
Revista Nº146 | ver
Junio de 2015 | Nº 146 de sus tareas diarias en el laboratorio Volumen 55 Nº 146 Junio de 2015 ISSN: 0558/1265 Uruguay 469, piso 2º B C1015ABI Buenos Aires, Argentina Tel: (54-11) 4373-0462 / 8900 (54-11) 4372-7389 Fax:(54-11) 4374-3630 www.safybi.org Si usted está buscando calidad y precisión en el laboratorio, nosotros lo tenemos. Descúbralo usted mismo y conozca nuestras balanzas, sistemas de agua, productos para el manejo de líquidos, control de calidad microbiológico y soluciones de filtración, respaldados por un equipo de servicio accesible a nivel mundial para ayudar a mantener el alto nivel de desempeño de sus productos Sartorius. Revista Revista SAFYBI | Lo ayudamos a cumplir con los requerimientos Revista SAFYBI SAFYBI: CLAPROMEDIS en Palabras del Presidente ANMAT: Uso racional de medicamentos Sartorius Argentina S.A. Tel. +54.11.4721.0505 [email protected] Visite nuestra revista online en www.sartorius.com/es Pesaje de Laboratorio Control de Calidad Microbiológico Manejo de Líquidos Filtración de Laboratorio Agua para Laboratorio Servicio www.safybi.org CONICET: Entrevista al Director de Vinculación Tecnológica USP: Materiales de ensayo clínico durante la distribución Revista Safybi COMITÉ EDITOR Comisión Directiva Presidente: Dr. Federico E. Montes de Oca Vicepresidente: Dra. Mirta B. Fariña Secretario: Dr. Germán C. Fernández Otero Prosecretaria: Dra. Susana B. Muñoz Director: Dra. Magdalena Nannei Consejo Asesor: Dr. Federico E. Montes de Oca Dr. Alberto García Dra. Mirta B. Fariña Dr. Germán C. Fernández Otero Dr. Alberto Grimoldi Corresponsal de Asuntos Universitarios: Dra. Susana B. Muñoz Tesorero: Dr. Claudio Vilariño Administración: Paula Mosquera Protesorero: Dr. Bernardo Gutman Fotografía: Marcela Marinangeli Vocales Titulares: Dra. Marta Fasanella Dr. Leonardo Fullone Dr. Alberto García Dra. María Celeste González Dr. Alejandro A. Meneghini Dr. Luis Moyano Diseño y diagramación: Virginia Gallino Comercialización: DKsiclo Group Cel: (011) 15-4474-2426 [email protected] Coordinación General: Lic. Doris Kaplán Vocales Suplentes: Dr. Mario Malaspina Dr. Augusto Pich Otero Dra. Viviana Ureña Comités de expertos Ver información actualizada en la página 10 Uruguay 469 2º B C1015ABI Bs. As., Argentina Tel.: (54-11) 4373-0462 / 8900 Tel.: (54-11) 4372-7389 Fax: (54-11) 4374-3630 [email protected] / www.safybi.org Cambios de domicilio, teléfono y correo electrónico Se ruega a los socios que notifiquen inmediatamente a la Secretaría todo cambio de domicilio, número telefónico o e-mail con el objeto de que no se interrumpan las comunicaciones de la Institución ni se envíen publicaciones a direcciones no vigentes. ISSN 0558-7265 Inscripta en el Registro Nacional de Propiedad Intelectual Nº 5221617 Propietario y Publicación Trimestral de la Asociación Argentina de Farmacia y Bioquímica Industrial, Uruguay 469 2º B, Buenos Aires, Argentina. Está incorporada al servicio de información bibliográfica internacional Pharmaceutical Abstracts Service. SAFYBI es publicada en Buenos Aires. Circula sin cargo entre profesionales y empresas asociadas a SAFYBI. Las opiniones vertidas en artículos y traducciones son exclusiva responsabilidad de los Señores Autores. Producción integral: DKsiclo Group E-mail: [email protected] Preimpresión e impresión: Artes Gráficas Buschi Ferré 2250/52 - C1437FUP C.A.B.A., Argentina SUMARIO SAFYBI 6Editorial 8 Palabras del Presidente Clapromedis: una iniciativa regional para impactar en salud 10 Comités de expertos ANMAT 14 Uso Racional de Medicamentos VOL 55 - No 146 - Junio de 2015 Conicet 16 Entrevista al Mg. Juan Carlos Soria Director de Vinculación Tecnológica (a/c) CONICET USP 18 Protección de la calidad de los materiales de ensayo clínico durante la distribución: estudios requeridos y momentos de aplicación Robert H. Seevers, PhDa y Rafik H. Bishara, PhDb Artículos Investigación y desarrollo Nuestros anunciantes 30 ¿Cuánto cuesta lanzar tarde? ¿Por qué tantas empresas fracasan en el desarrollo de productos? Lic. Eduardo Francisco Navarro 56 Aptar: Válvula con Bolsa de Aptar Pharma 38 Ophthalmic Squeeze Dispenser (OSD): ¿Un tamaño sirve para todos? Degenhard Marx, Matthias Birkhoff Packaging 48 Tapas plásticas para envases Aníbal H. San Juan Recursos Humanos 50 ¿Cómo es trabajar en un Laboratorio Farmacéutico en Suiza? Farmac. Lucas Fiorenza 58 Hitec: Calibración de contadores de partículas en aire Rodolfo Díaz, Favio Wainstein 64 Ionics: Inversión y crecimiento 65 IP&QA Solutions: Un nuevo concepto en soluciones para la industria farmacéutica 66 Sartorius: Una cuestión de seguridad Marcus Schütte 71 Tews: Medición de humedad y densidad con tecnología de resonancia de microondas Foto de tapa: Soluciones Integrales Sartorius para sus Procesos Biotecnológicos, desde la semilla hasta escala productiva. Ambr250 - Biostat STR - Sartoclear Dynamics Increased flexibility in microbial processes. Our single-use fermentor system. The Xcellerex™ XDR-50 MO fermentor system is purpose-built for microbial cultures and delivers performance you can trust under tough conditions. With a robust design, the single-use technology enables rapid batch-to-batch changeover for increased process efficiency and flexibility. upstream downstream single-use services When it comes to increasing facility utilization, we are a partner like no other. GE works. www.gelifesciences.com/BioProcess GE and GE monogram are trademarks of General Electric Company. Xcellerex is a trademark of General Electric Company or one of its subsidiaries. © 2014–2015 General Electric Company – All rights reserved. First published Apr. 2014. GE Healthcare Bio-Sciences AB, Björkgatan 30, 751 84 Uppsala, Sweden. Safybi Editorial Bienvenidos a esta edición de la Revista Safybi, cuando ya se está a muy pocas semanas de la EXPOFYBI 2015, que incluye el III Congreso Latinoamericano de Farmacia y Bioquímica Industrial, el XIV Congreso Argentino de Farmacia y Bioquímica Industrial y la XII JorFyBI. Los esperamos los días 4, 5, 6 y 7 de agosto, en el Centro Costa Salguero, Pabellón 6. Se debe destacar especialmente en este número, un artículo sobre la participación de nuestro Presidente, el Dr. Federico Montes de Oca en la convención de la USP. Agradecemos nuevamente la participación de la ANMAT con una nota sobre El uso racional de medicamentos donde nos introduce, entre otros aspectos, en la definición de la OMS y en el amplio espectro de actividades que abarca el mismo. Es para nosotros un motivo de genuino orgullo, presentar una entrevista que nos concediera el Director de Vinculación Tecnológica (a/c) del CONICET, Mg. Juan Carlos Soria, en el cual comenta las características de esta prestigiosa institución argentina y de las posibilidades de colaboración con la industria farmacéutica. Esperamos que este sea la primera de una serie de artículos para explorar y comentar campos de interés común. El tema de las Buenas Prácticas de Distribución (GDP por sus siglas en inglés) ha sido dominante en las regulaciones de distintas agencias regulatorias, tanto en América Latina como en Estados Unidos y Europa. Sin embargo, la consideración de la aplicación de las GDP en las muestras para ensayos clínicos no está contemplada específicamente y el artículo de estímulo que presenta la USP justamente tiene por objeto tratar ese tema y presentar un panorama claro de la situación. 6 revista safybi, junio 2015 La innovación es una de las cuestiones que más se debaten actualmente a nivel gubernamental y empresarial dentro del área de Investigación y Desarrollo. El Lic. Eduardo Francisco Navarro, experto en gestión de proyectos para la industria farmacéutica y cosmética, tratará la situación de cómo hacer efectiva la innovación, comenzando, en este primer artículo, por el análisis de la causa raíz de los fracasos en este campo. Siguiendo con el tema de la innovación en el área de Investigación y Desarrollo y su relación con el mercado, Degenhard Marx y Matthias Birkhoffse de Aptar Pharma presentan el diseño de un envase que representa una nueva oportunidad para los productos oftálmicos. En el área de Packaging, Aníbal H. San Juan, Jefe de Ingeniería de Empaque de Laboratorios Craveri y coordinador en temas de packaging para SAFYBI, demuestra los desafíos de una tapa plástica y comenta la nueva Norma D7860 de la ASTM que estandariza una serie de ensayos para tapas. El Farmac. Lucas Fiorenza es un colega que está viviendo una interesantísima experiencia laboral en Suiza y se ha prestado gentilmente para compartirla con todos nosotros a fin de que podamos evaluar similitudes y diferencias cuando trabajamos en entornos a los cuales no estamos habituados. Les agradeceríamos enviar sus comentarios a [email protected] Magdalena Nannei Director de la Revista Safybi Palabras del Presidente Clapromedis: una iniciativa regional para impactar en salud SAFYBI ha adherido recientemente a la Confederación Latinoamericana para la Investigación y Producción de Medicamentos e Insumos para la Salud, CLAPROMEDIS. Esta iniciativa surge de la necesidad detectada por varias asociaciones profesionales vinculadas al desarrollo y la producción de medicamentos e insumos para la salud de promover la búsqueda de soluciones comunes para Latinoamérica, frente a los permanentes desafíos que enfrenta la región. Observando el escenario de la región Latinoamericana, encontramos que las nuevas y crecientes exigencias regulatorias, la creciente competencia de los países asiáticos, la dificultad de las empresas para generar innovación o para participar en el mercado de los productos biotecnológicos, podemos comprender que muchos de estos factores ya están afectando y afectarán el desarrollo y producción de medicamentos e insumos para la salud en los países de nuestra región. CLAPROMEDIS tiene como visión, constituirse en “el foro regional de debate y discusión de ideas que fortalezcan el crecimiento y la sustentabilidad de la industria farmacéutica de los países latinoamericanos”, compartiendo conocimientos y experiencias de países que ya han superado o están superando dificultades, generando un punto de encuentro entre profesionales, autoridades sanitarias, científicos, académicos y referentes internacionales para cada uno de los temas a desarrollar. El objetivo planteado es unir a las Asociaciones profesionales de la Industria Farmacéutica, Universidades, Colegios Profesionales, Autoridades Sanitarias, Instituciones de Investigación y toda otra Organización relacionada con los objetivos de esta confederación latinoamericana, con el fin de compartir experiencias y estrategias que nos permitan continuar desarrollando tanto la actividad como a sus profesionales tanto de los ámbitos oficiales y académicos como del privado. Latinoamérica es una región con una realidad muy diferente a la europea o a la de América del Norte, sin embargo, la mayoría de la normativa que regula la actividad de la salud proviene de los países centrales. Es imperativo trabajar en foros técnicos que permitan una discusión informada de los complejos aspectos de nuestra actividad considerando las limitaciones de la región. Tenemos que ser conscientes de que nuestra población requiere insumos para la salud seguros y eficaces pero que también tenemos una deuda social que los países centrales tienen mucho más controlada: la accesibilidad. Realidades distintas conformadas por disímiles niveles de educación, presupuesto, infraestructura sanitaria, acceso al medicamento, entre otros, deben ser consideradas en forma integral para generar una política de salud que incluya una estrategia respecto a los medicamentos y demás insumos. Ser soberanos como países es también poder atender las necesidades de nuestra población y entre ellas se encuentro el acceso a medicamentos seguros y eficaces. Desarrollar las capacidades de la región para producir medicamentos y otros insumos para la salud en forma local es potenciar la matriz de investigación, desarrollo, industria y 8 revista safybi, junio 2015 capacidad exportadora en forma sinérgica. Ese ciclo virtuoso rebalsa en beneficios para la sociedad toda. En este marco es que SAFYBI hospeda el III Congreso Latinoamericano de Farmacia y Bioquímica Industrial organizado por CLAPROMEDIS, su XIV Congreso Argentino de Farmacia y Bioquímica Industrial, JORFYBI 2015 y EXPOFYBI 2015. El congreso científico tiene el sello de CLAPROMEDIS. Los diferentes ejes temáticos son abordados desde una perspectiva regional. Así es como las áreas de asuntos regulatorios, registro de medicamentos para enfermedades huérfanas y productos biológicos tienen como elementos destacados las mesas redondas donde podremos compartir la forma en la que distintos países de la región abordan la temática específica. Estarán presentes representantes de las distintas agencias regulatorias de la región como ANMAT (Argentina), COFEPRIS (México), ISP (Chile), ANVISA (Brasil), DIGEMID (Perú), entre otros. Hemos hecho un esfuerzo muy importante en abordar aquellos temas que actualmente ofrecen una complejidad singular. Entre ellos se destacan los productos biosimilares. La región y especialmente la Argentina, tienen una capacidad para el desarrollo de productos biosimilares que resulta en una inigualable ventaja competitiva a nivel global. Argentina tiene productos biosimilares (interferón, filgrastim, eritropoyetina) en el mercado hace más de 20 años. La historia ha demostrado que ha sido posible permitir acceder a la población a medicamentos basados en ingeniería de ADN recombinante seguros y eficaces aún sin un marco regulatorio existente al momento del registro. La responsabilidad y la capacidad científica y técnica de las compañías farmacéuticas y su equipo profesional dirigido por su Director Técnico como así también de la Autoridad Regulatoria, han sido los andamiajes de un desarrollo que nos debe llenar de orgullo. La biotecnología es el área de mayor desarrollo en la industria farmacéutica actual y lo será en el futuro aún más. En el 2014, cinco de las diez moléculas más vendidas en el mundo son de origen biotecnológico con un volumen en ventas de alrededor de 46.000 millones de dólares. Como profesionales de la salud, es nuestro deber conocer los aspectos científicos en los que se basa la biosimilaridad de los productos obtenidos por ADN recombinante. El futuro de la industria de la región depende de que el cuerpo de profesionales de la salud tenga un conocimiento científico basado en ciencia para que sea capaz de discernir entre un argumento que funciona como barrera de protección a un mercado multimillonario pero que a su vez limita el acceso a la población y drena los presupuestos de salud de las economías regionales de un argumento científico que asegura la seguridad y eficacia del producto. Crear el espacio para esta discusión científica y técnica es el objetivo de CLAPROMEDIS a nivel regional y de SAFYBI a nivel local. Sumar conocimientos, favorecer el intercambio de experiencias y la discusión científica en el marco de nuestras propias realidades para finalmente impactar en la salud de nuestra población. Dr. Federico Montes de Oca aptar.com/pharma Bag on ValVe Integridad del producto garantizada por la tecnología Máxima protección del contenido Fácil de usar y muy conveniente Fabricado para satisfacer los más altos estándares Delivering solutions, shaping the future. SAFYBI Comités de Expertos Aseguramiento de la Calidad Coordinador: Dr. Alejandro Meneghini Dr. Gustavo Hernán Aguirre Dra. Silvina Bessone Dr. Martín Dobovsek Dr. Guillermo Alberto Duda Dr. Hernan Martínez Abal Dra. Diana Oldani Dr. Pablo Ponziani Dra. Cecilia Sobrero Asuntos Regulatorios Coordinador: Dra. Viviana Ureña Dr. Víctor Capellino Dr. Ricardo Díaz Dra. Miriam Juarez Dr. Roberto Kuktosky Dra. Graciela Luque Dra. Carina Rismondo Dra. Nayla Gabriela Sabbatella Dra. Andrea Simanski Dr. Leonardo Fullone Dra. María Teresa Manzolido Dra. Virginia Peluffo Dr. Mauricio González Dra. María Laura Ciciliani Dra. Carolina Sian 10 revista safybi, junio 2015 Biotecnología Coordinador: Dr. Augusto Pich Otero Co-coordinador: Dr. Eduardo Spitzer Dr. Federico Montes de Oca Dr. Claudio Vilariño Dra. Laura Riera Dr. Esteban Fuentes Dra. Cristina Wiege Dr. Julio Vega Higiene, Seguridad y Gestión Ambiental Coordinador: Dr. Jorge V. Fernández Dr. Martín Dobovsek Dr. Pablo Ponziani Dra. María Alejandra Dumani Materias Primas Farmacéuticas Coordinador: Dr. Carlos Chiesa Farm. y Bioq. Melina Bisio Leandro Pintos (Estudiante Farmacia) Dr. Daniel Roberto Vega Bioq. Farm. María Emilia Callisto Sonia Faudone - Lic. en Química (UNC) Magister en Cs. Químicas Farm.Viviana Graciela Dabbene - Dra. en Ciencias Químicas Dra. Cecilia Beatriz Sobrero Microbiología Coordinador: Dra. Marta Fasanella Dra. Stella Maris Stagnaro Dr. Antonio Horacio Frade Dr. Gustavo García Dra. Juana Flota Productos Médicos y Esterilización Coordinador: Dra. Andrea Induni Dra. María Celeste González Dr. Damián Ramírez Spadaro Dra. María del Carmen Graziano Dra. Norma Maggia Química Analítica Coordinador: Dr. Bernardo Gutman Dr. Hernán Invenenato Dr. Eduardo E. Lopez Dra. Adriana Segall Dra. Silvia Chiarelli Dra. Alicia Gentile Tecnología Farmacéutica Coordinador: Dr. Jorge Ferrari Dr. Germán Fernández Otero Dr. Daniel Ventura Dra. Magdalena Nannei Dra. Laura Maurizio Dr. Fabian De Bonis Dr. Jorge Budzvicky Dra. Rosana Kelman Dr. Sebastian Barber Dr. Rodolfo Díaz Dr. Gabriel Monsalvo Dr. Javier Luca Dr. Javier Barber Distribución y Operadores Logísticos Coordinador: Dra. Susana B. Muñoz Dr. Pablo Álvarez Dra. Norma Amaya Dr. Pablo Bedo Dra. Liliana Kuharo Dr. Luis Moyano Dr. Germán Jaurena Dr. Mauricio Rittiner Dr. Juan Rolandi Dr. Sergio Di Trano Dra.. Mónica Pampim Dra. Estela Torres Dr. Cristian Troilo Comité de Tecnología Informática Coordinador: Dr. Gustavo Lago Dr. Pablo Martín Koval Dr. Martín Dobovsek ANMAT Uso racional de medicamentos Una manera eficaz de contribuir con la salud de la población El uso adecuado de los medicamentos, considerando a estos como un bien social, es indispensable para mejorar la calidad de vida. Los actores involucrados en esta gestión (dispensadores, prescriptores y población) intervienen con sus acciones en la comunicación e información correctas y necesarias para el beneficio de todos. Cuando los medicamentos, por diversa razones, no son usados de la manera apropiada pueden interferir con la salud y convertirse en un riesgo. Las estadísticas de la Organización Mundial de la Salud (OMS) muestran que “en el mundo más del 50 % de todos los medicamentos se recetan, se dispensan o se venden en forma inadecuada. Al mismo tiempo, alrededor de un tercio de la población mundial carece de acceso a medicamentos esenciales (entendiendo por tal a aquellos que cubren las necesidades de atención de salud prioritarias de la población) y el 50% de los pacientes los toman de forma incorrecta” - Promoción del uso racional de medicamentos: componentes centrales, OMS-Septiembre 2002. Tal como fue definido por la (OMS), se entiende por Uso Racional de los Medicamentos (URM) “la condición por la cual los pacientes reciben la medicación adecuada a sus necesidades clínicas, en las dosis correspondientes a sus requisitos individuales, durante un período de tiempo adecuado y al menor costo posible para ellos y para la comunidad (OMS, 1985)”. Es importante destacar que el URM abarca las actividades de adquisición, producción, distribución, almacenamiento, dispensa, prescripción y utilización, entendiéndolas como parte de una misma cadena. Por lo dicho anteriormente es indispensable concientizar sobre la prescripción adecuada de medicamentos. El proceso de prescripción racional, comprende un conjunto de aspectos que le permitirán al prescriptor apropiarse de las herramientas para pensar por su cuenta, comprender por qué en su país o en su institución se han elegido determinados protocolos de tratamiento habitual, y le enseñará cómo hacer el mejor uso de ellos. El uso incorrecto de fármacos abarca: •• la polifarmacia (consumo de varios medicamentos), •• el uso excesivo de antibióticos e inyecciones, •• la prescripción no ajustada a directrices clínicas, •• y la automedicación inapropiada Estas son las principales complicaciones que se encuentran con mayor frecuencia dentro del uso incorrecto de los medicamentos y traen aparejadas importantes consecuencias a nivel de la salud pública. 