
del grupo del usuario.
• lrwxrwxrwx — Un link simboplico. Todos los links simbólicos tienen permisos ‘postizos’. Los permisos
reales se mantiene en el archivo vinculado al que apunda el enlace simbólico.
• drwxrwx--- — Un directorio. El propietario y los miembros del grupo del usuario pueden accesar al directorio y crear, renombrar y eliminar archivos. Los miembros del grupo del usuario puede acceder al directorio
pero no puede crear, borrar o renombrar archivos.
chmod
Para cambiar el modo (permisos) de un archivo o directorio, se utiliza el comando chmod. Hay que tener en consideración de que sólo el propietario del archivo o el superusuario puede cambiar el modo de un archivo o directorio. Con
chmod se puede utilizar números octales y una representación simbólica.
Octal
0
1
2
3
4
5
6
7
Binario
000
001
010
011
100
101
110
111
Modo
—
–x
-w-wx
r–
r-x
rwrwx
chmod Anotación simbólica:
• u — Abreviación para usuario, representa al porpietario del archivo o directorio.
• g — Propitario de grupo.
• o — Abreviación para otros, pero implica a ‘todo el mundo’.
• a — Abreviación para todos, la combinación de u, g y o
74
Chapter 1. Índice
biote-2016, Release
Exemplos de notación simbólica de chmod:
• u+x — Agrega permisos de ejecución para el usuario.
• u-x — Elimina permisos de ejecución del usuario.
• +x — Agrega permisos para el propietario, grupo y todos el mundo. Equivale a a+x.
• o-rw — Elimina permisos de lectura y escritura para cualquier persona, además del propietario y grupo del
propietario.
• go=rw — Asigna al propietario del grupo y a todos además del propietario a que tenga permiso de lectura y
escritura. Si el propietario del grupo o todos los demás tenían premisos de ejecución, es removido el permiso.
• u+x,go=rx — Agrega permisos de ejecución para el propietario y configura los permisos para el grupo y
todos los demás para que puedan leer y ejecutar.
su
El comando su se usa para iniciar una shell como otro usuario. La sintaxis es de la siguiente manera:
su [-[l]] [user]
Si se uncluye la opción -l (también se puede abreviar -), la shell que se obtenga será la especificada para dicho
usuario. Eso significa que el entorno del usuario se cargará y se cambiará al directorio home del usuario. Si no se
especifica usuario, se asume que se requiere usar el superusuario.
zorbax@beatrix~:$ su Password:
root@beatrix~:#
1.5. Terminal
75
biote-2016, Release
sudo
El comando sudo es como su, de hecho Ubuntu configura a sudo para que le permita ejecutar comandos como
otro usuario (generalmente superuser), pero en un ámbito más controlado que su. El acceso con sudo no requiere la
contraseña del superusuario, sólo basta la contraseña de usuario.
sudo file /etc/shadow /etc/shadow: ASCII text
sudo less /etc/shadow # :O
chown
El comando chown se utiliza para cambiar el propietario y el propietario de grupo de un archivo o directorio. Para
este comando se requieren privilegios de superusuario. La sintaxis de este comando es:
chown [owner][:[group]] file...
El comando chown puede cambiar el propietario de un archivo y/o el grupo propietario dependiendo del primer
argumento del comando. Este comando es de mucha utilidad cuando se accede a servidores remotos donde uno no
tiene acceso a todos los archivos y se tiene que interactuar con diferentes propietarios.
Ejemplos
• bob — Cambia el propietario actual del archivo a bob.
• bob:users — Cambia el propietario de un archivo de su propietario actual al usuario bob y cambia el
propietario del grupo al grupo users.
• :admins — Cambia el propietario de grupo al grupo admins. El propietario del archivo sigue sin modificaciones.
• bob: — Cambia el propietario de archivos del propietario actual al usuario bob y cambia el propietario de
grupo al login de grupo del usuario bob.
1.6 Entorno
El shell mantiene información durante la sesión de shell, el entorno. Los datos almacenados en el entorno es usado por los programas para determinar ciertos datos acerca de nuestra configuración. La mayoría de los programas
utiliza archivos de configruación para almacenar las configuraciones del programa, algunos otros buscan los valores
almacenados en el entorno para ajustar su comportamiento.
1.6.1 Variables de entorno
El shell almacena dos tipos de datos en el entorno. Hay variables de entorno y variable de shell. Las variables de
shell son datos colocados por bash y las variables de entorno son todas las demás. El shell también puede almacenar
algunos otros datos, como alias y funciones.
Para visualizar las variables del entorno:
printenv | less
Para visualizar una variable en particular del entorno:
echo $PATH
echo $HOME
76
Chapter 1. Índice
biote-2016, Release
1.6.2 Variables shell
Es una manera simple de referir datos en la memoria, los cuales pueden ser usados y modificados con base en la
estructura del script. Por ejemplo, puedo definir una variable con un valor:
name="Otoniel"
echo "Hello $name"
name="world!"
echo "Hello $name"
name=`whoami`
figlet "Hello $name"
1.6.3 Funciones de shell, alias
¿Cómo se determina los parámetros del entorno?
Cuando iniciamos sesión en el sistema, el bash inicia y lee una serie de scripts de configuración llamados archivos
de inicio, los cuales definen el comportamiento del entorno compartido por todos los usuarios. Esto es seguido por
archivos de inicio en nuestro directorio de home que define nuestro entorno personal.
Shell que requiere y no requiere inicio de sesión
Una shell de inicio de sesión es aquella en la cual se solicita nuestro nombre de usuario y la contraseña, por ejemplo
cuando iniciamos una sesión en una consola virtual. Una shell que no requiere inicio de sesión ocurre cuando lanzamos
una sesión en terminal en interfaz gráfica.
Archivos de inicio para shell que requiere inicio de sesión:
• /etc/profile — El archivo de configuración global que aplica para todos los usuarios.
• ~/.bash_profile — El archivo de inicio personal de cada usuario.
• ~/.bash_login — Si no encuentra el archivo ~/.bash_profile, bash intentará leer este otro archivo.
• ~/.profile — Si no encuentra ~/.bash_profile y tampoco a ~/.bash_login, bash intentará de
leeer este archivo. Este es el default para distribuciones basadas en Debian, como Ubuntu.
Archivos de inicio para shell que no requiere inicio de sesión:
• /etc/bash.bashrc — Archivo global de configuración que aplica para todos los usuarios.
• ~/.bashrc — Archivo de inicio personal de cada usuario. Puede ser usado para extender o anular configuraciones globales.
Funciones
Una función del shell es una subrutina, un bloque de código que implemente un grupo de operaciones, incorporada
en el entorno. Son de mucha utilidad cuando se definen en los archivos de inicio del shell y permite realizar algunas
operaciones de manera mucho más sencilla.
Ejemplos
1.6. Entorno
77
biote-2016, Release
extract () {
if [ -f $1 ] ; then
case $1 in
;;
*.tar.bz2) tar xjf $1
tar xzf $1
;;
*.tar.gz)
bunzip2 $1
;;
*.bz2)
.rar)
rar
x
$1
;;
*
gunzip $1
;;
*.gz)
tar xf $1
;;
*.tar)
tar xjf $1
;;
*.tbz2)
.tgz)
tar
xzf
$1
;;
*
unzip $1
;;
*.zip)
uncompress $1
;;
*.Z)
echo "'$1' cannot be extracted via extract()" ;;
*)
esac
else
echo "'$1' is not a valid file"
fi
}
ziprm () {
if [ -f $1 ] ; then
unzip $1
rm $1
else
echo "Need a valid zipfile"
fi
}
tarrm () {
if [ -f $1 ] ; then
tar -zxvf $1
rm $1
else
echo "Need a valid tarfile"
fi
}
psgrep() {
if [ ! -z $1 ] ; then
echo "Grepping for processes matching $1..."
ps aux | grep $1 | grep -v grep
else
echo "!! Need name to grep for"
fi
}
Alias
Otro elemento del entorno son los alias, que como su nombre lo indica son comandos que podemos definir a nuestra
conveniencia. Los alias definidos para el usuario se pueden visualizar con:
alias
Ejemplos
Todos los alias se tienen que agregar a los archivos de inicio, generalmente ~/.bashrc.
78
Chapter 1. Índice
biote-2016, Release
alias
alias
alias
alias
alias
alias
alias
alias
alias
alias
alias
alias
alias
..='cd ..'
...='cd ../../...'
Oblaka='cd ~/Documentos/Oblaka'
Dropbox='cd ~/Documentos/Dropbox'
R='R -q'
cd..='cd ..'
cp='cp -iv'
df='df -h'
dt='date +"%T"'
fastqc2='fastqc --nogroup -f fastq'
ffind='find . -iname'
freedom='su -c "free -h && sync && echo 3 > /proc/sys/vm/drop_caches && free -h"'
h='history'
alias
alias
alias
alias
alias
alias
ll='ls -ltrhFGg'
ls='ls -G --color=auto'
lsd='ls -l | grep "^d"'
lsf='ls -l | grep "^\-"'
lt='ls -trh'
mv='mv -iv'
alias
alias
alias
alias
alias
poweroff='su -c "systemctl poweroff"'
reloadbash='source $HOME/.bashrc'
restart='su -c "systemctl reboot"'
shcpan='perl -MCPAN -e shell'
update='sudo apt-get update && sudo apt-get upgrade"'
Para impementar los cambios generados en ~/.bashrc se utiliza source:
source $HOME/.bashrc
#or
source ~/.bashrc
1.6.4 Editores de texto
Para editar los archivos del shell, así como otros archivos, ya sea de configuración o personales, se utilizan programas
llamados editor de texto. Un editor de texto es diferente a un procesador de texto (Microsoft Office, LibreOffice,
OpenOffice, Ashampoo Office) en que permite soporta sólo texto, sin ningún elemento que adicione formato ( \n, \r
, \t ). Hay editores de texto con interfaz gráfica: gedit, kate, pluma, etc.) y editores de texto en la terminal (nano,
vim, etc.).
El más sencillo de utilizar es nano:
nano /path/to/file
Las opciones básicas son (el acento circunflejo es equivalente de Ctrl + :
^X
^O
Cerrar el fichero mostrado / Salir de nano
Escribir el fichero actual a disco
Ahora, a practicar un poco. Agrega los alias y las funciones al archivo .bashrc.
1.6. Entorno
79
biote-2016, Release
1.7 Preprocesamiento
1.7.1 QC
FastQC
Para los pasos siguientes utilizaremos el siguiente set de prueba con lecturas pair-end de Illumina.
mkdir $HOME/dataset && cd $HOME/dataset
wget http://148.247.152.169/data/16data.tar.gz
extract 16data.tar.gz && rm 16data.tar.gz
cd 16data
tree
Cada archivo fastq tiene cuatro lineas:
1. Nombre de la secuencia (header - id del secuenciador, coordenadas del spot, flow-cell, adaptador, etc.)
2. Secuencia
3. Espaciador (+)
4. Valores de calidad: Q Score - alfanumérico.
Dentro del folder $HOME/dataset/16Sdata/Fastq se encuentran los archivos que utilizaremos para el análisis
de secuencias provenientes de amplicones. Para evaluar la calidad se podría conderar los archivos de manera individual
pero eso consumiría ‘mucho tiempo’, por lo que es mejor tratarlo como un sólo archivo. Concatenamos todos los
archivos fastq en un solo y ejecutamos el comando fastqc
cd Fastq/
cat *.fastq > all.fq
fastqc --nogroup -f fastq all.fq
El resultado se encuentra en el archivo *html el cual se puede abrir con Firefox/Chrome/Opera.
1. Estadística básica
80
Chapter 1. Índice
biote-2016, Release
La estadística básica de las secuencias incluye el nombre del archivo, cuántas secuencias contiene, si algunos reads
fueron etiquetados con una mala calidad. En este caso tenemos 1,080,000 secuencias, con un tamaño promedio de
150, un GC de 55% y ninguna lectura de mala calidad.
2: Calidad de secuencia por base
La calidad de secuencia por base muestra la calidad de cada nucleótido. del 1 al 150 en este caso. Muestra esta
información usando un gráfico de caja y bigotes para mostrarnos cuánta variación hay entre los reads. Idealmente
desearíamos que todo el gráfico se concentrara en la región verde; lo cual se consideraría muy buena calidad. No es
deseable que el gráfico se localice en las regiones naranja y roja. CUando el cuartil más bajo de cualquier posición es
menor a 10 o la media es menor que 25, el módulo dará un warning. Cuando el cuartil más bajo de cualquier posición
es menor a 5 o la media es menor que 20, el módulo dará un fail en este paso del control de calidad.