14 revista safybi, junio 2015 Se puede indicar que dentro de estas consecuencias se encuentran: •• La resistencia a los antimicrobianos, •• las reacciones adversas a los medicamentos, •• los errores de medicación, •• el desperdicio de recursos, •• la pérdida de confianza del paciente, etc. La prescripción de un fármaco es un proceso lógico deductivo, basado en información global acerca de un problema de salud. Como en todo acto médico, la prescripción de los psicofármacos debe ser precedida por la adecuada anamnesis, para llegar así en principio a un diagnóstico presuntivo, y de acuerdo a los cánones internacionales indicar el psicofármaco adecuado para ese padecimiento. En base a esto, se proponen los siguientes pasos para pensar el proceso de prescripción: •• definir el o los problemas del paciente, •• especificar los objetivos terapéuticos, •• diseñar un esquema terapéutico apropiado para el paciente, •• escribir la receta (iniciar el tratamiento) según lo dispuesto por Ley de promoción de la utilización de medicamentos por un nombre genérico N° 25649, •• brindar información, instrucciones y advertencias, •• supervisar la evolución del tratamiento Todo paciente tiene derecho a recibir información complementaria, clara y comprensible, así como la manera de utilizar el fármaco, su dosis, el intervalo de administración y la duración del tratamiento. El URM permite la obtención de importantes beneficios para la salud constituyéndose en una herramienta sanitaria. n CONICET Entrevista al Mg. Juan Carlos Soria, Director de Vinculación Tecnológica (a/c) CONICET En este número tenemos la satisfacción de presentar la entrevista que realizara la Directora de la Revista SAFYBI al Director de Vinculación Tecnológica del CONICET, con el fin de facilitar la divulgación de las actividades del CONICET relacionadas con la industria farmacéutica humana y veterinaria argentina, y por ese medio fortalecer emprendimientos mutuos orientados a mejorar la calidad de vida de la población de nuestro país. –Mg. Soria, con el propósito de dar conocer los desarrollos en los que está involucrado el CONICET y que interesan de manera sustancial a la industria farmacéutica, le agradecería nos indique, a manera de introducción, qué es el CONICET, cuáles son sus objetivos primordiales y en qué áreas focaliza sus investigaciones. –El CONICET, Consejo Nacional de Investigaciones Científicas y Técnicas, es la principal institución dedicada a la promoción científico-tecnológica en la Argentina. Su objetivo es la generación de conocimientos y la transferencia de los mismos a la sociedad y la formación de recursos humanos altamente calificados para llevar a cabo esas actividades de generación y transferencia de conocimientos. El propósito no es solo el avance del conocimiento científico y tecnológico, sino del desarrollo de la sociedad y la mejora de la calidad de vida de las personas. Para dar una idea de la envergadura del CONICET se debe considerar que a final del año 2014, contaba con alrededor de 8.500 investigadores, 9.500 becarios doctorandos y post-doctorandos y 2.500 profesionales de apoyo, es decir una organización de prácticamente 20.000 personas destinadas a generar conocimiento y desarrollar tecnología a lo largo y lo ancho del país. Su actividad se desarrolla principalmente en las ciencias agrarias, ingeniería y de materiales, ciencias biológicas y de la salud, ciencias exactas y naturales y finalmente ciencias sociales y humanidades. –¿Cuáles son los tipos de profesionales y las características de los integrantes del CONICET? –El CONICET incluye por una parte a los investigadores, que son quienes están abocados a generar conocimientos. Sus propósitos son las tareas de Investigación y Desarrollo, definidas por el Consejo como el trabajo intelectual y experimental llevado a cabo en forma sistemática para incrementar el volumen de conocimientos, incluido el conocimiento del hombre, la cultura y la sociedad y el uso de éstos para derivar nuevas aplicaciones. Estos Investigadores se dedican tanto a la Investigación como al Desarrollo Tecnológico. 16 revista safybi, junio 2015 El Personal de Apoyo está formado por personas con conocimientos y experiencia técnica que brindan su apoyo a las tareas de investigación y desarrollo. Por otra parte, se encuentran los becarios CONICET. Los Becarios pueden acceder a dos tipos de becas en diversas modalidades de convocatoria: las llamadas Becas Doctorales que están destinadas a realizar un plan de investigación original que forma parte de su tesis de doctorado, y las conocidas como Becas Postdoctorales. Es a través de las becas que el CONICET fomenta la formación de nuevos investigadores. –¿Tiene el CONICET convenios de investigación con otras instituciones? Nos parece interesante que nuestros lectores conozcan los mismos, los propósitos de colaboración y las razones que llevaron a establecer dicha colaboración. –El CONICET está formado por más de 220 Unidades Ejecutoras, es decir institutos o centros de investigación, de las cuales el 90% son de “doble dependencia”. Esto implica que existe un acuerdo con otra institución para su creación y gestión. Solo para nombrar alguna de esas Unidades Ejecutoras de Doble Dependencia relacionada con el área de salud, se puede mencionar al IBR, Instituto de Biología Molecular y Celular de Rosario, el cual está conformado por el CONICET y la Universidad Nacional de Rosario, así como también el Instituto de Investigación para el descubrimiento de Fármacos de Rosario. A su vez desde estas unidades, en conjunto con otros socios, han conformado Plataformas Tecnológicas en el área Biología Estructural y Metabolomica (PLABEM) y en el descubrimiento, diseño racional y desarrollo preclínico de sustancias químicas y biológicas con potencial terapéutico (SEDIPFAR) para brindar amplia gama de servicios de interés para la industria farmacéutica. También podemos hacer referencia al Centro de Investigaciones y Desarrollo en Inmunología y Enfermedades Infecciosas, este último formado por CONICET y la Universidad Católica de Córdoba que cuenta entre otros temas de investigación con una plataforma para el desarrollo anticuerpos monoclonales con aplicaciones diagnósticas y para tratamiento y el desarrollo de vacunas de administración oral. Existen incluso algunas Unidades Ejecutoras de Triple Dependencia, como sucede con el CIFASIS, Centro Internacional Franco Argentino de Ciencias de la Información y Sistemas, del cual participan el CONICET, la Universidad Nacional de Rosario y la AMU, Aix Marseille Université de Francia. Para este caso, que se dedica al área de la Informática, temas interesantes para los procesos industriales son por ejemplo, “Informática aplicada a ingeniería de procesos y control tolerante a fallas” o “Procesamiento digital de señales e imágenes, aplicaciones”. Nuestros investigadores como nuestros Centros de Investigación son nuestro principal activo para lograr desarrollos científicos y tecnológicos. En los últimos años, CONICET se ha planteado la importancia que implica que estos conocimientos y desarrollos puedan ser transferidos y que sean un motor para impulsar el desarrollo del país. –¿Cuál es el desafío más importante que enfrenta el CONICET en términos de vinculación y transferencia tecnológica? –Uno de los mayores desafíos es que el conocimiento que se genera en CONICET impacte positivamente a la sociedad, es decir que el conocimiento pueda contribuir en dar respuesta a los problemas de la sociedad, tanto de índole productiva como social. Para ello el Consejo ha desarrollado un conjunto de instrumentos para facilitar el proceso de transferencia de tecnología. A modo de ejemplo podemos mencionar, los Convenios de Investigación y Desarrollo, a través de los cuales se desarrollan proyectos en conjunto con empresas u otras organizaciones con el objetivo de desarrollar nuevos o mejores productos; también existen los Convenios de Licencia o Transferencia de Tecnología, donde el CONICET autoriza a un tercero el uso de una tecnología ya desarrollada por el mismo CONICET; luego están los Convenios de Asistencia Técnica, dónde el CONICET a través de un grupo de profesionales brinda su experiencia y conocimiento para asistir a una empresa u organismo público es una determinada temática. Por otra parte, se encuentran los Servicios Tecnológicos de Alto Nivel (STAN), los cuales son de rápido acceso y fácil contratación, los mismos se tratan de servicios tecnológicos ofrecidos que pueden o no requerir el uso de equipamiento e infraestructura del Consejo. Asimismo, se han desarrollado instrumentos que permiten a las partes llegar a un proyecto conjunto como los Convenios de Confidencialidad, y los Convenios de Transferencia de Material. De todas maneras, más que hacer una lista exhaustiva de instrumentos a disposición para la vinculación y transferencia tecnológica, lo que nos interesa es conocer el proyecto específico que necesita ejecutar la empresa o institución contraparte, y en función de ello buscamos contar con la mejor herramienta para poder llevar adelante el proyecto. En este sentido, CONICET ha desarrollado nuevas modalidades de colaboración para la ejecución de proyectos y hoy participa de varias decenas de consorcios públicos privados, plataformas tecnológicas e incluso ha conformado una empresa, Y-TEC, junto a YPF para el desarrollo de tecnologías asociadas al área energética. –Como pregunta final de esta entrevista le agradecería nos indique si usted considera que la actividad científica en Argentina es de alto nivel internacional. En caso que la respuesta sea afirmativa, cuáles son los elementos que apoyan esta posición. –La respuesta es sí, la ciencia en nuestro país es de alto nivel de calidad y tiene reconocimiento internacional. Me parece que una forma clara para demostrar este punto es revisar las estadísticas: por ejemplo en las estadísticas del Scimago Institutions Ranking (http://www.scimagoir.com), organismo que presenta recursos de apreciación de la ciencia para evaluar universidades e instituciones focalizadas en la investigación, el CONICET figura en la posición 79 sobre más de 5.100 instituciones de todo el mundo. Creo que estos datos hablan claramente de la importancia de la investigación científica dentro de nuestro país, de su reconocimiento internacional y señala en forma preponderante el rol del CONICET en la misma. n junio 2015, revista safybi 17 USP Protección de la calidad de los materiales de ensayo clínico durante la distribución: estudios requeridos y momentos de aplicación Estímulo al proceso de revisión Los artículos de estímulo no necesariamente reflejan las políticas de la USPC o del Consejo de Expertos de la USP Robert H. Seevers, PhDa y Rafik H. Bishara, PhDb Resumen Las guías actuales sobre los requisitos de almacenamiento para medicamentos se tratan en tres capítulos generales de la USP: 1) Buenas Prácticas de Distribución para Excipientes Farmacéuticos a Granel ‹1197›; Buenas Prácticas de Almacenamiento y Distribución para Medicamentos ‹1079› y Calidad de Productos Biotecnológicos: Pruebas de Estabilidad de Productos Biotecnológicos o Biológicos ‹1049›. Además, los capítulos ‹1197› y ‹1079› tratan las guías sobre los procesos de distribución de medicamentos, junto con una actualización propuesta al capítulo ‹1079› para agregar el capítulo Buenas Prácticas de Distribución ‹1083›. El propósito de este artículo de Estímulo es examinar las prácticas de almacenamiento y distribución de materiales de ensayo clínico, los cuales representan un subgrupo de medicamentos, además de proveer lo siguiente: 1) un resumen de las pruebas de estabilidad estándar y otras pruebas de estabilidad física (p.ej., análisis de congelación/descongelación y de impacto y vibración) que son necesarias para sustentar los procesos de almacenamiento y distribución para materiales de ensayos clínicos; 2) una recomendación para incluir una declaración independiente sobre transporte/distribución en el etiquetado del envase del medicamento que pueda ser usada por entidades que manipulan, almacenan o distribuyen los materiales de ensayo clínico. La revisión futura de los capítulos generales mencionados anteriormente y de cualquier otra revisión que trate la distribución o el almacenamiento de productos puede resultar favorable para aquellos que requieran la información presentada en este artículo de Estímulo. Introducción En años recientes (1–4), se ha prestado importante atención a la calidad de los medicamentos en su recorrido desde el fabricante hasta el paciente (5–8). Existen guías de organismos reglamentarios, de la USP y de la Organización Mundial de la Salud (OMS) a disposición sobre este tema (9–12). No obstante, estos esfuerzos se han centrado casi exclusivamente en los medicamentos aprobados. La sec- 18 revista safybi, junio 2015 ción Alcance del capítulo ‹1079› indica lo siguiente: “Aunque este capítulo no pretende tratar el almacenamiento y la distribución de ingredientes farmacéuticos activos (API), excipientes, productos radioactivos, reactivos, disolventes, dispositivos médicos, gases medicinales o materiales para ensayos clínicos cuyos requisitos de almacenamiento pudieran no haberse definido todavía (p.ej., medicamentos para ensayos clínicos de Fase I), los principios generales descritos en este capítulo pueden ser útiles si se aplican de manera selectiva o íntegra”. (2). Este artículo de Estímulo tiene el propósito de ampliar los conceptos de las Buenas Prácticas de Distribución (BPD) para materiales de ensayo clínico/productos medicinales para investigación y representa una actualización de un documento previo presentado por Bishara (13). Existen varias diferencias clave con respecto a la situación de los medicamentos aprobados que deben tenerse en consideración al cuestionarse la forma de aplicar las Buenas Prácticas de Distribución a los materiales de ensayo clínico/ productos medicinales para investigación. Lo primero y más importante es que la fase de ensayo clínico del ciclo de vida de un medicamento es un periodo de cambios significativos. Cuando se inicia el desarrollo, por lo regular se cuenta con un conjunto de datos muy limitado con respecto a la estabilidad del medicamento, la susceptibilidad a los extremos de temperaturas, la humedad y las condiciones físicas dinámicas tales como agitación, caída y presión. A medida que aumenta el conocimiento sobre el medicamento, se presenta la posibilidad de otros cambios, incluidos cambios en la formulación y en la forma farmacéutica—incluso si un fármaco se usa en una forma salina u otra, o como base libre. Mientras todo esto sucede, el número de pacientes en ensayos clínicos expuestos a un medicamento aumenta de la Fase 1 a la Fase 2 y a la Fase 3, a menudo en órdenes de magnitud. Finalmente, no debe olvidarse que una amplia mayoría de medicamentos que ingresan en ensayos clínicos no pasaran dicha etapa. Por consiguiente, resulta impráctico e inapropiado esperar un conjunto de datos de sustento para materiales de ensayo clínico/productos medicinales para investigación comparable a los que se pueden observar con