3: Calidad de secuencias de flowcell
Es un heatmap del la calidad del flowcell mostrando celdas individuales. Si la figura es un sólido azul brillante la
calidad del flowcell es consistentemente buena. Si hay parches de un azul más claro o de otro color, hay un problema
asociado con alguna de las celdas ( como una burbuja o una mancha ) y esto puede corresponder con un decremento
en la calidad de la secuencia en dichas regiones. En la gráfica se observan parches azul claro que indicaría problemas
potenciales con la secuenciación en esas lineas. Sin embargo, en este caso las lecturas son lo suficientemente buenas
para pasar el control de calidad; sería más preocupante si hubieran sido celdas color naranja o rojo.
1.7. Preprocesamiento
81
biote-2016, Release
4: Score de calidad por secuencia
El score de calidad por secuencia representa la calidad de cada lectura. El eje y es el número de secuencias, y el eje x
es la escala Phred, la cual está basada en una escala logarítmica. Un Predscore de 30 indica un ranto de error de 1 base
en 1000, o una exactitud de 99.9%, mientras que un Phred score de 40 indica un rango de error de 1 base en 10,000,
o una exactitud de 99.99%. Con un valor debajo de 27 se obtendrá un warning y por debajo de 20 se dará un fail. En
nuestro ejemplo, el promedio de calidad es 37, lo cual es muy bueno.
5: Contenido de secuencia por base
El contenido de secuencias por base muestra, para cada posición de cada secuencia, la composición de bases como
82
Chapter 1. Índice
biote-2016, Release
porcentaje para A, T, C y G. Este módulo obtendrá un warning si el contenido de base varía más del 10% en cualquier
posición, y la muestra obtendrá fail si hay más de 20% de variación en cualquier posición, como en el ejemplo de las
secuencias que estamos utilizando. Sin embargo, FastQC está diseñado para secuencias de genomas completos, pero
nosotros utilizamos secuencias de 16S como input. Aunque tenemos una falla en este módulo, no es que haya algo
malo con nuestras secuencias, es sólo que estamos usando secuencias que están enriquecidas en ciertas bases, más que
secuencias completamente aleatorias provenientes de una secuenciación de genoma completo.
6: Contenido de GC por secuencia
El contenido de GC por secuencia muestra el contenido total para todos los reads de acuerdo a la “distrbución teórica”
de GC’s. El pico de la linea roja corresponde a la media de contenido GC de las secuencias, mientras que el pico de
la linea azul corresponde a la media teórica de contenido de GC. Nuestro contenido de GC debería estar distribuido
normalmente. Es de esperar cambios en los picos ya que el contenido de GC varía entre organismos, cualquier otra
curva con una curva normal podría ser indicativo de contaminación.
La forma del pico de la figura de arriba se debe al hecho de que hemos enriquecido una secuencia específica, así que
esperaríamoa que la mayoría tenga aproximadamente el mismo contenido de GC. Se observa un warning si el 15%
total de las secuencias caen fuera de la distribución normal. Se obtendrá un fail si más del 20% de las secuencias están
fuera de la distribución normal. Los fails son generalmente debidos a contaminación, frecuentemente por secuencias
de adaptadores.
1.7. Preprocesamiento
83
biote-2016, Release
7: Contenido de N por base
El contenido de N por base muestra cualquier posición de las secuencias que no han sido ‘llamadas’ como A,T,C o G.
Idealmente el contenido de N por base sería una linea plana en 0% sobre el eje y, indicando que todas las bases han
sido ‘llamadas’. Se recibe un warning si el contenido de N es igual o mayor de 5%, y obtendrá un fail si el contenido
de N es igual o mayor a 20%. Nuestras secuencias muestran el resultado ideal para este módulo.
8: Distribución de longitud de secuencias
Este módulo simplemente muestra la longitud de cada secuencia en la muestra. Dependiendo de la plataforma de
secuenciación usada la gráfica puede variar. Para secuenciación con Illumina, cada lectura debería ser del mismo
84
Chapter 1. Índice
biote-2016, Release
tamaño, con variación aceptable de una o dos bases. Para otras plataformas, se puede obtener una cantidad considerable
de variación en el tamaño de las lecturas. Este módulo mostrará un warning si hay cualquier variación en la longitud
de las secuencias, el cual puede ser ignorado si se sabe que es normal para el tipo de datos que se tiene. Una falla es
este módulo significa que al menos una secuencia tiene longitud de 0. Nuestro ejemplo pasa este módulo ya que todas
las secuencias tienen una longitud de de 150 sin variación alguna.
9: Niveles de duplicación de secuencias
La gráfica de los niveles de duplicación de secuencias muestran en el eje x, el número de veces que una secuencia está
duplicada, y en el eje y el porcentaje de secuencias que muestran ese nivel de duplicación. Normalmente un genoma
tendrá un nivel de duplicación de secuencias de 1 a 3 para la mayoría de las secuencias, con sólo un puñado de lecturas
teniendo un nivel más alto que este; la linea debería tener la forma inversa a una gráfica log. Un alto porcentaje de
duplicación de secuencias es un indicativo de contaminación. Como utilizamos datos provenientes de secuenciación
de 16S los datos parecen un poco confusos, el resultado observado es normal considerando las secuencias que tenemos.
Este módulo nos dará un warning si más del 20% de las secuencias son duplicadas, y dará fail si más del 50% de las
secuencias están duplicadas. Un warning o fail puede ser resultado de artefactos de PCR.
1.7. Preprocesamiento
85
biote-2016, Release
10: Secuencias sobre-representadas
Si alguna secuencia se calcula que representa más del 0.1 % del genoma completo será etiquetada como una secuencia
sobre-representada y se obtendrá un warning. La presencia de secuencias qque representan más del 1% del genoma
dará como resultado que el módule falle, como se observa en este ejemplo. Estas secuencias sobre-representadas son
observadas porque estamos viendo datos de 16S; si no vieramos estas secuenciassobre-representadas estaríamos en
serios problemas.
11: Contenido de adaptadores
Este módulo busca secuencias específicas de adaptadores. Una secuencia que representa más del 5% del total causará
un warning en este módulo, y una secuencia que represente más del 10% del total causará una falla. Nuestro ejemplo
no muestra contaminación con secuencias de adaptadores, lo cual es ideal. Si existiera un número significativo de
secuencias de adaptadores, se debe utilizar un programa para recortarlos y realizar el análisis de calidad nuevamente.
86
Chapter 1. Índice
biote-2016, Release
12: Contenido de K-mer
En una librería completamente al azar, se podría observar cualquier k-mer de manera equitativa en cada posición (del
1-150 en este caso). En este módulo se reportará cualquier k-mer que esté enriquecido en algún sitio en particular.
Si algún k-mer está enriquecido en un sitio específico con una p < 0.01, se obtendrá un warning. El módulo fallará
si el k-mer está enriquecido en algún sitio con una p < 10^-5. Las secuencias de ejemplo fallan este módulo debido
a que manejamos lectura de 16S. Al igual que con las secuencias sobre-represntadas esperamso ver k-mer*s bien
representados por la alta conservación de las regiones 16S. En lectura no enriquecidas, es común ver *k-mers de
secuencias altamente representadas cerca del principio de las secuencias si los adaptadores están presentes.
1.7. Preprocesamiento
87
biote-2016, Release
## NOTA: Borrar el archivo concatenado anteriormente:
rm all.fastq
FastQC Documentation.
1.7.2 Ensamble de lecturas
Pandaseq
Las secuencias que tenemos son secuencias de amplicones de 16s rRNA obtenidas con MiSeq. Utilizaremos Pandaseq
para unir la lectura forward y reverse en una sola secuencia. Ejecutamos el comando:
mkdir pandaseq_merged_reads
pandaseq -F -f C01D01_1.fastq -r C01D01_2.fastq -w pandaseq_merged_reads/C01D01_pd.fastq -g pandaseq_
Veamos la estructura del comando:
• pandaseq: script de pandaseq.
• -F: habilitar archivo de salida en formato fastq.
88
Chapter 1. Índice
biote-2016, Release
• -f: archivo de lecturas forward.
• -r: archivo de lecturas reverse.
• -w: escribe la salida en formato fastq en el archivo *pd.fastq en el directorio pandaseq_merged_reads.
• -g: archivo log del proceso. Si no se habilita esta opción el log saldrá en standard output (2>) haciendo un
poco más lento el proceso.
• -B: las secuencias input no contienen barcode.
• -A: el algoritmo utilizado para el ensamble.
• -L: especifica la longitud máxima de las lecturas ensambladas, (el tamaño del amplicón 150X150 de Illumina
es de 253 bp)
• -O: especifica la cantidad de sobrelapamiento permitido entre las secuencias forward y reverse.
• -t: score de calidad entre 0 y 1 que cada secuencia debe de tener para que se mantenga en el archivo final.
No es necesario repetir el comando para cada par de secuencias. Aplicaremos lo visto en la sección for-do-done.
Copia en un editor de texto, guarda como *.sh, cambia el modo a ejecutable con chmod y ejecútalo en la carpeta
Fastq, especificanto el número de procesadores a utilizar $1.
Para verificar la lista de archivos que usaremos en el loop:
ls *fastq | cut -d\_ -f1 | sort | uniq
Ahora, lo enviamos a una lista:
ls *fastq | cut -d\_ -f1 | sort | uniq > list.txt
#!/bin/bash
for i in $(<list.txt)
do
pandaseq -F -T $1 -f ${i}_1.fastq -r ${i}_2.fastq -w pandaseq_merged_reads/${i}_pd.fastq -g ${i}_
rm *log list.txt
done
O en una sola linea:
for i in $(<list.txt); do pandaseq -F -T 4 -f ${i}_1.fastq -r ${i}_2.fastq -w pandaseq_merged_reads/$
Para revisar que el ensamble de las secuencias se haya hecho de forma correcta revisaremos los archivos y haremos
nuevamente un análisis de calidad con FastQC:
cd pandaseq_merged_reads && cat *pd.fastq > all.pd.fq
fastqc --nogroup -f fastq all.pd.fq
Comparar lecturas, antes y después:
#Cuento cuántas secuencias tengo en el archivo C01D01_1.fastq
expr $(cat C01D01_1.fastq | wc -l ) / 4
> 10000
#Cuento cuántas secuencias tengo y de qué longitud en el archivo C01D01_1.fastq
cat C01D01_1.fastq | awk '{if(NR%4==2) print length($1)}' | sort -n | uniq -c
> 10000 150
#Cuento cuántas secuencias tengo en el archivo C01D01_2.fastq
expr $(cat C01D01_2.fastq | wc -l ) / 4
> 10000
1.7. Preprocesamiento
89
biote-2016, Release
#Cuento cuántas secuencias tengo y de qué longitud en el archivo C01D01_2.fastq
cat C01D01_2.fastq | awk '{if(NR%4==2) print length($1)}' | sort -n | uniq -c
> 10000 150
expr $(cat C01D01_pd.fastq | wc -l ) / 4
> 7193
#Cuento cuántas secuencias tengo y de qué longitud en el archivo de panda C01D01_pd.fastq
cat C01D01_pd.fastq | awk '{if(NR%4==2) print length($1)}' | sort -n | uniq -c
> 7193 253
1.7.3 Recorte de lecturas
En este caso en particular, las secuencias no tienen adaptador y tienen la misma longitud. En otros casos, como en
lecturas provenientes de 454 y Ion Torrent, es necesario cortar las secuencias a una misma longitud y eliminar los
adaptadores.
Trimmomatic
Como script:
for i in *fastq
do
name=$(basename "$i" .fastq) #Define la variable name con el nombre del archivo, sin la extensión
trimmomatic SE -phred33 $i $name.trimm.fastq MILEN:150 CROP:200 #Single End, input, output, longitu
done
Como una sola linea:
for i in *fastq; do name=$(basename "$i" .fastq); trimmomatic SE -phred33 $i $name.trimm.fastq MINLEN
1.7.4 Sub-muestra
Una de las limitantes de los análisis de 16S es el hardware con el que se dispone para realizar los análisis. Para fines
prácticos utilizaremos sólo 1000 lecturas por muestra. Si es posible, realizaremos el mismo análisis con el total de
lecturas, si la infraestructura de cada computadora lo permite.
Usaremos Seqtk para obtener sub-muestras.
Elegiremos 1000 lecturas al azar de cada una de las muestras. En el folder pandaseq_merged_reads ejecutamos
la siguiente linea:
seqtk sample -s 100 C01D01_pd.fastq 1000 > C01D01_pd1k.fastq
Para todas las muestras escribe el código para for-do-done y que el output sea una carpeta llamada
subset_qiime. El siguiente paso lo realizaremos con dicha secuencias.
ls *pd.fastq | cut -d\_ -f1 | sort | uniq
> list_pd.txt
for i in $(<list_pd.txt); do seqtk sample -s 100 ${i} 1000 > ${i}\_pd1k.fastq ; done
#Crear un subdirectorio y mover las lecturas obtenidas con seqtk
mkdir subset_qiime && mv *pd1k.fastq subset_qiime
90
Chapter 1. Índice
biote-2016, Release
1.7.5 QIIME
QIIME™ (chime) - Quantitative Insights Into Microbial Ecology- Es una linea de procesos bioinformáticos de codigo
abierto para realizar análisis de microbioma amplicones de 16S rRNA, provenientes de diferentes plataformas de
secuenciación.