un producto comercializado. Existe un aspecto adicional a considerar con respecto a la calidad de los materiales de ensayo clínico/productos medicinales para investigación. La destrucción de una carga de un medicamento aprobado como consecuencia de su exposición a variaciones de temperatura que están fuera de las variaciones aceptables, generará pérdidas monetarias significativas. Si una carga de materiales de ensayo clínico/ productos medicinales para investigación no puede usarse por la misma razón, pueden presentarse retrasos en la consecución de la investigación, lo que puede llevar a retrasos correspondientes en la aprobación del nuevo medicamento. En este caso, los pacientes tendrían que esperar más tiempo para obtener un medicamento necesario, además de que las compañías podrían perder cantidades aún mayores de ingresos. En este momento, las guías reglamentarias disponibles para materiales de ensayo clínico (14–16) no abarcan las buenas prácticas de distribución. El documento provisto por Bishara (13), citado anteriormente, se posiciona como el resumen disponible más actualizado. En general, el enfoque basado en riesgos propuesto para otros asuntos relacionados con el desarrollo farmacéutico (17, 18) puede extenderse de manera apropiada a la distribución. Los principios descritos en este artículo de Estímulo tienen como propósito su aplicación a los materiales de ensayo clíCarpe-ad-CatLab 200x140-F.pdf 1 26/01/15 08:55 nico, es decir, a los medicamentos terminados; sin embargo, también pueden usarse para fármacos, productos intermedios y formas farmacéuticas terminadas a granel. Proceso de desarrollo El desarrollo se inicia con el descubrimiento—hallazgo de moléculas que bloquean una enzima particular o que se unen a un receptor específico, u otros parámetros in vitro que indican que las moléculas pueden tener un efecto benéfico sobre un estado de enfermedad. Una vez que una molécula presenta un desempeño prometedor, es necesario desarrollar métodos analíticos para detectarla y valorarla. Para esto, se realiza un análisis de estrés en el que la molécula se expone al calor, a la oxidación, a ácido fuerte, a base fuerte, a la luz y a otras variables que permiten a los científicos desarrollar los patrones de degradación de la molécula y asegurar que la valoración detectará, no sólo la molécula madre, sino también sus productos de degradación y metabolitos más probables (19). A medida que la molécula se mueve en lo que se conoce como Fase 1, se continúa analizando en animales y, posteriormente, en humanos. Antes de iniciar dichas pruebas, por lo regular se cuenta con algunos datos de estabilidad del fármaco (ingrediente farmacéutico activo) en las condiciones de almacenamiento pretendidas. Asimismo, se puede contar con datos más limitados para el U AR • Amplio rango de pH (0-12,0) . OS AES EV ALFA NUCTOS • Farmacopea de Estados Unidos (En versiones: Libro, Online, CD). OD PR Soluciones integrales, incorporando innovación, calidad y servicio desde 1939 • Estándares para ICP y absorción Atómica. • Guía de Reactivos Cromatográficos. • Gran variedad de tamaños de partícula, entre 3 y 10 µ • Compendio de Suplementos Dietarios. • Mayor vida últil de las columnas. • Estándares de Referencia. • Alta eficiencia y resolución de las columnas. • Indicadores. • Flexibilidad para el desarrollo de métodos. • Catalizadores. • Cursos online. • Food Chemical Codex (FCC): Compendio de Normas reconocido internacionalmente para determinar la pureza y la identidad de ingredientes alimenticios Servicios de entregas regulares e inmediatas. CARPE SCHEIDER & CIA S.A. Godoy Cruz 2769 5 Piso. C1425FQK - CABA Tel.: +54 11 4776-0477 Fax +54 5276 - 9813 • Mejor sensibilidad para LC-MS . • Cromatografía Iónica. • Ácidos y Bases. • Soluciones Buffers. • Reactivos. • Mayor duración de las columnas. Bajos costos, amplia variedad de columnas para desarrollos. Servicios de entregas regulares e inmediatas. [email protected] www.carpescheider.com.ar AÑOS revista safybi 19 USP medicamento en las condiciones de almacenamiento pretendidas. ¿Cuál es el volumen de datos de estabilidad con los que se debe contar para comenzar los ensayos en seres humanos? Resulta difícil proveer una respuesta que se ajuste a todos los casos. Una estabilidad forzada suficiente es esencial para determinar las vías probables de degradación y para asegurar que los métodos analíticos sean adecuados. Cuando un fármaco ha demostrado ser estable durante muchos meses, pueden ser aceptables los estudios de estabilidad del medicamento simultáneos con los ensayos clínicos. Este es el caso particular cuando el medicamento es el mismo fármaco con modificaciones menores o nulas. Cuando existe un lote de desarrollo del fármaco o el medicamento fabricado mediante el proceso destinado a la fabricación del material de ensayo clínico, es probable que existan datos de estabilidad disponibles para dicho lote durante un periodo prolongado, los cuales deberían ser aplicables al material de ensayo clínico. El análisis de estabilidad estándar incluye el almacenamiento en la condición de largo plazo y una condición acelerada. Para productos a temperatura ambiente, la condición de largo plazo es 25 °C/60% de humedad relativa, y la condición acelerada es 40 °C/75% de humedad relativa. Muchos productos a temperatura ambiente se someten a estabilidad a 30 °C para contemplar su uso en los países de la Zona 20 revista safybi Climática 4 (20). Para productos refrigerados, las condiciones son 5 °C y 25 °C, respectivamente (19). Los estudios de largo plazo y acelerados no son las únicas pruebas requeridas para algunas moléculas o formas farmacéuticas. Cuando los datos de estabilidad indiquen riesgos debidos a las variaciones de temperatura, resulta apropiado contar con más datos para el medicamento antes de comenzar con los estudios en humanos. Por ejemplo, se puede colocar una partida para ensayo clínico o un lote de desarrollo representativo en estabilidad, y tan pronto como estén disponibles al menos 1 mes de datos aceptables de estabilidad acelerada, la partida o lote puede ser liberado para el ensayo clínico. El estudio de estabilidad continúa al mismo tiempo que el ensayo clínico. Esta práctica de estabilidad simultánea es necesaria para evitar los retrasos y los gastos que se generarían si se necesitasen datos de estabilidad más extensos antes de iniciar el ensayo clínico. De esta forma, el beneficio de iniciar el ensayo de manera prematura se equilibra en función del riesgo de que los datos de estabilidad adicionales pudieran presentar resultados o tendencias de estabilidad inaceptables, en cuyo caso se retiraría la partida del ensayo clínico. Para productos biológicos y para formas farmacéuticas sólidas orales de liberación modificada, puede existir un riesgo para la calidad por la exposición a condiciones de USP congelación. En el caso de productos biológicos, el riesgo es la aglomeración de las biomoléculas en solución, la cual podría llevar a la pérdida de potencia y a un aumento en la inmunogenicidad. Para formas farmacéuticas sólidas orales de liberación modificada, en los que la liberación de fármacos se controla mediante un recubrimiento polimérico, es posible que la congelación ocasione la fisura del recubrimiento. Esto puede generar una falta de confiabilidad en el patrón de liberación del fármaco y posiblemente la liberación masiva de la dosis. Por estas razones, se puede realizar un estudio de congelación-descongelación a los medicamentos de Fase 1 que se consideren en riesgo (8). El análisis de estabilidad que se realizó para la Fase 1 es informativo, pero si la formulación del medicamento ha cambiado al pasar a la Fase 2, como a menudo sucede, se requieren nuevos estudios. En la Fase 2, se analiza un medicamento en pacientes para determinar la dosificación apropiada y para obtener más información sobre la forma en que el medicamento se manipula in vivo. Lo anterior no necesariamente se aplica al fármaco, incluso si la vía sintética ha cambiado, a menos que una nueva impureza tenga el potencial de incrementar la velocidad de degradación (por ejemplo, un catalizador metálico). En la Fase 2, el medicamento puede analizarse nuevamente en las condiciones de largo plazo y aceleradas. Además, para productos biológicos y para formas farma- 22 revista safybi, junio 2015 céuticas sólidas orales de liberación modificada es apropiado realizar un estudio de congelación/descongelación. Debido a que la Fase 2 por lo general requiere un tiempo mayor y se aplica a más sujetos en más sitios de investigación que la Fase 1, existe la necesidad de una cadena de suministro para distribución más compleja. Por lo tanto, puede ser ventajoso realizar estudios de variación de temperatura que vayan más allá de la condición de estabilidad acelerada. Por ejemplo, en el caso de un medicamento refrigerado, la exposición a una temperatura de 30 °C durante varias horas no puede ser comprendida simplemente observando los resultados de estabilidad acelerada realizados a 25 °C. Por otra parte, un medicamento refrigerado puede comenzar a mostrar un cambio significativo a 25 °C, en cuyo caso analizar variaciones a temperaturas más altas no proveerían información útil. En los casos en los que un medicamento haya presentado cambios limitados en condiciones aceleradas, puede ser útil realizar estudios cíclicos de temperatura o de variaciones de temperatura (8). Los desafíos de la Fase 3 son interesantes y demandantes. El ensayo clínico se amplía para evaluar la seguridad y la eficacia del fármaco en un número significativo de pacientes. La estabilidad necesita sustentar los ensayos de la Fase 3 y el desarrollo de los datos requeridos para el registro. Si la formulación del medicamento ha cambiado desde la Fase 2, lo cual no es inusual, es necesario realizar USP Tituladores automáticos Autosampler hasta 18 muestras Karl Fischer Volumétricos y Coulométricos La más amplia gama de equipamiento de mesada para: pH, EC, DO, Fotómetros multiparamétricos, etc. Electrodo con tecnología Bluetooth inteligente Servicio Técnico Stand H5 Hanna Instruments Argentina S.A. Saavedra 1023 – Ciudad Autónoma de Buenos Aires, Argentina Tel.: (011) 4308-1904 / 1905 / 4807 [email protected] www.hannaarg.com 24 revista safybi, junio 2015 Sistema de Gestión de Calidad nuevos estudios de estabilidad de largo plazo y acelerados. Es durante la Fase 3 cuando se obtienen los datos de estabilidad para el registro. Este proceso requiere una cantidad significativa de tiempo. La norma ICH Q1A (19) indica que la expectativa son 12 meses de datos de estabilidad de largo plazo y 6 meses de datos acelerados al momento de la presentación de una solicitud de comercialización. Una mayor cantidad de datos de estabilidad permitirán una mayor vida útil para el medicamento aprobado (21, 22). Durante la Fase 3, los estudios adicionales sustentan la distribución del medicamento y es necesario realizarlos para obtener datos para el registro. Estos estudios pueden incluir el análisis de congelacióndescongelación de la formulación comercial pretendida (para productos biológicos y para formas farmacéuticas sólidas orales de liberación modificada), variaciones de temperatura y estudios cíclicos de temperatura (8). Para los productos biológicos, las autoridades reglamentarias esperan ver incluida la validación del transporte del medicamento en la validación general de la fabricación del medicamento. Se ha experimentado un cambio de perspectiva en la pasada década con respecto a este tema. Antes, la validación del transporte se revisaba como parte de la inspección del sitio de fabricación del medicamento previa a la aprobación; en la actualidad, la expectativa reglamentaria es revisar dicha información en las Secciones S.2.5 y P.3.5 Validación del Proceso en la presentación de solicitud para comercialización. Se debe tomar en cuenta que dicha expectativa abarca el fármaco y el medicamento. Lo anterior puede generar desafíos debido a que el envasado comercial final de un medicamento, en particular los envasados secundario y terciario tales como cartones y multi-empaques, podría no determinarse hasta una etapa muy tardía del proceso. Los impactos, la vibración y la presión también son riesgos para los productos biológicos formulados como líquidos. El análisis para establecer los riesgos para un medicamento particular es importante y puede realizarse usando protocolos estándar de la American Society for Testing Materials (Sociedad Estadounidense para el Análisis de Materiales) o de la International Safe Transport Association (Asociación Internacional de Transporte Seguro) (23, 24). Es importante recordar que el análisis en la Fase 3 es justo eso, un análisis. Muy a menudo, los medicamentos incumplen las expectativas clínicas en dicho momento. Cuando un medicamento incumple en la Fase 3, todo el esfuerzo invertido para determinar los riesgos para la calidad se transforma en dinero y recursos perdidos. Por esta razón, es necesario equilibrar los riesgos de un fracaso clínico durante la Fase 3 en función del riesgo de contar con un conjunto de datos de estabilidad incompleto o de un retraso en la presentación de la solicitud para comercialización. Miembro de CISQ Federation ISO 9001 Sistema de Calidad Certificado ISO 9001:2008 La Tabla 1 provee una perspectiva general sobre el análisis y presenta los momentos típicos en los que se realizan ciertas pruebas. Tabla 1.: Desarrollo del Fármaco: momento y tipo de prueba que se realiza para sustentar la distribución DescuFase 1 brimiento Fase 2 Fase 3 Presentación X X X X Ciclo de temperatura X X X X Desviación de temperatura X X X X Presión X X Impacto y vibración X X Prueba Estrésa X Congelación/descongelación b X a El análisis de estrés se define como la exposición de un fármaco o medicamento a extremos de pH, oxidantes, luz y temperatura (19). b El análisis de congelación/descongelación se realiza en productos biológicos y algunas moléculas pequeñas, en particular parenterales y formulaciones de liberación modificada (8). Número de lotes analizados Durante la fase de investigación del ensayo clínico, las pruebas descritas en la Tabla 2 por lo regular se llevan a cabo únicamente en un solo lote de fármaco o medicamento; a menudo, únicamente se encuentra disponible un lote. En el caso de la estabilidad para el registro, la expectativa es que se realice el análisis de largo plazo y acelerado en al menos tres lotes (19). Se debe considerar lo siguiente: El número de lotes de material que deben usarse para realizar el análisis de congelación-descongelación, de variación de temperatura, de ciclos de temperatura, de impacto, de vibración y de presión. El tipo de datos de validación del transporte que deben proveerse para sustentar una solicitud de comercialización. Aunque en la actualidad no existe una guía reglamentaria disponible sobre este tema, parece razonable y apropiado usar un número correspondiente de lotes para dichas pruebas como en el momento en que los resultados formarán parte de la solicitud para comercialización. Por ende, los autores recomiendan aplicar especial cuidado y análisis estadístico al decidir cómo se debe analizar una gran cantidad de lotes. Validación del transporte de productos biológicos Si se examinan las interacciones reglamentarias en la década pasada con respecto al tema de la validación del transporte de productos biológicos, en particular para los Estados Unidos, se observará que durante la parte temprana de dicho periodo, las firmas recibieron observaciones en el formulario 483 de la FDA durante las inspecciones previas a la aprobación, cuando las autoridades reglamentarias consideraron los datos de validación de transporte como faltantes o inadecuados. En años más recientes, la tendencia cambió a compromisos posteriores a la aprobación en lugar de observaciones 483. Se puede concluir que, al menos en el caso de los Estados Unidos, la expectativa de las autoridades reglamentarias ha cambiado de evaluar la validación del transporte para productos biológicos durante la inspección del sitio de fabricación previa a la aprobación (tanto para medicamen- Say hello to streamlined sterile connections in seconds, anywhere Say goodbye to bulky, expensive tube welders Available in multiple sizes, Kleenpak™ sterile connectors are rigorously validated under worst-case conditions, so you can be sure the connection remains sterile every time... even outside a controlled air environment. w: www.pall.com/allegro e: [email protected] Providing Flexible Solutions © 2010 Pall Corporation. Pall, , and Kleenpak are trademarks of Pall Corporation. The ® indicates a trademark registered in the USA. GN10.3621 junio 2015, revista safybi 25 USP Tabla 2.: Productos aprobados en EE.UU. con recomendaciones para transporte en la sección “Modo de Suministro” Medicamento Declaración INYECCIÓN DE AMINOÁCIDOS: solución acetato de lisina, leucina, fenilalanina, valina, isoleucina, metionina, treonina, triptofano, alanina, arginina, glicina, histidina, prolina, ácido glutámico, serina, ácido aspártico y tirosina Almacenar en el cartón cerrado; no exponer la solución a la luz hasta el momento de su uso. Se debe minimizar la exposición de los productos farmacéuticos al calor. Evitar el calor excesivo. Se recomienda almacenar el producto a 20 –25 °C (68 –77 °F). (Ver el capítulo USP ‹659› Requisitos de Envasado y Almacenamiento, Temperatura Ambiente Controlada.) La exposición breve a temperaturas superiores a 25 °C durante el transporte y el almacenamiento no afectará de manera adversa el producto. No se debe usar una solución que se haya congelado. CYANOKIT: hidroxocobalamina, inyección, polvo, liofilizado, para solución Forma liofilizada: Almacenar a 25 °C (77 °F); variaciones permitidas a 15°–30 °C (59–86 °F). (Ver las Advertencias y Requisitos Generales USP, Temperatura Ambiente Controlada). El Cyanokit puede exponerse durante periodos cortos a variaciones de temperatura comunes durante el transporte [15 días sometido a temperaturas que van de 5° a 40 °C (41° –104° F)], transporte en el desierto [4 días sometido a temperaturas que van de 5° a 60 °C (41°–140 °F)] y ciclos de congelación/descongelación [15 días sometido a temperaturas que van de -20° a 40 °C (-4° a 104 °F)]. Solución reconstituida: Almacenar hasta por 6 horas a una temperatura de no más de 40 °C (104 °F). No congelar. Desechar cualquier porción no usada después de 6 horas. Marcaine® 0,5% con epinefrina 1:200 000, inyección (como bitartrato; Clorhidrato de Bupivacaína y Epinefrina, Inyección, USP) Almacenar a 25 °C (77 °F). Se puede almacenar durante periodos breves durante el transporte a temperaturas de no más de 30 °C (86 °F). No permitir que se congele. VIVACAÍNA: clorhidrato de bupivacaína y bitartrato de epinefrina, solución para inyección Almacenar a 25 C (77 F). Se puede almacenar durante periodos breves durante el transporte a temperaturas de no más de 30 °C (86 °F). No permitir que se congele. Xalatan® latanoprost, solución oftálmica Almacenamiento: Proteger de la luz. Almacenar los frascos sin abrir en refrigeración a 2°–8 °C (36°–46 °F). Durante el transporte al paciente, el frasco puede mantenerse a temperaturas de hasta 40 °C (104 °F) durante un periodo no mayor a 8 días. Una vez que se abre el frasco, se puede almacenar a temperatura ambiente hasta 25 °C (77 °F) durante 6 semanas. tos como fármacos) al envío de datos de validación del transporte en la Solicitud de Autorización para Productos Biológicos. Se debe tener en cuenta que, en el caso del transporte, el término «validación» se usa en su sentido más amplio, el que significa demostrar en ciclos repetidos que un material puede ser transferido de un sitio a otro en condiciones controladas de temperatura. Este control se puede lograr mediante transportes con temperatura controlada (camiones, aviones), contenedores de transporte calificados, líneas de transporte cuidadosamente monitoreadas o, más probablemente, una combinación de estos. La validación de un proceso de fabricación es una situación en la que se controlan múltiples variables; la validación de un proceso de transporte es aquella en la que no se tiene control sobre variables tales como el clima, el tráfico y los retrasos en aduanas, y en la que, como resultado, el sistema sebe ser lo suficientemente robusto para tolerar una variabilidad importante en las condiciones. El Documento Técnico Común de la ICH (Common Technical Document o CTD, por sus siglas en inglés) fue creado antes de que se generara la expectativa de proveer datos de validación del transporte en el documento de registro (25). A pesar de esto, el Documento Técnico Común ha 26 revista safybi, junio 2015 provisto información en la que se considera apropiado proveer resultados de validación del transporte para el fármaco en la sección S.2.5 Validación del Proceso, y para el medicamento en P.3.5 Validación del Proceso. Implicaciones para el etiquetado del producto Si un medicamento cuenta con toda esta información, se debe determinar si existe una forma adecuada y efectiva de comunicarla a un nivel apropiado para diversas audiencias tales como pacientes, conductores de camiones, personal del almacén, entre otros. Una posibilidad es a través de la sección “Modo de Suministro” (How Supplied) de la etiqueta del producto. En la actualidad, esta sección presenta una declaración de almacenamiento recomendado, que por lo regular indica “Almacenar a 2 °– 8 °C (36 – 46 °F)”. Debería ser posible agregar las condiciones de transporte recomendadas, tales como las indicadas en la Tabla 2, lo cual se ha aplicado en diversos productos actualmente aprobados en los Estados Unidos. Una práctica similar ha sido la presentada por la Asociación Internacional de Transporte Aéreo (International Air Transport Association o IATA) en el Capítulo 17 de las Reglamentaciones para Cargas Perecederas (26), el cual indica el uso de una etiqueta independiente de tiempo/temperatura para el Monitoreo, registro y emisión de alarmas Testo Saveris • Integración de sondas de temperatura y humedad, así como transmisores de cualquier otra variable (ej. presión diferencial en salas limpias - punto de rocío en aire comprimido). • Combinación de sensores inalámbricos o Ethernet en un mismo sistema. • Operación autónoma, segura y eficiente. • Emisión de alarmas al superar valores límite, en forma sonora, óptica, SMS y/o e-mail. • Opción de soft de cumplimiento CFR21 parte 11. www.testo.com.ar/saveris Nuestro laboratorio de calibración y servicio técnico ofrece y garantiza un soporte adecuado y permanente. Av. Directorio 4901 - C1440ASB Buenos Aires, Argentina Tel.: (011) 4683-5050 - Fax: (011) 4683-2020 [email protected] - www.testo.com.ar USP transporte de materiales sensibles a la temperatura, incluyendo productos farmacéuticos. Conclusión Es importante proteger la calidad de los materiales para ensayo clínico durante el proceso de distribución. Existe una obligación ética para asegurar que los pacientes de ensayos clínicos reciban medicamentos de alta calidad. Además, existe una obligación económica para asegurar que los datos derivados de los ensayos clínicos sean válidos. Se han establecido recomendaciones en este artículo de Estímulo que cada organización debe adaptar a sus situaciones particulares. Las firmas deben capturar sus expectativas en los Procedimientos Operativos Estándar de Desarrollo de modo que los equipos las conozcan. Este artículo de Estímulo ha establecido recomendaciones para pruebas requeridas para sustentar los procesos de almacenamiento y distribución para los materiales de ensayos clínicos y proveer una práctica es- Referencias 1.USP. USP37–NF32, Buenas Prácticas de Distribución para Excipientes Farmacéuticos Granel ‹1197›. Rockville, MD: Convención de la Farmacopea de los Estados Unidos; 2014. 2.USP. USP37–NF32, Buenas Prácticas de Almacenamiento y Transporte ‹1079›. Rockville, MD: Convención de la Farmacopea de los Estados Unidos; 2014. 3.USP. USP37–NF32, Calidad de Productos Biotecnológicos: Pruebas de Estabilidad de Productos Biotecnológicos ‹1049›. Rockville, MD: Convención de la Farmacopea de los Estaods Unidos; 2014. 4.USP. Buenas Prácticas de Distribución ‹1083› [versión preliminar]. Pharmacopeial Forum. 40(2); 2015 [en línea, sólo en inglés]. 5. Parenteral Drug Association. PDA Technical Report 39. (TR 39) Guidance for temperature controlled medicinal products: maintaining the quality of temperaturesensitive medicinal products through the transportation environment. Bethesda, MD: Parenteral Drug Association; S2(1) revised 2007. https://store.pda.org/ProductCatalog/Product.aspx? ID=1154 6. Parenteral Drug Association. PDA Technical Report No. 46. (TR 46) Last mile: guidance for good distribution practices for pharmaceutical products to the end user. Bethesda, MD: Parenteral Drug Association; 2009. https://store.pda.org/productcatalog/Product. aspx?ID=1223 7. Parenteral Drug Association. PDA Technical Report 52. (TR 52) Guidance for good distribution practices (GDPs) for the pharmaceutical supply chain. Bethesda, MD: Parenteral Drug Association; 2011. https://store.pda.org/ProductCatalog/Product.aspx?ID=1200 8. Parenteral Drug Association. PDA Technical Report 53. (TR 53) Guidance for industry: stability testing to support distribution of new drug products. Bethesda, MD: Parenteral Drug Association; 2011. https://store.pda.org/ProductCatalog/Product.aspx?ID=1201 9. European Commission. Guideline on good distribution practice of medicinal products for human use. Official J Eur Union. 2013. http://eurlex.europa.eu/LexUriServ/LexUriServ.do? uri=OJ:C:2013:343:0001:0014:EN:PDF 10. Health Canada. Health Products and Food Branch Inspectorate. Guidelines for temperature control of drug products during storage and transportation (GUI0069). Health Canada; revised 2011. http://www.hcsc.gc.ca/dhp mps/alt_formats/pdf/compliconform/gmpbpf/ docs/GUI0069eng.pdf 11. Medicines and Healthcare Products Regulatory Agency. Rules and guidance for pharmaceutical manufacturers and distributors 2014 the ‘Orange Guide’. London: Pharmaceutical Press; 2014. http://www.mhra.gov.uk/Publications/Regulatoryguidance/Medicines/Othermedicinesregulatoryguidance/CON2030291 12. WHO. WHO Technical Report Series, No. 957, 2010, Annex 5. Good distribution practices for pharmaceutical products. Geneva, Switzerland: World Health Organization; 2010. http:// www.who.int/medicines/publications/TRS957_2010.pdf 13. Bishara RH. Good cold chain management practices for clinical trial materials/investigational medicinal products. AmPharm Outsourcing. 9(2), pp. 14–20 (2008). 14. FDA. Guidance for industry. Content and format of investigational new drug applications (INDs) for phase 1 studies of drugs, including wellcharacterized, therapeutic, biotechnology 28 revista safybi, junio 2015 tándar para compartir la forma en que un medicamento, incluyendo materiales de ensayo clínico, debe manipularse en los procesos de distribución para uso por parte de entidades a lo largo de la cadena de suministro. Se han provisto cinco ejemplos de etiquetado de “Modo de Suministro” aprobados por la FDA para demostrar esta práctica. Se sabe que esta información será necesaria a medida que el producto se mueve a lo largo de la cadena de suministro, y proveer el etiquetado aprobado por la FDA se considera una práctica razonable para comunicar los requisitos que sustentarán una investigación en caso de que se presenten variaciones de temperatura o de otro tipo de una entidad a otra. n Reconocimiento Los autores desean agradecer a la Dra. Mary Foster, Presidenta del Comité de Expertos de Envasado, Almacenamiento y Distribución de Medicamentos de la Farmacopea de los Estados Unidos (USP) por su revisión y comentarios durante la preparación de este artículo. derived products. 1995. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM074980.pdf 15.European Commission. Repeal of EU, DIRECTIVE 2001/20/EC, on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use, 2001. http://ec.europa.eu/health/files/clinicaltrials/2012_07/ proposal/2012_07_proposal_en.pdf 16. FDA. Guidance for industry. INDs for phase 2 and phase 3 studies: chemistry, manufacturing, and controls information. 2003. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070567.pdf 17.International Conference on Harmonisation. ICH Q8(R2) Pharmaceutical development. 2009. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/ Q8_R1/Step4/Q8_R2_Guideline.pdf 18. International Conference on Harmonisation. ICH Q9 Quality risk management. 2005. http:// www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/ Q9_Guideline.pdf 19. International Conference on Harmonisation. ICH Q1A(R2) Stability testing of new drug substances and products. 2003. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1A_R2/ Step4/Q1A_R2 Guideline.pdf 20.Seevers RH, SchantzShirley RD. Regulatory strategy for longterm stability conditions to support submission in zone IV countries. Pharm Outsourcing. 2011; 12(3):44–51. 21. International Conference on Harmonisation. ICH Q1E Evaluation for stability data. 2003. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1E/ Step4/Q1E_Guideline.pdf 22.International Conference on Harmonisation. ICH Q5C Stability Testing Of Biotechnological/Biological Products. 1995. http://www.ich.org/fileadmin/Public_Web_Site/Training/ ASEAN_Q5C_workshop_May_2011/SESSION_Ia_ICH_Q5C.pdf 23. ASTM D416909 Standard practice for performance testing of shipping containers and systems. 2009. http://www.astm.org/Standards/D4169.htm 24. International Safe Transit Association. ISTA 3 series: general simulation performance tests. 2008. http://www.ista.org/forms/3Aoverview.pdf 25. International Conference on Harmonisation. ICH. The common technical document. 2000. http://www.ich.org/products/ctd.html 26. International Air Transport Association. IATA. In: Perishable cargo regulations. 7th ed. Chapter 17. 2013. http://www.iata.org/publications/Pages/perishablecargo.aspx a Principal Regulatory Scientist, Eli Lilly and Company. b Technical Advisor and Chair, Pharmaceutical Cold Chain Interest Group (PCCIG), Parenteral Drug Association (PDA). Dirigir la correspondencia a: Desmond Hunt, PhD, Enlace Científico Sénior, USP, 12601 Twinbrook Parkway, MD 208521790; 301.816.8341; email [email protected]. Investigación y Desarrollo ¿Cuánto cuesta lanzar tarde? ¿Por qué tantas empresas fracasan en el desarrollo de productos? Lic. Eduardo Francisco Navarro Una de las características clave de las firmas innovadoras es su capacidad para desarrollar nuevos productos exitosos en el mercado. Sin embargo, una investigación de 2012 de la American Productivity and Quality Control indica que en promedio sólo el 50% de los nuevos lanzamientos alcanzan sus objetivos económico-financieros (ventas, market share y rentabilidad). El 25% más exitoso logra hasta 2,5 veces mejor performance que el 25% de la base. En esta serie de artículos, se brindarán algunas pautas para ayudar a organizaciones de diversos tamaños a gestionar esta compleja actividad. Ver Fig.1. 1) Introducción al desarrollo de nuevos productos Un creativo no necesariamente es un innovador. Por sí sola, la idea no es suficiente. Para poder hablar de innovación, es necesario llevarla a la práctica y que produzca algún tipo de efecto (positivo o negativo) en el ámbito al cual está orientada. Innovar es, entonces, dar vida a una idea. Un producto (o servicio) exitoso en un mercado es fruto de una (o varias) muy buenas ideas que han sido desarrolladas gracias a procesos. De esto se trata la innovación en productos. 1.1) Los tipos de innovación en productos Adhiriéndonos a una de las corrientes de estudio de esta disciplina (Crawford & Di Benedetto, 2006)1 se utilizarán en forma indistinta las expresiones innovación en productos y desarrollo de productos para definir cualquiera de los siguientes casos: Productos que son nuevos para el mundo o productos realmente nuevos. En su momento, el teléfono celular o el motor de combustión interna. Productos nuevos para la empresa o nuevas líneas de producto. Por ejemplo, el iPhone o la línea de tés saborizados de Starbucks –Tazo. Agregados a líneas de producto existentes. Por ejemplo, el tren de alta velocidad ACELA a los servicios existentes de trenes de Amtrak o las botas de lluvia a la línea de calzado deportivo Figura 1: Facturación y ganancias de nuevos productos (lanzados de Lacoste. dentro de los últimos tres años). Muestra: 211 empresas. Año de análisis: 2012. Fuente: Cooper, Robert G. & Edgett, Scott (March-April 2012). Best practices in the idea to launch process and its governance. Research and technology management. Pp. 43-54. Mejoras incrementales o revisiones a productos existentes. Por ejemplo, el nuevo iPhone 6 o las actualizaciones permanentes que se le realizan al proceso de fertilización in-vitro. Reposicionamientos de productos. Por ejemplo, la aspirina para evitar problemas cardiovasculares o el yoga como método para combatir el estrés. Reducciones de costos. Por ejemplo, cambios de proveedores de materias primas, material de empaque o de procesos de tercerización. 1.2) ¿Por qué es importante el desarrollo de productos? ¿Por qué suele decirse que una organización no innovadora ve amenazada su subsistencia? 30 revista safybi, junio 2015 Investigación y Desarrollo Diversos factores han ido variando con el correr de los Fuente: Elaboración propia. tiempos. Entre los principales, se encuentra el acortamiento del ciclo de vida de los productos motivado por la rapidez de la innovación, la gran cantidad y globalización de competidores y la creciente velocidad de cambio de las sociedades. Según una investigación de la consultora McKinsey titulada Innovación y Liderazgo2 (basada en una encuesta a 700 ejecutivos a nivel mundial), la innovación es uno de los tres principales factores que explicarán el crecimiento, el desempeño y la valuación de las compañías en los próximos tres a cinco años. Figura 2: Secuencia virtuosa para el desarrollo de nuevos productos. 1.3) El portafolio de productos Una organización podría tener un excelente proceso de innovación, ejecutado por los mejores profesionales, coordinados por el mejor gerente de proyectos y, sin embargo, sólo obtendría pobres resultados si desarrollara los productos equivocados. Precisamente, una debilidad común en muchas organizaciones reside en el proceso de selección de los nuevos productos que deben integrar el portafolio. Con esto, nos referimos a la inexistencia o pobre definición de criterios para identificar las buenas ideas de productos y los proyectos con potencial de éxito o a la inacción para desactivar proyectos que no deberían seguir adelante. En un estudio de la consultora estadounidense NewProd, se descubrió que la priorización de los proyectos es una de las áreas más débiles.3 En la figura 2 se muestra en forma resumida la secuencia ideal para conseguir resultados de alta calidad en el Proceso de Desarrollo de Productos. 2) Las causas frecuentes de fracaso en el desarrollo de productos De acuerdo con una investigación de Cooper4 sobre 211 empresas, alrededor de 25% de la facturación y ganancias de una empresa corresponde a productos que no existían tres años antes (Ver Fig. 1). En este mismo trabajo, se destaca que los mejores en prácticas de innovación logran casi un 50 por ciento de su facturación y ganancias del portafolio de nuevos productos. Es muy típico encontrar a las compañías debatiéndose 32 revista safybi, junio 2015 en idas y vueltas, marchas y contramarchas y tiempos que parecen nunca terminar, para dar con un producto que logre ser exitoso en el mercado. 