Archivo de metadatos
El archivo de metadatos (mapping file) es generado por el usuario, en un formato *tsv. Este archivo contiene toda
la información de las muestras necesaria para realizar el análisis de datos. Para este tutorial usaremos el archivo
fasting_map_combine que está en $HOME/dataset/16Sdata/MappingFile. A continuación se muestra las
primeras lineas del archivo. La primer columna siempre es el #SampleID, seguido de las secuencias barcode para
cada muestra y el adaptador para el secuenciador. Las demás columnas corresponden a los diferentes metadatos que se
van a utilizar en el análisis, ya sea tratamiento, réplica, descripción, entre otros. Los nombres de los metadatos deben
estar en caracteres alfanuméricos.
#SampleID
C01.R1.D01
C01.R1.D02
C01.R1.D03
C02.R1.D01
C02.R1.D02
C02.R1.D03
BarcodeSequence LinkerPrimerSequence
GGAGACAAGGGA
C01
R1
AATCAGTCTCGT
C01
R2
AATCCGTACAGC
C01
R3
ACACCTGGTGAT
C02
R1
TATCGTTGACCA
C02
R2
TTACTGTGCGAT
C02
R3
Sample
D01
D02
D03
D01
D02
D03
Replicate
C01D01_pd.fna
C01D02_pd.fna
C01D03_pd.fna
C02D01_pd.fna
C02D02_pd.fna
C02D03_pd.fna
Extraction
C01_D01
C01_D02
C01_D03
C02_D01
C02_D02
C02_D03
Formato Mapfile:
• Debe de ser un archivo delimitado por tabulador (*.tsv)
• La primera columna debe ser “#SampleIDs”
• SampleIDs alfanumerico o puntos. No debe contener ‘_’
• La última columna debe ser “Description”
• Pueden ponerse columnas intermedias, las que sean necesarias
• Si se realiza QC con QIIME es necesario tener las columnas “BarcodeSequence” y “LinkerPrimer” en segunda
y tercera columna, respectivamente
Demultiplexing y filtrado de calidad de lecturas
cd $HOME/dataset/16Sdata/Fastq/pandaseq_merged_reads/subset_qiime
ln -s ../../../MappingFile/fasting_map_combine .
mkdir -p 00_raw/{a_qual,b_fasta}
Fastq -> Fasta
Los archivos se convierten a FASTA. Esto también lo puede hacer Pandaseq, sólo basta quitar la opción -F que
habilida el output en Fastq, y obtendremos archivos Fasta por default. También se puede convertir utilizando con el
script convert_fastaqual_fastq.py de QIIME.
for i in ./*fastq
do
echo "Converting $i"
convert_fastaqual_fastq.py -c fastq_to_fastaqual -f $i -o .
mv *qual 00_raw/a_qual
1.7. Preprocesamiento
91
InputF
biote-2016, Release
mv *fna 00_raw/b_fasta/
mv $i 00_raw/
done
Demultiplexing
El demultiplexing es asociar a qué archivo corresponde los barcodes, los cuales corresponden a los amplicones que se
secuenciaron; para ello utilizamos el archivo fasting_map_combine.
add_qiime_labels.py -i 00_raw/b_fasta/ -m fasting_map_combine -c InputFileName -o combined_fasta
count_seqs.py -i combined_fasta/combined_seqs.fna
1.7.6 Summary
Get the dataset
mkdir dataset
cd dataset
wget 148.247.152.169/data/16data.tar.gz
tar -zxvf 16data.tar.gz
cd 16data/Fastq
Concatenate file
cat *fastq > all.fq
QC
fastqc --nogroup -f fastq all.fq
Get the list for Pandaseq
ls *fastq | cut -d\_ -f1 | sort | uniq > list.txt
rm *zip
Run Pandaseq for all samples
mkdir pandaseq_merged_reads
for i in $(<list.txt); do pandaseq -F -T 4 -f ${i}_1.fastq -r ${i}_2.fastq -w pandaseq_merged_reads/$
QC for pandaseq output
cd pandaseq_merged_reads
cat *pd.fastq > all_pd.fq
fastqc --nogroup -f fastq all_pd.fq
rm *zip
Subsample
for i in *pd.fastq; do name=$(basename "$i" _pd.fastq); seqtk sample -s 100 $i 1000 > $name\_pd1k.fas
92
Chapter 1. Índice
biote-2016, Release
Qiime dir
mkdir qiime_subset
mv *pd1k.fastq qiime_subset
cd qiime_subset
ln -s ../../../MappingFile/fasting_map_combine .