2.1) La evidencia del fracaso El primer paso para construir un área eficaz de desarrollo de productos consiste en comprender los problemas que enfrenta la organización en esta función. Aunque ésta aún no posea un proceso o área formal de innovación será interesante que analice esta evidencia para estar en alerta durante la implementación formal y a lo largo de las primeras experiencias. Tiempos de desarrollo de productos muy prolongados y discontinuación del desarrollo en etapas avanzadas. Ambos resultan en alta incertidumbre, lo que impacta directamente en una dificultad extrema de los Gerentes de productos para planificar tanto su portafolio a mediano y largo plazo como las campañas de lanzamiento en consonancia con las necesidades de los mercados. Atributos del producto diferentes a los esperados. Cuando el desarrollo se aproxima a las etapas finales, es frecuente que los atributos de productos difieran parcialmente con lo esperado por los Departamentos de Marketing y/o la alta gerencia. Esto suele generar cambios de último momento que implican tensiones y conflictos entre los miembros dedicados al desarrollo de productos y otros niveles y sectores, así como retrasos en la disponibilidad. Clientes insatisfechos o baja demanda. El producto no es bien recibido en el mercado o por el público objetivo, produciendo gran cantidad de reclamos y un bajo nivel de demanda. Decisiones y cambios permanentes. Muchos productos en desarrollo caen en un círculo vicioso, envueltos en un ir y venir de decisiones cambiantes, que no permite su lanzamiento en el tiempo requerido por el mercado. Pobre calidad del trabajo. La calidad de los entregables y la creatividad a lo largo de las etapas de desarrollo suele ser pobre. Alto nivel de frustración y descontento. Los miembros de los equipos de desarrollo se sienten frustrados porque el lanzamiento de nuevos productos se vuelve un objetivo en extremo difícil de alcanzar y por los constantes “retrabajos” que deben realizar a lo largo del proceso. Desconocimiento de la figura de cliente interno. Los responsables de generar e impulsar las ideas, generalmente los Gerentes de Producto, no son tratados como clientes sino como un área más de la organización. Bajo compromiso con la innovación. Los miembros demuestran mayor compromiso con las áreas funcionales a las que pertenecen que con los equipos de desarrollo de nuevos productos. Investigación y Desarrollo 2.2) Causas raíz de los fracasos En el apartado anterior, hemos presentado los principales síntomas que evidencian el fracaso en los procesos de desarrollo de productos. Ahora bien, ¿cuáles son las principales causas que provocan estos problemas? Inexistencia de una estrategia de nuevos productos y tecnológica O puede faltar la comunicación fehaciente a todos los niveles de la organización. Falla o no existe el análisis de alineación de la nueva idea o concepto de producto con dicha estrategia. Ausencia o deficiencias en el proceso de diseño y desarrollo de productos En ocasiones, la organización carece de un proceso de desarrollo formal, éste no ha sido consensuado entre las áreas o personas involucradas o su calidad es pobre. También pueden existir fallas en la comunicación a los involucrados. Falta de gates o aprobaciones a lo largo del proceso. En consonancia con el punto previo, los chequeos permanentes y en momentos específicos del proyecto para verificar variables claves del nuevo producto son fundamentales. Son aquellas que nos permiten “parar” o “dar autorización” para seguir adelante. Lo que suele suceder es que estas gates no están incorporadas dentro del proceso, no son realizadas o, si se hacen, no son demasiado rigurosas como para eliminar un proyecto inviable. Falta de metodología para gestionar los proyectos No se aplica una metodología probada para hacer la evaluación, planificación y seguimiento de los proyectos, ni para liderar los equipos, ni para generar una mayor integración de los involucrados que suelen pertenecer a áreas en competencia. Fallas en los estudios de mercado. Los estudios de mercado no se realizan en profundidad (o directamente no se realizan), dando como consecuencia una pobre orientación al mercado y entendimiento de las necesidades y deseos de los clientes. Requerimientos no consensuados, incompletos o inexistentes (Moving target). Con el pretexto de avanzar rápidamente sobre el nuevo desarrollo se omite, al inicio del proyecto, la discusión, evaluación de factibilidad y formalización de los requerimientos del nuevo producto a desarrollar. Esto suele resultar en varios puntos de vista entre los involucrados que causan cambios en las características de los productos en etapas avanzadas del mismo, con los “retrabajos”, mayores tiempos y recursos que esto implica Falta de respaldo (sponsorship) de la Alta Gerencia La consecuencia última que genera este problema impacta directamente sobre la función vital de innovación e 34 revista safybi implica que la organización no contará con un portafolio razonable de productos que evolucione año tras año. La inexistencia de respaldo o el desinterés de la alta dirección permean en los cuadros de management inferiores dando el “ejemplo” sobre qué es importante y qué no lo es dentro de los objetivos a cumplir. Las secuelas más visibles serán la imposibilidad o altísima dificultad a la hora de implementar el proceso de desarrollo y las mejores prácticas asociadas al mismo y la reticencia y bajo compromiso de la gente para con esta función y el área que lidere el esfuerzo. Manejo ineficiente de los recursos humanos y materiales. Dentro de esta categoría ingresan varios problemas. La mayoría, se refiere a una estructura organizativa e incentivos (económicos y de evaluación de desempeño) que hacen que los integrantes de los equipos de proyectos prioricen su lealtad y compromiso al área funcional en detrimento de los procesos de desarrollo. Falta de empowerment y reconocimiento de los logros. En empresas donde el involucramiento de la alta gerencia en temas cotidianos y en detalles es usual, los especialistas que componen el equipo de proyectos pueden encontrarse en inferioridad de condiciones para sostener su punto de vista técnico. Asimismo, los equipos pueden enfrentarse con un escaso reconocimiento tanto al final del desarrollo como a medida que se van cumpliendo los hitos del proyecto. Doble función incompatible. Los Gerentes de Productos no pueden realizar las actividades de gestión de los proyectos en forma permanente y focalizada porque deben dedicar la mayor parte de su tiempo a las actividades de gestión de las líneas de productos existentes para asegurar su rentabilidad. Falta de integración de los miembros del equipo. Aunque la multidisciplinariedad de los equipos pide “desesperadamente” actividades de integración, éstas no suelen ser la norma en las organizaciones. Entrenamiento de los colaboradores. Deficiencia en la actualización (capacitación) de conocimientos y prácticas de los miembros de los equipos dedicados a esta actividad. Gran poder de la estructura funcional. Este es un franco inhibidor de la eficiencia, la sinergia de equipo y fuente de las llamadas actitudes “de silo” (por analogía con la aislación que tiene en el campo un silo de otro, son compartimientos sin comunicación entre sí). Las probabilidades de fracaso se incrementan en compañías con estructuras basadas en áreas funcionales opuestas y que compiten en vez de una estructura matricial formalizada, basada en la colaboración. Los gerentes funcionales, en ciertos casos, no “compran” la idea porque les resta tiempo a sus recursos humanos para dedicarse a actividades del área funcional. revista safybi 35 Investigación y Desarrollo Estructura espacial y cercanía de los miembros de los equipos. Si bien pudiera parecer un tema menor, la interacción personal de los miembros durante el desarrollo de los proyectos es un factor importante para el éxito del trabajo en equipo. Sin embargo, puede encontrarse una estructura espacial de las oficinas no adecuada y miembros diseminados por varias locaciones que entorpecen la fluidez del proceso. Las dificultades o el desafío principal a los que se enfrentará quien decida liderar la iniciativa son diversos, sin embargo, la mayoría de ellos pueden englobarse bajo el paraguas “cambio en la cultura de trabajo” donde aparecen gran cantidad de variables blandas a tener en cuenta. Y como es sabido los cambios culturales son los más complejos y los más lentos porque requieren que la gente se adapte, pierda el miedo y el escepticismo a lo nuevo. Quien deba encarar este proceso de cambio tendrá que ser muy versátil para adaptarse a perfiles sumamente heterogéneos, y flexible para moverse hábilmente entre diferentes niveles de la organización. El premio es grande: ser parte del selecto grupo de compañías más innovadoras y obtener beneficios por encima de la media, en temas tan importantes como rentabilidad, crecimiento de mediano y largo plazo, posibilidad de competir en mercados más exigentes, y, al mismo tiempo, ser una organización que atrae y retiene a los mejores profesionales. ¿Un interesante círculo virtuoso, no? 36 revista safybi, junio 2015 ¿Cómo lograr este cambio tan complejo? En el próximo artículo se presentan las mejores prácticas y recomendaciones para su correcta implementación en tres áreas fundamentales: 1) estrategia y cultura, 2) recursos humanos y 3) procesos y metodologías. n Referencias 1Crawford, Merle C.; DiBenedetto Anthony (2003). New Products Management, 7th Edition, Boston: Irwin/McGraw-Hill. 2Joanna Barsh, Marla Capozzi y Jonathan Davidson, Innovation and Leadership, McKinsey, January 2008. 3Cooper, Robert G. (2001). Winning at New Products: Accelerating the Process from Idea to Launch, 3rd ed. Reading, MA: Perseus Books, Pág 215. 4 Cooper, Robert G. & Edgett, Scott (March-April 2012). Best practices in the idea to launch process and its governance. Research and technology management. Pp. 43-54. Eduardo F. Navarro es Licenciado en Comercio Internacional y obtuvo su Maestría en Administración de Empresas en la Universidad Di Tella. Posee una experiencia de 17 años como Project Manager tanto en empresas locales como en multinacionales. Está especializado en la Gestión de proyectos de desarrollo de productos y remediaciones con foco en la Industria Farmacéutica y Cosmética, habiendo dedicado los últimos 10 años a esta actividad. Ha creado, reestructurado y administrado oficinas de proyectos para desarrollo de nuevos productos, liderando equipos interdisciplinarios locales y multiculturales tanto 100% internos como con estructura de desarrollo tercerizada. En su haber se cuenta la dirección de más de 50 proyectos. Es tutor de tesis del MBA en la Universidad Di Tella desde el 2006. Ha dictado cursos y talleres universitarios y también in-company sobre gestión de la Innovación, desarrollo de productos y proyectos. En los últimos 10 años ha publicado una serie de artículos sobre temas de su especialidad. Más información en http://ar.linkedin.com/in/eduardofrancisconavarro/es Materias Primas para la Industria Farmacéutica Elaboración de Cápsulas de Gelatina Blanda dromex.com Investigación y Desarrollo Ophthalmic Squeeze Dispenser (OSD): ¿Un tamaño sirve para todos? Degenhard Marx, Matthias Birkhoff Introducción ¿Cuáles son los desafíos? La discusión sobre el uso de conservantes en las formulaciones oftálmicas aún despierta controversias, pero cada vez más la evidencia apoya el uso de fórmulas oftálmicas sin conservantes para el tratamiento de enfermedades crónicas. Por ejemplo, el ojo seco, una condición en la cual la película lagrimal es deficiente, cada vez más se reconoce como una enfermedad inflamatoria. En este caso, los conservantes pueden empeorar la situación. Para el tratamiento de los síntomas leves, se recomienda el uso de gotas artificiales no irritantes y sin conservantes, mientras que para los casos más graves el uso de un principio anti-inflamatorio se convertirá en el tratamiento estándar. Lo mismo se aplica para el glaucoma, que requiere un tratamiento de por vida. Baudoine et al han demostrado que los riesgos de glaucoma para pacientes que experimentan efectos secundarios graves aumentan con la cantidad de medicamentos oftálmicos con conservantes que se aplican en paralelo1. Los productos combinados que reducen la cantidad de medicamentos necesarios, o el uso de formulaciones oftálmicas sin conservantes, ciertamente ayudarán a los pacientes que lo padecen, y también aumentará la adherencia al programa de tratamiento prescripto. Por lo menos en Europa, las autoridades han reconocido el problema y la Agencia Médica Europea (EMA) impulsa, por ejemplo, el uso de formulaciones oftálmicas sin conservantes, más recientemente en el borrador de pautas para medicina pediátrica 2. Reconociendo la tendencia hacia fármacos tópicos sin conservantes, en 2006 Aptar Pharma comenzó el desarrollo del Ophthalmic Squeeze Dispenser (OSD), un dispositivo multidosis diseñado para formulaciones oftálmicas sin conservantes [3]. El sistema está diseñado en forma de dispensador a presión para asegurar su aceptación. El primer producto comercial registrado bajo la Directiva Europea de Dispositivos Médicos que usa un OSD como envase principal fue el VISMED Multi, un producto de lágrimas artificiales del experto en cuidados oculares TRB Chemedica (Suiza), presentado en el mercado en agosto de 2011. A la fecha más de 45 OSD se venden como sistemas multidosis sin conservantes para varios productos, y muchos proyectos nuevos están en camino. La pregunta clave que responderemos en esta revisión es cómo un único dispositivo puede adecuarse a un amplio rango de formulaciones para diferentes condiciones y enfermedades. Las formulaciones oftálmicas pueden ser muy diferentes entre sí y su comportamiento es muy difícil de predecir. Las lágrimas artificiales que se utilizan para complementar la película lagrimal no contienen ingredientes activos, pero varían en sus propiedades reológicas, como la viscosidad, de baja a alta viscosidad, dependiendo de las necesidades de los pacientes. Las formulaciones oftálmicas que contienen fármacos activos, por ejemplo para el tratamiento del glaucoma, pueden contener muchos compuestos auxiliares para estabilizar la formulación y el contenido del ingrediente farmacéutico activo puede variar de µg a pocos mg por ml. El gotero siempre debe dispensar el tamaño de gota correcto, la formulación debe ser estable para almacenar y usar en un período determinado y la fuerza para dispensar la gota debe permanecer en un rango razonable. ¿Qué se debe considerar cuando se coloca una formulación en el OSD? 38 revista safybi, junio 2015 Cómo funciona el OSD Para comprender el sistema, su flexibilidad así como sus límites técnicos, es útil explicar cómo funciona. El sistema sigue un enfoque mecánico puro para evitar la contaminación microbiana del producto y usa la llamada “tecnología de punta sellada”, una tecnología de válvula de salida diseñada específicamente, y luego filtración estéril del aire de venteo. Como cualquier otro envase gotero convencional, el OSD debe ser sostenido en una posición angular o completamente invertida para dispensar las gotas. El sistema se purga apretando la botella hasta que se dispense la primera gota. Durante el proceso de accionamiento (apretar la botella), la presión dentro del sistema aumenta y el líquido es forzado a través del canal de líquido en un espacio entre el dispensador a presión y el aplicador. Cuando la presión del líquido excede la fuerza de resorte de la válvula de la punta sellada, la parte central del dispensador a presión se fuerza hacia atrás, se abre el orificio y se dispensa el líquido. El formato exterior especial del orificio permite que el producto forme y dispense una gota. Cuando cae la presión por debajo de un umbral definido, la válvula de salida con punta sellada se cierra de inmediato con un movimiento hacia afuera. Esta función se regula automáticamente y evita el reflujo hacia el sistema y por lo tanto la contaminación microbiana potencial. Sistema Vanquish Sistema XRS Sistemas de Nanoflujo Sistemas de Alta Resolución Sistema Estándar Sistema Básico • Económico y confiable • UHPLC a 620 bar • Flujos de hasta 10 ml/min • UHPLC a 620 bar • Flujos de hasta 10 ml/min • Gran flexibilidad: Diversidad de configuraciones, HPLC y UHPLC 620 bar • RSLCnano/ Easy-nLC • 20 nl/min – 50 ml/ min hasta 800 bar con RSLCnano • 20-2000 nl/min hasta 1000 bar con Easy-nLC • Capacidad de flujos directos continuos • RSLC / BioRS • UHPLC con bombas cuaternaria y binaria • 1000 bar hasta 5 ml/min • Velocidad de muestreo de 200 Hz • Flujos de trabajo avanzados con capacidad dual: HPLC y UHPLC • UHPLC con bomba cuaternaria RSLC especialmente optimizado para LC/MS • Presiones de hasta 1250 bar • Flujos de hasta 2 ml/min • Muestreador automático abierto opcional 1000 bar 1250 bar • La mayor capacidad en término de presiones (hasta 1500 bar) • La menor dispersión merced al diseño • Mayor capacidad de muestras: hasta 4 gradillas en el muestreador automático • Cargador/ alimentador con capacidad para 9 gradillas de viales o pocillos profundos/ 20 gradillas de pocillos bajos • Dos modos de termostatización de columnas para encarar distintos desafíos analíticos • Detección y sensibilidad sin paralelo, linealidad hasta 3 UA 1500 bar Revoluciona su experiencia en HPLC y UHPLC Representante autorizado Tel.: (011) 4704-0865 - [email protected] / www.sol-analiticas.com Investigación y Desarrollo Imagen 1: Componentes del OSD. El mecanismo de válvula hermética accionada por