mkdir -p 00_raw/{a_qual,b_fasta}
Fastq to fasta
for i in *pd1k.fastq; do echo "Converting $i" ; convert_fastaqual_fastq.py -c fastq_to_fastaqual -f $
1.8 Asignación taxonómica
1.8.1 Flujo de trabajo de QIIME
Using QIIME to analyze 16S rRNA gene sequences from microbial communities
1.8. Asignación taxonómica
93
biote-2016, Release
94
Chapter 1. Índice
biote-2016, Release
1.8.2 Asignación de OTUS
De Novo
Asignación de OTUs:
pick_de_novo_otus.py -i combined_fasta/combined_seqs.fna -o otus_de_novo
Identificación de secuencias quimeras:
wget http://greengenes.lbl.gov/Download/Sequence_Data/Fasta_data_files/core_set_aligned.fasta.imputed
identify_chimeric_seqs.py -m ChimeraSlayer -i otus_de_novo/pynast_aligned_seqs/combined_seqs_rep_set_
rm core_set_aligned.fasta.imputed
Filtrar tabla de OTUS a partir de una tabla de OTUS:
filter_otus_from_otu_table.py -i otus_de_novo/otu_table.biom -o otus_de_novo/otu_table_non_chimeric.b
Ordenar tabla de OTUS:
cat fasting_map_combine | cut -f1 > sample_id
sort_otu_table.py -i otus_de_novo/otu_table_non_chimeric.biom -o otus_de_novo/otu_table_de_novo_sorte
Gráficos de asignación taxonómica:
## Absoluta
echo "summarize_taxa:absolute_abundance True" >> qiime_parameters_abs
echo "plot_taxa_summary:type_of_file svg" >> qiime_parameters_abs
echo "plot_taxa_summary:dpi 200" >> qiime_parameters_abs
echo "plot_taxa_summary:chart_type bar" >> qiime_parameters_abs
summarize_taxa_through_plots.py -i otus_de_novo/otu_table_de_novo_sorted.biom -o taxa_summary_abundan
## Relativa
echo "plot_taxa_summary:type_of_file svg" >> qiime_parameters_rel
echo "plot_taxa_summary:dpi 200" >> qiime_parameters_rel
echo "plot_taxa_summary:chart_type bar" >> qiime_parameters_rel
summarize_taxa_through_plots.py -i otus_de_novo/otu_table_de_novo_sorted.biom -o taxa_summary_abundan
Closed
Asignación de OTUs:
echo "pick_otus:enable_rev_strand_match True" >> $PWD/otu_picking_params_97
echo "pick_otus:similarity 0.97" >> $PWD/otu_picking_params_97
pick_closed_reference_otus.py -i $PWD/combined_fasta/combined_seqs.fna -o $PWD/otus_closed_ucr97/ -p
Ordenar tabla de OTUs:
cat fasting_map_combine | cut -f1 > sample_id
sort_otu_table.py -i otus_closed_ucr97/otu_table.biom -o otus_closed_ucr97/otu_table_closed_sorted.bi
Gráficos de asignación taxonómica:
1.8. Asignación taxonómica
95
biote-2016, Release
## Abstoluta
echo "summarize_taxa:absolute_abundance True" >> qiime_parameters_abs
echo "plot_taxa_summary:type_of_file svg" >> qiime_parameters_abs
echo "plot_taxa_summary:dpi 200" >> qiime_parameters_abs
echo "plot_taxa_summary:chart_type bar" >> qiime_parameters_abs
summarize_taxa_through_plots.py -i otus_closed_ucr97/otu_table_closed_sorted.biom -o taxa_summary_abu
## Relativa
echo "plot_taxa_summary:type_of_file svg" >> qiime_parameters_rel
echo "plot_taxa_summary:dpi 200" >> qiime_parameters_rel
echo "plot_taxa_summary:chart_type bar" >> qiime_parameters_rel
summarize_taxa_through_plots.py -i otus_closed_ucr97/otu_table_closed_sorted.biom -o taxa_summary_abu
Open
Asignación de OTUS:
echo "pick_otus:enable_rev_strand_match True" >> ucrss_params
echo "align_seqs:min_length 65" >> ucrss_params
pick_open_reference_otus.py -i $PWD/combined_fasta/combined_seqs.fna -r $HOME/bin/qiime/db/gg_13_8_ot
Ordenar tabla de OTUS:
sort_otu_table.py -i otus_open_ucrss_usearch97/otu_table_mc2_w_tax.biom -o otus_open_ucrss_usearch97/
Gráficos de asignación taxonómica:
## Abstoluta
echo
echo
echo
echo
"summarize_taxa:absolute_abundance True" >> qiime_parameters_abs
"plot_taxa_summary:type_of_file svg" >> qiime_parameters_abs
"plot_taxa_summary:dpi 200" >> qiime_parameters_abs
"plot_taxa_summary:chart_type bar" >> qiime_parameters_abs
summarize_taxa_through_plots.py -i otus_open_ucrss_usearch97/otu_table_mc2_w_tax_open_sorted.biom -o
## Relativa
echo "plot_taxa_summary:type_of_file svg" >> qiime_parameters_rel
echo "plot_taxa_summary:dpi 200" >> qiime_parameters_rel
echo "plot_taxa_summary:chart_type bar" >> qiime_parameters_rel
summarize_taxa_through_plots.py -i otus_open_ucrss_usearch97/otu_table_mc2_w_tax_open_sorted.biom -o
1.8.3 Diversidad
Parámetros:
echo "alpha_diversity:metrics chao1,fisher_alpha,goods_coverage,observed_species,shannon,simpson,simp
96
Chapter 1. Índice
biote-2016, Release
De Novo
Diversidad alfa:
alpha_rarefaction.py -i otus_de_novo/otu_table_de_novo_sorted.biom -m fasting_map_combine -o adiv_de_
Rarefacción única:
biom summarize-table -i otus_de_novo/otu_table_de_novo_sorted.biom -o otus_de_novo/otu_table_summary_
number=`cat otus_de_novo/otu_table_summary_table | grep Min | cut -d\: -f2 | sed 's/^ //g' | cut -d\.