resorte en el orificio se denomina “punta sellada”. sin problemas, el diámetro y longitud del canal de flujo entre la botella y el espacio junto al orificio son importantes. La longitud de este canal de flujo puede variarse, para que la resistencia del sistema se adapte a la viscosidad de la formulación. La longitud del canal interno puede establecerse en múltiples pasos para obtener los mejores resultados para una formulación dada. Esta característica se denomina control de flujo. El control de flujo óptimo para cada formulación se determina mediante experimentos en el laboratorio usando sistemas completos con diferentes valores de control de flujo predeterminados. Fuerza de activación Durante el accionamiento en posición angular o invertida, el contenido de la botella o producto toma contacto con el filtro. El lado de la membrana del filtro que se encuentra hacia la formulación es altamente hidrofóbico, lo que evita que la membrana se humedezca, o que alguna parte de la formulación sea forzada a través del filtro. Al dispensar gotas se reduce el volumen dentro del envase; en consecuencia se requiere un equilibro del contenido del mismo. Cuando el envase vuelve a su forma inicial, se equilibra la diferencia de presión ingresando aire al envase a través del filtro, evitando la contaminación microbiana de la formulación. Esta función es independiente de la orientación del dispositivo. Imagen 2: (1) El OSD se activa como un gotero convencional: al apretar la botella se dispensará una gota. (2) Al apretar la botella, la formulación forzará la apertura de la punta sellada en el orificio (flecha roja) y se formará y dispensará una gota. (3) Al finalizar la activación cuando la presión en el envase disminuya, el resorte cerrará la punta sellada de inmediato con un movimiento hacia afuera. La fuerza de restauración de la botella impulsará el aire de venteo dentro de la botella a través del filtro estéril. La fuerza de activación para goteros convencionales para formulaciones con conservantes con orificio abierto depende principalmente de la rigidez de la botella, que está determinada por el espesor de la pared y el material utilizado (por ejemplo, polietileno más blando o polipropileno más rígido). En comparación con los diseños de botellas abiertas, la fuerza de activación del OSD está influida por tres factores: 1. función de punta sellada; 2. nivel de líquido de la botella y 3. rigidez de la botella. Como se describió anteriormente, se debe generar una presión determinada dentro del sistema para que se abra la punta sellada. En una primera generación de OSD, la fuerza de activación era bastante alta para proporcionar un sello hermético real del orificio. La mejora continua y la optimización del producto han disminuido significativamente la fuerza para activar y, por tanto, han mejorado el cumplimiento y conveniencia del paciente. Imagen 3: Fuerza de activación usada inicialmente en el diseño de punta sellada en comparación con las configuraciones mejoradas usando una botella de 10 ml llena (barras azules) o casi vacía (barras rojas). ¿Cómo asegurar una activación sin problemas? Al apretar el envase, la presión fuerza la formulación a través de una ruta de líquido específica dentro del espacio detrás de la punta del dispositivo, que posteriormente abre la válvula de salida con punta sellada. Para una activación 40 revista safybi, junio 2015 Un factor importante que influye sobre la fuerza de activación es el nivel de líquido dentro de la botella. Una botella llena requiere menos presión para dispensar una gota que una que está por vaciarse. En una botella casi vacía se debe Especialistas en Indicadores Biológicos Ajustándose a las necesidades de los distintos procesos industriales ◆ IB en tiras de esporas secas Esporas en suspensión ◆ IB Autocontenidos ◆ Población a requerimiento del usuario ◆ IB en tiras customizadas ◆ Suspensiones customizadas ◆ Tiras de esporas secas customizadas ◆ Kits para determinación poblacional ◆ Gaskets para indicadores biológicos ◆ Incubadoras para cultivo ◆ Paños para áreas clasificadas clase ISO 4.5 ◆ Productos desarrollados solo con cepas ATCC® certificados por F.D.A. y A.N.M.A.T. Llerena 3192, CABA, Argentina Tel.: (54-11) 4524-7878 / 7979 [email protected] www.2ms.com.ar Comercialización de insumos para el monitoreo microbiológico en la industria Farmacéutica, Cosmética y Alimenticia Investigación y Desarrollo comprimir más aire antes de que se alcance la presión de apertura de la válvula de salida. Por lo tanto, se recomienda usar un tamaño de botella cercano al volumen de llenado. Comenzando con la botella estándar de PEBD de 10 ml; ahora hay disponible una botella ovalada o redondeada de 5 ml que cumple con los requisitos del mercado. Las botellas para el OSD están fabricadas de polietileno blando. El espesor de las paredes se optimizó para balancear la fuerza de activación baja, la transmisión de vapor de agua y la fuerza de restauración suficiente para permitir la ventilación del sistema. Compatibilidad con la función Al manipular líquidos en dispositivos de dispensado, siempre existe el riesgo potencial de interacciones que pueden impedir la estabilidad de la formulación o el funcionamiento del sistema completo. Tales problemas de compatibilidad potenciales deben considerarse temprano en el desarrollo y las pruebas que tratan tales problemas son obligatorias. El mayor riesgo potencial es la absorción de los ingredientes activos por el material del sistema de cierre del envase. El sistema usa un material de elastómero termoplástico (TPE) para la válvula de salida. Aptar identificó diferentes materiales elastoméricos que se pueden usar en casos de que el material estándar genere preocupaciones de compatibilidad. te pequeño de resina TSE que se desliza sobre el filtro. Esta medida evita que la formulación entre en contacto con la membrana del filtro sin afectar la ventilación del sistema. Otro tema relacionado con la botella es su procedimiento de esterilización. El procedimiento estándar para la esterilización de las botellas de polietileno de baja densidad, es la radiación gamma con más de 25 kGy. De acuerdo con la bibliografía, la esterilización por radiación puede tener un efecto sobre el material expuesto, a pesar de que el polietileno está bien adecuado para este procedimiento. La esterilización de óxido de etileno (EtO) puede usarse como una alternativa para la botella. Debido a su diseño complejo, la parte de gotero del OSD necesita esterilización por radiación. Para el OSD, se estableció un proceso de esterilización gamma validado de acuerdo con la norma ISO 11137-1 y 2. Imagen 5: Botellas para OSD fabricadas de PEBD, de forma ovalada de 5 ml (izquierda), redondas de 5 ml (media) y redondas de 10 ml (derecha). Imagen 4: Los dispensadores a presión para el OSD fabricados de diferentes materiales flexibles y la válvula de protección del filtro opcional (inferior). Cristalización alrededor del orificio Como se describió anteriormente, cuando se usa el OSD, el filtro se sumergirá en la formulación. Su membrana hidrofóbica evitará que la formulación genere una función incorrecta. Esto es verdadero para la mayoría de las formulaciones. Pero, también existen formulaciones conocidas por una tensión superficial baja, que permiten que la membrana se humedezca o incluso ingrese en los poros del filtro. Si esto sucede, normalmente la ventilación del sistema se restringe y la fuerza de restauración del envase no es suficiente para devolverlo a su forma inicial. Como resultado, la fuerza de apretar requerida aumentará paso a paso para las activaciones siguientes. La solución técnica para tales formulaciones es el uso de la llamada válvula de protección de filtro, un componen- 42 revista safybi, junio 2015 Como se describió anteriormente, al final de la activación, la válvula de punta sellada se cierra con un movimiento hacia afuera, dejando algo de formulación visible en el orificio. Esta gota restante (aproximadamente de 3 a 7 µl) se secará en las siguientes 4-6 horas, incluso si se coloca la tapa de protección. Para algunas formulaciones el material restante de ese líquido presenta el riesgo de bloquear el orificio debido a la cristalización después de unos días. Este riesgo potencial puede evitarse introduciendo un material de recubrimiento poroso en la tapa de protección. Cuando se vuelve a colocar la tapa de protección sobre el OSD después del uso, el material de recubrimiento tomará la gota restante y la extenderá sobre el material en una superficie mayor. Esto acelerará el secado y así reducirá el potencial de crecimiento de microbios y evitará la cristalización alrededor del orificio. Debe probarse cuidadosamente si esta solución técnica es adecuada para una formulación particular. El impacto de una absorción potencial de la gota, así como la velocidad de secado debe ser entendido; También el número previsto de dosis por día debe ser tomado en consideración. Líder en equipamiento de laboratorios Hei-VAP Industrial Hei-VAP o Sistema de evaporación continuo de 20 litros. o El mayor rendimiento con la mejor seguridad. o Panel táctil resistente a químicos. o Evaporación inteligente. o Panel desmontable. o Variedad en set de vidrios. Vacuubrand Hei-Torque Tecnología de sistemas de vacío o Bombas de membrana resistentes a químicos. o Bombas rotativas de paleta. o Puesto de bomba de alto vacío. o Redes de vacío local para laboratorios. Nueva serie de agitadores a varilla o Potencia máxima a un nivel de ruido menor. o Panel táctil intuitivo. o Apto para trabajo continuo. Lauda Kern El profesional en balanzas o Balanzas analíticas, de precisión e industriales. o Determinadores de humedad. La temperatura más exacta del mundo o Equipamiento de Frío/ Calor. o Rango de -90ºC a 300ºC. o Enfriadores de circulación para el servicio continuo. www.icientifica.com Representamos las marcas más prestigiosas del mundo Integre su laboratorio con los mejores equipos, al precio adecuado y con el asesoramiento técnico indispensable para que sus análisis y procesos sean de excelencia. Saavedra 1021 – Ciudad Autónoma de Buenos Aires, Argentina Tel.: (011) 4308-5170 - líneas rotativas [email protected] Sistema de Gestión de Calidad Miembro de CISQ Federation ISO 9001 Sistema de Calidad Certificado ISO 9001:2008 Investigación y Desarrollo Imagen 6: Nueva línea de ensamblado totalmente automatizada para el OSD en un cuarto limpio clase ISO 7. Conclusión El mayor uso de dispositivos multidosis sin conservantes para formulaciones oftálmicas comenzó en Europa para las lágrimas artificiales. Los pacientes y consumidores apreciaron la manipulación conveniente e intuitiva del OSD, propiedades que son importantes para un mayor éxito del dispositivo. Para los fabricantes farmacéuticos la rentabilidad del OSD es otro aspecto importante. El sistema es flexible para cumplir con el desafío de las diferentes formulaciones oftálmicas. Sin embargo, se necesita una guía profesional y provista por Aptar Pharma para encontrar la configuración óptima. Las medidas técnicas provistas se probaron exhaustivamente antes de introducirlas al mercado. A pesar de que esto constituye una validación exhaustiva del sistema mismo, no puede eximir a los comercializadores de realizar la diligencia debida correspondiente sobre el producto terminado. Pero el trabajo calificado realizado por la compañía ayuda a mejorar este progreso significativamente y permite un menor tiempo para la comercialización. Siempre se prestó especial atención a la seguridad, obviamente con un énfasis especial en la integridad microbiana del sistema. Después de todo, nadie debe poner en riesgo un ojo. n Referencias 1. Baudouin C et al. Prevalence and risk factors for ocular surface disease among patients treated over the long term for glaucoma or ocular hypertension. Eur J Ophthalmol. Epub June 11, 2012 2. Guideline on pharmaceutical development of medicines for paediatric use, 3 January 2013, EMA/CHMP/QWP/805880/2012 Rev. 1 2 3. Marx D and Birkhoff M. New Devices for Dispensing Ophthalmic Treatments May Be the Key to Managing the Life Cycles of Established Products. Drug Delivery Technology 2010, 10 (9): 16-21 Matthias Birkhoff es el vicepresidente de Marketing de la División de Cuidado de la Salud (“CHC”, por sus siglas en inglés) de Aptar Pharma. En su cargo, Matthias es responsable del programa de Cuidado de la Vista de Aptar Pharma y coordina actividades de investigación y desarrollo, evaluación microbiológica y estrategias comerciales. Matthias inició su trayectoria profesional en el área de ventas farmacéuticas en una importante empresa farmacéutica multinacional antes de unirse a Aptar hace dieciséis años. Antes de desempeñarse en tareas de marketing y desarrollo comercial, Matthias tenía a cargo las ventas de la región de Asia Pacífico. Estudió medicina en la Universidad de Dusseldorf, Alemania. Dr. Degenhard Marx a continuación de sus estudios de Medicina Veterinaria y la exitosa finalización de sus tesis en la Universidad de Leipzig, ingresó en la investigación corporativa Arzneimittelwerke Dresden/Acta Medica en 1992. En el año 2001 ocupó la posición de Investigador Senior en Altana Pharma/Nycomed en Constanza, Alemania. Durante este tiempo en la industria farmacéutica recogió una amplia experiencia en el desarrollo de medicamentos antiinflamatorios y cardiovasculares. En el año 2008 fue nombrado Gerente de Desarrollo de Negocios de Ing. E. Pfeiffer, Division Pharma, que luego fue Aptar Pharma en 2010. Ocupa actualmente la posición de Asuntos Regulatorios de la división de Productos de Cuidados de la Salud de Aptar Pharma. 44 revista safybi, junio 2015 Logística Packaging Tapas plásticas para envases Anibal H. San Juan Ajustar lo necesario Es todo un desafio pasar de Tapa de Aluminio (la conocida Pilfer Proof), donde los ajustes de cierre se realizan modificando variables en linea como las presiones axiales que ejerce la platina del cabezal al comprimir la guarnición sobre la boca del envase y los cabezales de moleteado sobre la rosca y el cierre Pilfer, a comenzar a usar una Tapa Plastica donde no solo cuentan las condiciones de proceso sino tambien las caracteristicas de las roscas tanto del envase como de la tapa, sin olvidar tambien el tipo de material de la misma en orden a que no contenga polimero reciclado. En este caso se analizará la Tapa Plastica comenzando con detalles sobre las caracteristicas de la rosca donde la fuerza aplicada para el cierre siempre será mayor a la necesaria para su apertura, pudiendose encontrar en ésta, valores que son inferiores hasta un orden del 40 %. Los filetes de la rosca se denominan habitualmente cuerda y la misma en general, al menos para la industria Farma, debe ser continua y estar bien definida para lograr no solo un buen cierre sino ademas asegurar la hermeticidad del conjunto. Por esa razon es importante que los filetes de rosca, tanto del envase como de la tapa tengan el mismo angulo, sin dejar de lado su geometria o perfil pues existen diferentes terminaciones que permiten optimizar y mejorar el cierre del conjunto (si bien a veces es dificil de observar a simple vista, se puede advertir al realizar un corte tangencial). Claro esta que los filetes de las tapas, en sus dimensiones, tienen tambien sus limitaciones de acuerdo al sistema de expulsion desde el molde de inyección, ya sea por placas como por desenrosque pues en un caso la expulsión despues de la inyección es directa y los filetes generalme- 48 revista safybi, junio 2015 nete apareceran barridos una vez expulsada la tapa y en la otra, el sistema que permite expulsarla desenronscandola, los filetes tendran mejor definición y además pueden diseñarse con mayor diámetro que en el otro sistema. Pero sucede muchas veces que estando dimensionalmente en especificación, se observa que la tapa al cerrarse, toca con el hombro del envase evitando que el plano de la boca del mismo pueda tener contacto cierto con la guarnición de la tapa, produciéndose entonces falta de hermeticidad, lo que no se soluciona aplicando mas torque pues los filetes envase-tapa se montan generando luz entre ambos y produciendo el zafado de la misma. Esto se soluciona solamente dando mayor altura al pico del envase. Ya se ha mencionado la palabra torque, que se puede definir como la tendencia de una fuerza a producir la rotación y se representa como el producto de la fuerza y la distancia perpendicular de la línea de acción al eje de la rotación instantáneo, expresada por unidades de kg/cm o lb/in. Atención especial merecen las guarniciones de las Tapas pues también pueden incidir en el cierre según la fuerza de compresión con la superficie de la botella, ya que la fricción debido a un ajuste “apretado” entre esta y el envase puede producir un valor de torque que podría carecer de sentido. Se pueden diferenciar tres Tipos de Torques: •• Torque de Uso: Es el aplicado para cerrar. •• Torque de Retiro: Es el necesario para aflojar la tapa y comenzar el desenrosque. •• Torque de Desmontaje: Es el necesario para “pelar” los hilos de rosca de la tapa mientras se aplica el esfuerzo de uso. En cuanto a las tapas a rosca (y es una duda que habitualmente puede surgir al especificar las necesidades), se pueden identificar ciertas caractersticas que se expresan con dos números por ej., 24/410 o 24/415 (Fig. 1). •• El primero indica el diámetro de la rosca en milímetros y de acuerdo con el Glass Packaging Institute (GPI)/Society of the Plastics Industry (SPI) los diámetros de las roscas son expresados como Rosca 24, 28, 33, 38, y así sucesivamente. Estos números son una medida universal y son expresados en milímetros. •• El segundo indica la altura de la rosca. Figura 1: Identificación de tapas a rosca En la Fig. 1, los números 400, 410 y 415 tienen el siguiente significado: •• 400 - Cierre con una sola vuelta •• 410 - Cierre con una vuelta y media •• 415 - Cierre con dos vueltas A principios del corriente año, la ASTM emitió una nueva Norma D7860 (Torque estándar para medicamentos con tapa a prueba de niños) que facilita el desarrollo desde la Ingeniería de Empaque y permite normalizar los controles de Calidad por Recepción y a los de Línea durante el Envasado, asegurando que los envases salgan en condiciones óptimas de cierre, estandarizando así los Métodos de Prueba para la Medición de Torque. Si bien estos ensayos se realizan habitualmente siguiendo Técnicas de Control y Análisis propias o referenciales, la oportunidad de poder contar con esta normativa permite adecuar y tener el mismo criterio de ensayo frente a distintos proveedores de empaques y sistemas de cierre-tapado en líneas de envasado sea cual fuere el tipo de cabezales roscadores que existan en Planta (mecánicos o magnéticos). Como información adicional, la Norma D7860 fue desarrollada por el Subcomité D10.32 sobre Embalaje Resistente al Consumidor, Farmacéutico, Médico y Niño, parte del comité D10 de ASTM sobre Envases. n Nota : Esta presentación es una base de conocimiento y ayuda sobre la materia y deberá estudiarse cada material en desarrollo en cuanto a las condiciones de uso, producto y mercado destino. Aníbal H. San Juan – [email protected] Con formación en Química, se desempeñó como Jefe de Control de Calidad de Material de Empaque en Unilever, Johnson & Son y Jefe de Desarrollo de Packaging en Laboratorio Elea. Actualmente es Jefe de Ingeniería de Empaque en Laboratorio Craveri, participando en el IAE como Jurado del Concurso Estrella del Sur al Packaging y disertante del Módulo Packaging Farmacéutico del Curso de Técnico en Envases, además de coordinador y poniente de temas de packaging en Safybi. junio 2015, revista safybi 49 Recursos Humanos ¿Cómo es trabajar en un Laboratorio Farmacéutico en Suiza? Experiencias de un farmacéutico argentino Farmac. Lucas Fiorenza Hace ya unos años sentí ganas de hacer una nueva experiencia laboral y conocer como es trabajar en un laboratorio farmacéutico en Europa. Debido a mis conocimientos de alemán (viví algunos años en Alemania durante mi niñez) y como farmacéutico con más de 11 años de experiencia en distintos laboratorios de Argentina decidí iniciar una búsqueda de trabajo en un laboratorio de Alemania o Suiza. Comencé simplemente enviando mi curriculum por internet a varias búsquedas de personal en Alemania. Al principio con poco éxito, después de un tiempo empecé a recibir llamadas y mails y distintas propuestas. Varios intentos avanzaron bastante pero finalmente no se concretaron. Hasta que, después de superar varias entrevistas por Skype, acepte una propuesta para trabajar en Suiza. Llegué a Suiza, más precisamente a la ciudad de Basilea en Abril de 2014, con mis deseos de conocer y con un contrato con una empresa consultora por 6 meses. Basilea está enclavada de modo privilegiado en la frontera con Alemania y Francia, la región es conocida como la Dreiländereck (la esquina de los tres países) o el distrito de las 3 fronteras. Basilea, es la tercera ciudad de Suiza, y es el último puerto para la navegación fluvial sobre el Rin. Aquí nació un tenista famoso: Roger Federer. Pero también nacieron o vivieron en Basilea personalidades como Paracelso, Erasmo de Rotterdam, Daniel Bernoulli, Friedrich Nietzsche y un matemático notable, Leonhard Euler. Basilea posee la universidad más antigua de Suiza (fundada en 1493), se realiza ART Basel, una de las muestras de arte más destacadas de Europa y hace pocos meses se realizó, como todos los años, la exposición de relojes más grande del mundo: Baselworld. Actualmente, Basilea es el centro de las industrias química y farmacéutica de Suiza, a tal punto que la región es conocida también como el Bio-Valley. Las principales farmacéutiRathaus (año 1514), Casa cas de Suiza tienen su de la Municipalidad 50 revista safybi, junio 2015 Vista de Basilea y el l Rin desde la torres de la Catedral sede aquí. Desde las enormes Novartis y Roche, como también Bayer, GlaxoSmithKline, Johnson & Johnson, Clariant, Merck Sharp & Dohme entre otras. Esta concentración de industrias farmacéuticas brinda muchas oportunidades de trabajo a farmacéuticos y profesiones afines. Desde hace poco más de un año estoy aquí trabajando en una de las industrias farmacéuticas en la región de Basel Land, en la localidad de Aesch, situada a 10 km de la ciudad de Basilea. Al principio solo obtuve un permiso de residencia muy limitado. Cumplido 6 meses de contrato pasé a formar parte del personal de la firma con un contrato sin fecha límite. Esto permite obtener el permiso de residencia clase B que otorga casi los mismos derechos que a un ciudadano suizo excepto el derecho a votar. La empresa en la que estoy trabajando es una compañía farmacéutica suiza y tiene actualmente cerca de 600 empleados. Desarrolla, fabrica y distribuye internacionalmente productos farmacéuticos en distintas formas de administración. Es líder tecnológico en el área de administración avanzada de medicamentos en formas modificadas de liberación orales y sistemas transdérmicos para el cual también posee patentes. También funciona como tercerista fabricando para importantes clientes de toda la región. Me desempeño como jefe de Laboratorio de Control de Calidad de materias primas. Lidero un grupo de 10 Analistas y 1 Supervisor entre los cuales hay suizos, franceses, y alemanes. Una de las primeras dificultades con las que me encontré fue el idioma. Suiza tiene 4 idiomas oficiales (el alemán, el francés, el italiano y el romanche). En Basilea se habla alemán, pero el alemán suizo, una especie de dialecto de la Recursos Humanos región. Conocer el alemán es fundamental, pero se requiere un tiempo para poder entender el dialecto. Afortunadamente los suizos son en general muy educados y atentos y muchos cambian y se expresan en el alemán oficial cuando observan que el interlocutor no es suizo. Pero también reconocen que les genera un esfuerzo extra por eso prefieren usar su dialecto y luego de unos meses ya casi no me hablan en alemán puro. Pero ¿cómo es realmente trabajar todos los días en un laboratorio en Suiza? ¿Cuáles son las diferencias más importantes, más allá del idioma? Calidad Suiza en Medicamentos: una marca registrada El Laboratorio de Control de Calidad a nivel de instalaciones, equipamientos, material de vidrio y HPLCs no dista mucho de cualquier laboratorio importante de Argentina. Obviamente que se trabaja bajo estrictas normas GMP con lo cual hasta aquí se observan pocas diferencias. La mayoría de las materias primas se analizan siguiendo la Farmacopea Europea y se evalúa toda la monografía completa independientemente de la cantidad de lotes que ingresan. También todos los proveedores de materias primas son auditados regularmente estén en India, China o Europa. Las auditorias de clientes o autoridades sanitarias se desarrollan siguiendo los mismos pasos generales, así que respecto a este tema tampoco existe ninguna diferencia significativa con Argentina. Uno de los aspectos a destacar es la rapidez con que se gestionan los pedidos de compra, por ejemplo cualquier columna de HPLC no demora más de 5 días laborables para recibirla en el Laboratorio, como así también los reactivos. El horario normal de trabajo es de 7 a 16 hs, en realidad es flexi-time pero la mayoría trabaja en esa franja horaria. El ritmo de trabajo es intenso, y como todo sector que interacciona con la producción, el nivel de exigencia y de presión es alto. Llevo a cabo reuniones semanales con los analistas, donde discutimos los problemas y tareas del laboratorio. Para mi es importante que cada analista tenga unos minutos para hablar y que se genere un dialogo abierto. Un aspecto en el cual más tengo que trabajar es en la motivación ya que son muy sensibles al estrés. Muchos analistas se sienten sobre exigidos si tienen más de dos tareas a realizar durante el día. Otro aspecto notable es que los empleados Museo de la Historia de la Farmacia, Basilea 52 revista safybi, junio 2015 están muy atentos a los cambios de dirigencia, a la estrategia financiera, al balance de la empresa y la evolución general de los negocios. Los empleados tienen en cuenta todos estos aspectos y si no están de acuerdo simplemente renuncian. ¡Así de simple!, ya que están tranquilos que consiguen trabajo rápidamente y por 3 meses cobran seguro de desempleo que es igual al último ingreso recibido. Con lo cual se abre una nueva problemática a nivel de Jefatura debido a que no existen muchas herramientas para retener al personal si estos ya no comparten la estrategia que en los años siguientes va a llevar a cabo el Laboratorio. Uno de los aspectos que también me llamo la atención es un sistema computarizado para documentar los OOS (Out Of Specification), desvíos, control de cambios, las acciones correctivas y/o preventivas muy estructurado y con diferentes perfiles de usuario que hace más fácil la descripción, la investigación y el seguimiento. Actualmente el laboratorio comenzó a aplicar metodologías de 5 S, de mejora continua y medición de indicadores de desempeño (Six Sigma). Y debo reconocer que han comenzado a dar resultados notables, en lo que está relacionado con la organización del trabajo y orden del laboratorio. Gracias a la aplicación de varias de estas herramientas hemos podido mejorar la productividad por semana y por analista. El laboratorio ha realizado y continua realizando un importante esfuerzo por mejorar la comunicación intersectorial y el trabajo en equipo y hemos participado en varios Workshops sobre estos temas. La reciente valorización del franco respecto al euro, ha generado la necesidad de ajustar ciertos gastos y definir mejor los mercados donde comercializar los productos. Participo regularmente en reuniones con producción donde se plantean las próximas prioridades en función de la estrategia de fabricación más rentable y rápida con las materias primas a disposición. También asisto a reuniones con el Sector de Compras donde se plantean temas de precios, de las cantidades a adquirir y sobretodo la calidad de los APIs de diferentes proveedores. De esta forma estoy mejorando mi conocimiento de la estrategia general de compras y ventas de la empresa y de la complejidad propia de un mercado competitivo como el sector farmacéutico europeo. Pero creo que la diferencia más notable de trabajar en un Laboratorio farmacéutico en Suiza no está precisamen- te en el trabajo sino fuera de él, o sea en Suiza. Chocolate, relojes, queso, neutralidad, paraíso fiscal o magníficos paisajes: estas cosas pertenecen tal vez a las características más conocidas de Suiza. Pero la Suiza moderna ofrece más que solo estos lugares comunes. Suiza es hoy en día una de las economías más competitivas del mundo, la universidad ETH Zúrich posee la mayor cantidad de premios Nobel por titulados, tres ciudades suizas (Zurich, Basilea y Ginebra), figuran dentro de las diez ciudades con mejor calidad de vida, y se considera detrás de Japón, como segundo país con la esperanza de vida más alta del mundo. Pero Suiza es también conocido por sus alquileres no precisamente baratos, precios de las propiedades y costo de vida muy alto. Mucha gente aprovecha que Basilea este en la frontera con Alemania y Francia y realiza las compras en los supermercados de estos países y muchos otros optan directamente por vivir en Alemania o Francia y trabajar en Suiza. Basilea se destaca también por su red de transporte. No hay en Basilea red de subterráneo, pero cuenta con una extensa red de tranvías, buses y trenes. Un abono mensual de 76 francos suizos (aprox. 76 Eur.) permite utilizar sin límite todos los servicios. Para ir al trabajo, solo necesito caminar 400 metros hasta la parada del tren que circula con precisión suiza hasta la estación de Aesch. Cuando el tren llega a la estación de Aesch, los colectivos están esperando, los pasajeros bajan del tren y suben al colectivo. La regularidad del servicio es tal que todos mis horarios de fichada a la mañana en el trabajo solo presentan una diferencia de +/- 1 minuto. Disfruto mucho de practicar deporte y entreno triatlón. Poco después de mi llegada me incorporé al grupo de triatlón de Basilea. Aquí es posible salir a correr, y luego de pocos minutos se deja la ciudad y se está en medio de una pradera o en medio de un bosque… ¡son cosas muy asombrosas! Los circuitos de bicicleta también están perfectamente señalizados y uno puede realizar circuitos partiendo de Basilea y pasar por Francia, o por Alemania y llegar a la Selva Negra. Para la natación hay piletas revestidas de acero inoxidable, que pueden disfrutarse al aire libre desde abril hasta noviembre. Durante este tiempo, y mientras continuo incorporando conocimientos y nuevas experiencias, que espero poder aplicar (dentro de algunos meses) cuando decida regresar a la Argentina, he descubierto que la vida laboral dentro del Laboratorio es similar a cualquier laboratorio importante de Argentina, aunque afuera las diferencias son mayores. n Lucas Fiorenza es Farmacéutico egresado de la Universidad de Buenos Aires. Ha desempeñado diferentes funciones en Bayer Argentina SA, Schering AG Argentina/Newprod Farmacéutica, Laboratorios Richmond, Boehringer Ingelheim, Maprimed y Laboratorio Internacional Argentino en las áreas de Desarrollo Analítico, Estabilidad y Validación. Su expertise radica principalmente en el desarrollo de métodos de análisis para productos y principios activos y la validación de los mismos así como de los procedimientos de limpieza y estabilidad. Desde abril del año 2014 es Manager del Laboratorio de Materias Primas de Acino Pharma, con sede en Basilea, Suiza, liderando un importante grupo de trabajo. Email: [email protected] revista safybi 53 Nuestros anunciantes Válvula con Bolsa de Aptar Pharma Aptar Pharma anuncia el relanzamiento de sus actividades centradas en la tecnología Válvula con Bolsa (BOV) en su planta de manufactura de Radolfzell. La planta de Radolfzell (Alemania), totalmente renovada, constituye el centro experto de Aptar Pharma en sistemas BOV para aplicaciones farmacéuticas. Después de una importante inversión, el área de producción de 2300 m2 está ahora equipada con nuevas máquinas de moldeo por inyección e innovadoras líneas de montaje para cumplir con la creciente demanda proveniente de un mercado desafiante. Aptar es pionera en el desarrollo y la manufactura de esta tecnología de dispensación ecológica. La tecnología Válvula con Bolsa de Aptar Pharma otorga enormes beneficios a los productos farmacéuticos; por ejemplo, mejor uso, mejor conservación y mayor protección del producto. Los sistemas BOV de Aptar, meticulosamente desarrollados a lo largo de varios años, son ampliamente utilizados por empresas multinacionales de primera línea. «Aptar Pharma es el segmento comercial de AptarGroup dedicado a proporcionar soluciones innovadoras de dispensación de fármacos para responder a las necesidades de las empresas farmacéuticas, de atención médica y de biotecnolo- 56 revista safybi, junio 2015 gía, que están en constante evolución. Somos el líder mundial en bombas spray nasales para rinitis alérgica, descongestivos nasales y soluciones salinas nasales, y el líder mundial en válvulas de dosificación utilizadas en los inhaladores presurizados de dosis medida (pMDI) para el tratamiento del asma y la EPOC. Dado que tenemos un fuerte enfoque en aprovechar la innovación para ofrecer soluciones centradas en los pacientes, destinamos una importante cantidad de nuestros ingresos anuales a actividades de investigación y desarrollo. Con centros de manufactura en Argentina, Brasil, Colombia, México, China, Francia, Italia, Alemania, India, Suiza y Estados Unidos, podemos atender el mercado de la industria farmacéutica en todo el mundo. Ofrecemos un completo surtido de servicios con valor agregado que incluyen el desarrollo de soluciones personalizadas, asistencia en investigaciones y trabajo de laboratorio, y un departamento de asuntos regulatorios exclusivamente dedicado. Nuestro equipo de ventas y nuestros expertos técnicos brindan asistencia global a nuestros clientes. n Para obtener más información, comuníquese con nosotros en: aptar.com/pharma/contact» Nuestros anunciantes Calibración de contadores de partículas en aire Rodolfo Díaz, Favio Wainstein Introducción En la industria farmacéutica existen requerimientos específicos respecto de la calidad del aire en recintos donde se realizan ciertas operaciones del proceso productivo. Debido a esto, es cada vez más frecuente la utilización de contadores de partículas en aire para caracterizar objetivamente dicha calidad, y en los últimos tiempos con una marcada tendencia a la instalación de sistemas de monitoreo continuo. Los contadores de partículas en aire son instrumentos en los cuales se encuentra una combinación poco frecuente (al menos para instrumentos de proceso) de diversas disciplinas; en ellos conviven artilugios ópticos, neumáticos y electrónicos por citar los principales. Este hecho, sumado a la relativamente reciente publicación (año 2007) del primer estándar exhaustivo sobre el particular (ISO 21501 – “Determination of particle size distribution – Single particle light interaction methods”), resulta en que tanto los requerimientos del citado estándar como el análisis de los resultados obtenidos en una calibración resulten algo confusos. Este artículo se propone entonces clarificar algunos de los conceptos involucrados en la calibración de este tipo de instrumentos (sobre todo para los instrumentos diseñados posteriormente a la publicación de