single_rarefaction.py -i otus_de_novo/otu_table_de_novo_sorted.biom -o otus_de_novo/otu_table_de_novo
Diversidad beta:
beta_diversity_through_plots.py -i otus_de_novo/otu_table_de_novo_sorted_$number\.biom -m fasting_map
Diversidad beta Jackknifed:
jackknifed_beta_diversity.py -i otus_de_novo/otu_table_de_novo_sorted_$number\.biom -t otus_de_novo/r
Closed
Diversidad alfa:
alpha_rarefaction.py -i otus_closed_ucr97/otu_table_closed_sorted.biom -m fasting_map_combine -o adiv
Rarefacción única:
biom summarize-table -i otus_closed_ucr97/otu_table_closed_sorted.biom -o otus_closed_ucr97/otu_table
number=`cat otus_closed_ucr97/otu_table_summary_table | grep Min | cut -d\: -f2 | sed 's/^ //g' | cut
single_rarefaction.py -i otus_closed_ucr97/otu_table_closed_sorted.biom -o otus_closed_ucr97/otu_tabl
Diversidad beta:
beta_diversity_through_plots.py -i otus_closed_ucr97/otu_table_closed_sorted_$number\.biom -m fasting
echo "Jackknifed beta diversity"
jackknifed_beta_diversity.py -i otus_closed_ucr97/otu_table_closed_sorted_$number\.biom -t $HOME/bin/
Open
Diversidad alfa: ::
alpha_rarefaction.py -i otus_open_ucrss_usearch97/otu_table_mc2_w_tax_open_sorted.biom -m fasting_map
Rarefacción única:
biom summarize-table -i otus_open_ucrss_usearch97/otu_table_mc2_w_tax_open_sorted.biom -o otus_open_u
number=`cat otus_open_ucrss_usearch97/otu_table_summary_table | grep Min | cut -d\: -f2 | sed 's/^ //
single_rarefaction.py -i otus_open_ucrss_usearch97/otu_table_mc2_w_tax_open_sorted.biom -o otus_open_
Diversidad beta:
beta_diversity_through_plots.py -i otus_open_ucrss_usearch97/otu_table_mc2_w_tax_open_sorted_$number\
Diversity beta Jackknifed:
jackknifed_beta_diversity.py -i otus_open_ucrss_usearch97/otu_table_mc2_w_tax_open_sorted_$number\.bi
1.8. Asignación taxonómica
97
biote-2016, Release
1.8.4 Archivos biom
Convertir archivos BIOM a JSON:
biom convert -i otus_closed_ucr97/otu_table_closed.biom -o otus_closed_ucr97/otu_table_closed_json.bi
Convertir archivos BIOM a TSV:
biom convert -i otus_closed_ucr97/otu_table_closed.biom -o otus_closed_ucr97/otu_table_closed_tsv.bio
1.8.5 Alfa diversidad - Phyloseq
En el directorio de OTU closed
En R:
library(phyloseq)
library(ggplot2)
biomfile <- paste0("otu_table_closed_json.biom")
data.biom <- import_biom(biomfile, parseFunction=parse_taxonomy_greengenes)
sdfile <- paste0('../fasting_map_combine')
data.mapping <- import_qiime_sample_data(sdfile)
Global <- merge_phyloseq(data.biom, data.mapping)
GP <- prune_taxa(taxa_sums(Global) > 0, Global)
sample_variables(GP)
p = plot_richness(GP, x = "Description", color= "Description",
p + geom_point(size = 3, alpha = 0.4)
measures = c("Observed", "Chao1", "Sh
GPst = merge_samples(GP, "Sample")
sample_data(GPst)$Sample <- factor(sample_names(GPst))
p = plot_richness(GPst, x = "Sample", color = "Sample",
p + geom_point(size = 3, alpha = 0.4)
measures = c("Observed", "Chao1", "Shannon",
1.9 Asignación funcional
PICRUST es una herramiente diseñada para predecir contenido metagenómico funcional de un gen marcador o genomas completos. PICRUST consiste en dos implementaciones: inferencia de contenido de genes e inferencia metagenómica. La inferencia de genes es proporcionada de manera precalculada por lo que nos enfocaremos en la inferencia metagenómica a partir de datos de
El contenido de genes por muestra se realiza a partir de una tabla de OTUS y en una tabla de contenido de genes
basada en OTUS. Los archivos de entrade deben de provenir de una aproximación closed u open
1.9.1 Closed reference
NOTA: Picrust utiliza biom-format-1.3.1, mientras que QIIME 1.9 utiliza biom-format-2.1.5. Por esta
razón utilizaremos el entorno virtual de python picrust_env.
Convertir de la codificación de biom de HDF5 a JSON.
98
Chapter 1. Índice
biote-2016, Release
biom convert -i otus_closed_ucr97/otu_table_closed.biom -o otus_closed_ucr97/otu_table_closed_json.bi
Iniciamos picrust_env (para biom-format-1.3.1)
picrust_env
Normalización de tabla de OTUS
• PICRUSt: Normalizar tabla de OTUS por el número predicho de copias de 16S.
normalize_by_copy_number.py
-i
otus_closed_ucr97/otu_table_closed_json.biom
crust_closed/otu_table_closed_json_norm.biom
-o
pi-
Asignación de funciones para el metagenoma predicho
• PICRUSt: Predicción de KOs.
predict_metagenomes.py
-i
picrust_closed/otu_table_closed_json_norm.biom
crust_closed/otu_table_closed_json_norm_ko.biom
-o
pi-
• PICRUSt: Colapsar datos jerárquicaCollapse de KOs a niveles de rutas KEGG.
categorize_by_function.py -i picrust_closed/otu_table_closed_json_norm_ko.biom
KEGG_Pathways -o picrust_closed/otu_table_closed_json_norm_ko_L3.biom
-l
3
-c
Finalizamos el entorno virtual picrust_env
deactivate
• Convertir biom al formato de STAMP.
biom_to_stamp.py -m KEGG_Pathways picrust_closed/otu_table_closed_json_norm_ko_L3.biom > picrust_closed/otu_table_closed_json_norm_ko_L3.spf
1.9.2 Open reference
La única diferencia con respecto a closed reference es que open reference debe filtar la tabla de OTUS, manteniendo
sólo aquellos OTUS que hayan dado match con la base de datos de Greengenes. Es necesario realizar el siguiente
paso.
filter_otus_from_otu_table.py -i otus_open_us97/otu_table_mc2_w_tax_open.biom -o otus_open_us97/otu_t
Luego procedemos de la misma manera que con closed reference:
biom convert -i otus_open_us97/otu_table_mc2_w_tax_open_picrust.biom -o otus_open_us97/otu_table_mc2_
picrust_env
normalize_by_copy_number.py -i otus_open_us97/otu_table_mc2_w_tax_open_picrust_json.biom -o picrust_o
predict_metagenomes.py -i picrust_open/otu_table_mc2_w_tax_open_picrust_json_norm.biom -o picrust_ope
categorize_by_function.py -i picrust_open/otu_table_mc2_w_tax_open_picrust_json_norm_ko.biom -l 3 -c
deactivate
biom_to_stamp.py -m KEGG_Pathways picrust_open/otu_table_mc2_w_tax_open_picrust_json_norm_ko_L3.biom
1.9. Asignación funcional
99
biote-2016, Release
1.9.3 STAMP
Generar archivo de metadatos:
cat fasting_map_combine | awk -v OFS='\t' '{ print $4, $6}'
> metadata_stamp.tsv
Ejecutar STAMP y cargar los archivos generados: Load data > Profile file | Group metadata
file.
Profile file: otu_table_closed_json_norm_ko_L3.spf
Group metadata file: metadata_stamp.tsv
1.10 Install
mkdir -p $HOME/bin
mkdir -p $HOME/bin/R
cat <<EOF >> $HOME/.bashrc
if [ -d "\$HOME/bin" ] ; then
export PATH="\$HOME/bin:\$PATH"
fi
EOF
cat <<EOF >> $HOME/.bashrc
tar -zxvf () {
if [ -f \$1 ] ; then
case \$1 in
tar xzf \$1
;;
*.tar.bz2)
tar zxzf \$1
;;
*.tar.gz)
bunzip2 \$1
;;
*.bz2)
rar x \$1
;;
*.rar)
gunzip \$1
;;
*.gz)
tar xf \$1
;;
*.tar)
tar xjf \$1
;;
*.tbz2)
tar xzf \$1
;;
*.tgz)
unzip \$1
;;
*.zip)
uncompress \$1 ;;
*.Z)
echo "'\$1' cannot be tar -zxvfed via tar -zxvf()" ;;
*)
esac
else
echo "'\$1' is not a valid file"
fi
}
EOF
echo "export R_LIBS=\$HOME/bin/R" >> $HOME/.bashrc
echo "PATH=\"\$HOME/bin/perl5/bin\${PATH+:}\${PATH}\"; export PATH;" >> $HOME/.bashrc
echo "PERL5LIB=\"\$HOME/bin/perl5/lib/perl5\${PERL5LIB+:}\${PERL5LIB}\"; export PERL5LIB;" >> $HOME/.
echo 'export PS1="\[$(tput bold)\]\[\033[38;5;39m\]\u\[$(tput bold)\]\[\033[38;5;15m\]@\[$(tput bold)
source $HOME/.bashrc
FastQC Linux/Win
cd $HOME/bin
wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.5.zip
tar -zxvf fastqc_v0.11.5.zip && rm fastqc_v0.11.5.zip
chmod +x FastQC/fastqc && ln -s FastQC/fastqc .
100
Chapter 1. Índice
biote-2016, Release
Trimmomatic
wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.36.zip
tar -zxvf Trimmomatic-0.36.zip && chmod +x Trimmomatic-0.36/trimmomatic-0.36.jar
ln -s Trimmomatic-0.36/trimmomatic-0.36.jar trimmomatic
rm Trimmomatic-0.36.zip
Pandaseq
git clone http://github.com/neufeld/pandaseq.git/
cd pandaseq/
./autogen.sh && ./configure && make && sudo make install
sudo ldconfig && cd ..
rm -rf pandaseq
Si da un error de libpandaseq.so.[número], ejecutar:
sudo ldconfig
Seqtk
git clone https://github.com/lh3/seqtk.git seqTK && cd seqTK
sudo make && mv seqtk ..
rm -rf seqTK
Virtualenv
sudo pip install virtualenv
Dependencias R
cat <<EOF > install_packages.R
install.packages(c('ape', 'biom', 'optparse', 'RColorBrewer', 'randomForest', 'vegan', 'gplots'), rep
source('http://bioconductor.org/biocLite.R')
biocLite(c('DESeq2', 'metagenomeSeq', 'phyloseq', 'Heatplus'))
q()
EOF
Install R packages
Rscript install_packages.R
rm install packages.R
Dependencias QIIME:
mkdir -p $HOME/bin/python/ && cd "$_"
virtualenv qiime
virtualenv picrust
echo "alias qiime_env=\"source \$HOME/bin/python/qiime/bin/activate\"" | tee -a $HOME/.bashrc
echo "alias picrust_env=\"source \$HOME/bin/python/picrust/bin/activate\"" | tee -a $HOME/.bashrc
source $HOME/.bashrcq
qiime_env
pip install
pip install
pip install
pip install
pip -U
setuptools numpy
biom-format scipy pandas
'scikit-bio==0.2.3' 'cogent==1.5.3' 'matplotlib==1.3'
Descargar QIIME
1.10. Install
101
biote-2016, Release
cd $HOME/bin && mkdir -p qiime
wget https://github.com/biocore/qiime/archive/1.9.1.tar.gz
tar -zxvf 1.9.1.tar.gz && rm 1.9.1.tar.gz
cd qiime-1.9.1
Instalar QIIME
python setup.py build
python setup.py install --prefix=$HOME/bin/qiime
cd ..
echo "export PATH=\$HOME/bin/qiime/bin:\$PATH" >> $HOME/.bashrc
source $HOME/.bashrc
Descargar Greengenes 18_8
mkdir -p $HOME/bin/qiime/db && cd "$_"
wget ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_8_otus.tar.gz
tar -zxvf gg_13_8_otus.tar.gz
rm *tar.gz
Instalar QIIME deploy
cd $HOME/bin
git clone git://github.com/qiime/qiime-deploy.git
git clone git://github.com/qiime/qiime-deploy-conf.git
python qiime-deploy/qiime-deploy.py $HOME/bin/qiime_software/ -f $HOME/bin/qiime-deploy-conf/qiime-1.
rm -rf $HOME/bin/qiime-deploy $HOME/bin/qiime-deploy-conf
deactivate
source $HOME/.bashrc
Usearch61
cd $HOME/bin
wget http://148.247.152.169/data/usearch61.tar.gz
tar -zxvf usearch61.tar.gz
rm usearch61.tar.gz
STAMP
sudo apt-get install libblas-dev liblapack-dev gfortran
sudo apt-get install freetype* python-pip python-dev python-numpy python-scipy python-matplotlib pyth
sudo pip install STAMP
Picrust
picrust_env
mkdir picrust
pip install 'numpy==1.5.1'
pip install 'cogent==1.5.3' 'biom-format==1.3.1'
wget https://github.com/picrust/picrust/releases/download/1.0.0/picrust-1.0.0.tar.gz
tar -zxvf picrust-1.0.0.tar.gz
rm picrust-1.0.0.tar.gz
mkdir -p picrust-1.0.0/picrust/data
wget ftp://ftp.microbio.me/pub/picrust-references/picrust-1.0.0/ko_13_5_precalculated.tab.gz -P picru
wget ftp://ftp.microbio.me/pub/picrust-references/picrust-1.0.0/16S_13_5_precalculated.tab.gz -P picr
cd picrust-1.0.0
python setup.py install --prefix=$HOME/bin/picrust
102
Chapter 1. Índice
biote-2016, Release
cd ..
rm -r picrust-1.0.0
echo "export PATH=$HOME/bin/picrust/bin:\$PATH" >> $HOME/.bashrc
echo "export PYTHONPATH=$PYTHONPATH:$HOME/bin/picrust/lib/python2.7/site-packages" >> $HOME/.bashrc
git clone https://github.com/mlangill/microbiome_helper.git
echo "export PATH=$HOME/bin/microbiome_helper:\$PATH" >> $HOME/.bashrc
deactivate
source $HOME/.bashrc
1.10. Install
103

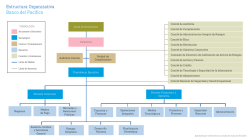

© Copyright 2026