esta norma). Alcance de la norma La norma ISO 21501 está dividida en cuatro partes, de las cuales la parte cuatro es la relevante para la finalidad del presente artículo (ISO 21501-4 “Light scattering airborne particle counter for clean spaces”). Tal como está expresado en la introducción de dicha norma, “el propósito de esta parte de ISO 21501 es proveer un procedimiento de calibración y un método de verificación para contadores de partículas, para minimizar la inexactitud en el resultado de medición realizada por un contador, así como las diferencias en los resultados medidos por instrumentos diferentes1 [sic]”. Principios básicos de un contador de partículas en aire Si bien la descripción detallada de los principios de funcionamiento de un contador de partículas en aire no es el objetivo del presente artículo, es conveniente realizar al menos una aproximación básica y extremadamente simplificada sobre cómo funcionan estos instrumentos. Asumiendo que el instrumento posee un sistema para la toma de muestras de aire, y que su diseño es tal que en 58 revista safybi, junio 2015 un instante dado hay una única partícula en la “zona de medición” del sensor del instrumento, dicha partícula será impactada por (en realidad se interpondrá en el camino de) un haz de luz coherente (láser) y refractará una cantidad de luz proporcional al diámetro de dicha partícula (figura 1). Fig. 1: Principio óptico Por medio de un artilugio óptico (en general uno o más espejos parabólicos de primera superficie), la luz refractada es colectada y concentrada sobre un área puntual de un elemento fotodetector, que a su vez forma parte de una matriz de fotodetectores. En la figura 2 se reproduce el esquema del banco óptico de un contador de partículas en aire real, donde pueden ubicarse los elementos señalados. Este elemento fotodetector convierte la energía lumínica proveniente de la luz reflejada por la partícula en una señal eléctrica conocida como pulso, cuya magnitud o “altura” está relacionada a la energía lumínica “vista” por el fotodetector y por ende, finalmente, al tamaño de la partícula. A continuación, dicha señal es clasificada según su “altura” en distintas categorías o “bines” –los cuales se corresponden efectivamente con los canales de un instrumento-, siendo el sentido físico de cada uno de estos bines “partículas de diámetro mayor o igual a n µm”. Este análisis de la altura de pulsos y su posterior clasificación en función de dicha altura se realiza por medio de un sistema electrónico Fig. 2: Banco óptico (vista superior) conocido como PHA por sus siglas en inglés (Pulse Height Analyzer = Analizador de Altura de Pulsos). Finalmente, asumiendo que la muestra analizada se realiza a un caudal conocido y durante un tiempo determinado, y que se contabilizaron los pulsos clasificados en cada categoría o “bin” durante la muestra, se obtiene una distribución del diámetro de partículas por unidad de volumen para la muestra analizada. Algunas definiciones y requerimientos específicos de ISO 21501-42 Uno de los puntos importantes de la norma es la definición unívoca de las partículas de calibración a utilizarse como “partícula esférica mono-dispersa de tamaño medio conocido, por ejemplo partículas de látex de poliestireno (PSL), trazable a un patrón internacional de longitud, y donde la incertidumbre estándar del tamaño medio de partícu- junio 2015, revista safybi 59 Nuestros anunciantes la es menor o igual al ± 2,5 %”. También se caracteriza el índice de refracción de tales partículas3. En la figura 3 puede verse la presentación habitual comercialmente obtenible. Fig. 3: Partículas de calibración Otros requerimientos de la norma son la determinación del caudal de muestra y/o de tiempo de muestreo según sea el caso, el máximo admisible de cuentas falsas, la máxima pérdida por coincidencia5 (coincidence loss), y el tiempo de respuesta luego de ser saturado en determinadas condiciones. Ejemplo real de calibración de tamaño – Canal de 1 µm En la figura 4 puede verse el diagrama de un sistema de calibración de contadores de partículas típico (el contador de referencia se utiliza exclusivamente para los ensayos de eficiencia de conteo, de ahí su representación con línea de puntos). Fig. 4: Sistema de calibración típico Otros parámetros relevantes definidos en la norma son: Eficiencia de conteo: relación entre el resultado medido por un contador de partículas y el obtenido por medio de un instrumento de referencia utilizando la misma muestra. La eficiencia de conteo debe ser de 50% ± 20% para partículas de calibración de un tamaño cercano al mínimo tamaño detectable, y debe ser 100% ± 10% para partículas de calibración con un tamaño de 1,5 a 2 veces mayor que el mínimo tamaño detectable. Resolución en tamaño: medida de la habilidad de un instrumento para distinguir entre partículas de diferentes tamaños. La resolución en tamaño debe ser menor o igual a 15% utilizando partículas de calibración de un tamaño especificado por el fabricante. En la figura 5 se reproduce la distribución obtenida durante la calibración del canal de 1 µm de un contador de partículas en aire comercialmente disponible y de amplia difusión en la industria. Fig. 5: Distribución de partículas mono-dispersas Verificación de las determinaciones de tamaño: El error en el mínimo tamaño de partícula detectable (el canal “más pequeño”), y en otros tamaños especificados por el fabricante (el resto de los canales) debe ser menor o igual a ± 10%. Para este requerimiento, la norma describe un método específico para realizar el ensayo. Intervalo de calibración: Como intervalo de calibración para un contador de partículas en aire, la norma recomienda un año o menos. Si bien no es frecuente en el ámbito de las calibraciones que se señalen intervalos de calibración en la normativa, posiblemente4 en este caso se haga la recomendación teniendo en cuenta la complejidad inherente de estos instrumentos y la presencia de elementos que –en caso de presentar envejecimiento– podrían afectar directamente los resultados de una medición. Un claro ejemplo de esta situación podría ser la disminución de potencia del haz de láser. 60 revista safybi, junio 2015 Este es el resultado de la calibración del canal de 1 µm. Durante el ensayo se utilizaron partículas de calibración de 0,994 µm ± 0,015 µm, y tal como puede observarse se obtuvo una distribución estadística que puede asumirse –sin introducir errores significativos- como normal. Nótese la conformación del diagrama: en el eje de abscisas se encuentran las categorías de alturas de pulsos observados (expresados en Voltios), y en el de ordenadas la cantidad de pulsos contabilizados en cada una de dichas categorías. A grandes rasgos, se procesan los datos de este gráfico para ubicar la mediana (que es aquella categoría que divide en partes iguales la totalidad de los pulsos contados dentro del intervalo analizado). La altura de pulso así identificada como mediana, expresada en Voltios, es entonces el valor calibrado de la altura de pulso correspondiente a partículas de 1 µm. Esta altura de pulso se denomina habitualmente como umbral o threshold del canal calibrado. El proceso descripto se reitera utilizando otros tamaños de partículas seleccionados según los canales del instrumentos; de tal modo se determinan los umbrales de todos los canales del instrumento. Suponiendo un contador de partículas de 4 canales, por ejemplo 0,3 µm, 0,5 µm, 1,0 µm y 5,0 µm, superponiendo los resultados puede construirse un diagrama conceptual como el de la figura 6. Del análisis de dicha figura, se desprende que para este ejemplo todas las partículas mayores o iguales a 0,3 µm serán contadas en el canal 1, todas las partículas mayores o iguales a 0,5 µm serán contadas en el canal 2, todas las partículas mayores o iguales a 1,0 µm serán contadas en el canal 3, y finalmente todas las partículas mayores o iguales a 5,0 µm serán contadas en el canal 4. Fig. 6: Instrumento de cuatro canales con sus respectivos umbrales Resultados de calibración En la figura 7 se reproduce parte de un certificado de calibración6 de un contador de partículas de última generación. En la sección titulada Threshold Information (información de umbrales), pueden verse los distintos valores obtenidos siguiendo el procedimiento descripto previamente7. De hecho, obsérvese para cada canal se señala el tipo de medición empleado para determinar el umbral correspondiente y el mismo ha sido el de obtener la distribución (nuevamente figura 5). Nótese que los valores Umbral Medido se expresan como adimensionales, esto se debe exclusivamente a los mecanismos digitales utilizados en el diseño del instrumento y cada uno de estos valores representa unívocamente el valor de tensión asignado a la “altura” del pulso que caracteriza cada uno de los canales del instrumento. junio 2015, revista safybi 61 Nuestros anunciantes Fig. 7 – Resultados de una calibración Threshold information Channel size (micron) Measured Threshold Received Threshold Measurement Type Ch 1: 0,30 Ch 2: 0,50 Ch 3: 1,00 Ch 4: 5,00 00825 05052 19445 02426 00863 05375 21148 01517 Distribution Distribution Distribution Distribution Parameter Results Allowed Range Expanded Uncertainty Pass/Fail Skipped Flow Rate Pre Zero Count Size Error @ 0,30 Size Error @ 0,50 Size Error @ 1,00 Size Error @ 5,00 Resolution @ 0,30 CE (%) @ 0,30 CE (%) @ 0,50 29,7 L/min 52,97 Cnt/m3 0,8% 3,0% 4,3 3,3% 5,9% 51,4% 98,3% 26,9 to 29,7 L/min 0 to 176,50 Cnt/m3 -10,0% to 10,0% -10,0% to 10,0% -10,0% to 10,0% -10,0% to 10,0% 0,0% to 15,0% 30,0% to 70% 90,0% to 110,0% n/a n/a 2,39% 2,18% 2,04% 2,65 n/a n/a n/a Pass Pass(*1) Pass (*2) Pass (*2) Pass (*2) Pass (*2) Pass Pass Pass Verification Results Note *1: Process staged intervals to achieve 95% UCL for stated concentration. Note *2: Sizing Error operational sign per ISO 21501-4 En la sección titulada Verification Results se reproducen los resultados obtenidos y se comparan contra los requerimientos de la norma ISO 21501-4; véanse por ejemplo los errores de tamaño obtenidos y los límites de aceptación (± 10 %), la eficiencia de conteo con partículas del mínimo tamaño detectable (señalado como CE(%) @ 0.30 ) con su límite de 30,0% a 70% (equivalente al 50% ± 20% requerido por la norma), y la eficiencia de conteo con partículas 1,5 a 2 veces más grandes que las mínimas detectables (señalado como CE(%) @ 0.50 ) con su límite de 90,0% a 110% (equivalente al 100% ± 10% requerido por la norma). También pueden observarse las determinaciones de caudal de muestra y de cuentas falsas o zero count. Conclusiones Referencias 1 Traducción de los autores. 2 Esta lista de definiciones y requerimientos no es exhaustiva, pero incluye aquellas que, a juicio del autor, resultan menos “intuitivas”. En los casos donde se indican requerimientos, estos son los que la norma impone a los contadores de partículas en aire para ser considerados aptos para su utilización en la clasificación de áreas limpias de acuerdo a la norma ISO 14644-1. 3 Alrededor de 1,59 a una longitud de onda de 589 nm. 4 Si bien es una suposición fundada, en la norma no se argumenta la recomendación. 5 Es una situación que se presenta cuando se supera la máxima concentración admisible de partículas para un instrumento, y se debe a que se incrementa la posibilidad de múltiples partículas en el volumen de sensado y/o saturación en el sistema electrónico. 6Si bien en la figura puede leerse que son los resultados de una verificación, en realidad pertenecen a una calibración en el sentido formal de la palabra. Esto se debe a que son los resultados obtenidos a la recepción del instrumento antes de realizar cualquier ajuste; en los Estados Unidos (de donde proviene el sistema de calibración utilizado) con frecuencia se denomina “calibración” a la acción de realizar ajustes. 7Nótese que de los canales 1 al 3 los valores de los umbrales –aún adimensionales– son crecientes y acompañan el crecimiento del tamaño de los canales, pero esto deja de ser así en el canal 4. Esto se debe pura y exclusivamente a cuestiones de diseño del instrumento (en este caso el canal de 5,0 µm es medido con una ganancia distinta –menor– al resto de los canales. Esto es sólo un artilugio electrónico interno del instrumento y no invalida en absoluto los razonamientos expuestos. Rodolfo Díaz Departamento de Investigación y Desarrollo, Hitec S.R.L. San Blas 2655 (C1416EFU), Buenos Aires, Argentina Email: [email protected] Se graduó de Ingeniero Electromecánico con orientación en Electrónica en la UBA. Posteriormente, se especializó en Optoelectrónica en la Universidad de Paris Nord y cursó la Maestría en Dirección Estratégica y Tecnológica en el ITBA y la Escuela de Organización Industrial de Madrid. En 1986 fundó HITEC SRL, empresa que desde hace más de 20 años se dedica a la Instrumentación y el Control de Procesos Farmacéuticos, y de la que actualmente es Socio Gerente. Ha dictado conferencias y participado como coautor en publicaciones científicas tanto a nivel nacional como internacional. Favio Wainstein Dirección de Metrología, Hitec S.R.L. San Blas 2655 (C1416EFU), Buenos Aires, Argentina Email: [email protected] Técnico en Electrónica. Cursó estudios de Ingeniería Electrónica en la Universidad Tecnológica Nacional (UTN), y Dirección de Empresas en la Universidad Argentina de la Empresa (UADE). Realizó numerosos cursos de especialización en diversas disciplinas relacionadas con el control industrial y la metrología, tanto en el país como en el exterior. Con veinte años de trayectoria en Hitec S.R.L., ha desempeñado distintas y muy variadas funciones técnicas dentro de la empresa. En la actualidad, y desde hace unos diez años, ejerce la Dirección Técnica de la Dirección de Metrología de la empresa. 62 revista safybi, junio 2015 Si bien el presente artículo no explora todos los aspectos del funcionamiento de un contador de partículas en aire de última generación (diseñado acorde a la norma ISO 21501-4) ni de su calibración, es un intento de presentar en términos sencillos algunos de los más importantes (a juicio de los autores). Habiendo sido introducida en el año 2007, la norma ISO 21501-4 al día de hoy se encuentra en un estado que puede juzgarse como maduro, por lo que entender los aspectos principales de dicha norma y su relación con el desempeño de un contador de partículas seguramente resulte de utilidad para quienes de una manera u otra estén relacionados con dichos instrumentos. n CRECIENDO EN ARGENTINA GC, GC-MS Y SPE ESTUFAS, INCUBADORAS, CÁMARAS CLIMÁTICAS, ETC. MICROONDAS HACIA EL LUGAR QUE MERECEN CON EL COMPROMISO, LA SERIEDAD, EL SERVICIO Y EL SOPORTE DE Tel: +54(11) 4795-1920 [email protected] www.cosmobio.com.ar Virginia Gallino Diseñadora gráfica Comunicación visual Fotografía [email protected] - www.virginiagallino.com.ar junio 2015, revista safybi 63 Nuestros anunciantes Inversión y crecimiento En los últimos diez años, Ionics S.A. ha crecido y evolucionado acorde con los cambios y exigencias impuestos por la realidad del país y del mundo. Se han realizado modificaciones para optimizar la infraestructura y combinado con nuestros clientes estrategias en el ámbito nacional e internacional. Se trabajó activamente en nuevos desarrollos de aplicaciones para nutracéuticos, suplementos dietarios, y alimentos que requieren mayor capacidad de tratamientos, y el crecimiento de aplicaciones biológicas para semillas de oleaginosas en la región nos ha llevado a ser creativos a la hora de atender esa demanda. El crecimiento de las operaciones a través del tiempo fue motivando la idea y el desarrollo de un proyecto para incrementar la capacidad de producción mediante la instalación de una nueva unidad radiante. Contra esa idea conspiraba el temor a realizar inversiones en Latinoamérica, región en la que a veces las políticas de estado, por equívocas, inconvenientes o de final incierto, incentivan la desinversión y la fuga hacia ambientes menos tormentosos. Desde comienzos de 2012, con un contexto inflacionario complejo y con la imposibilidad de girar dividendos al exterior se impulsó una estrategia para conservar el valor del patrimonio y las inversiones. Ionics asumió estos desafíos consolidando su cartera de clientes y mejorando no sólo el servicio y la atención sino la infraestructura de la planta: Se pusieron en marcha una serie de inversiones, entre las cuales cabe destacar el diseño y construcción de una nueva vía vehicular interna para facilitar el transito y maniobras de los camiones, permitiéndoles realizar todas sus operatorias circulando en un solo sentido. Esta vía vehicular fue el paso inicial hacia el proyecto de ampliación, ya que su trazado se adaptaba perfectamente al proyecto. Finalmente, a comienzos de 2013 se tomó la decisión de invertir en el negocio Ionics y concretar la instalación de la nueva unidad radiante. Todo el equipo Ionics participó en el diseño, aportando ideas y detalles a tener en cuenta, haciendo especial énfasis en los temas de seguridad radiológica así los de logística y circulación. En enero de 2014 el proyecto obtuvo la licencia de construcción por parte de la Autoridad Regulatoria Nuclear, que viabilizó el comienzo de la obra. El proyecto de ampliación propone una distribución simétrica y especular para los nuevos depósitos y el nuevo búnker con respecto a los existentes, tomando como eje la nueva vía vehicular, quedando así en el centro de las dos unidades radiantes y permitiendo un tránsito óptimo de vehículos y mercaderías, mientras se realiza el control de todas las operaciones desde una sala central única. Se contrató a Martorell Construcciones para la obra civil 64 revista safybi, junio 2015 aprovechando la amplia experiencia de esta empresa en unidades logísticas de envergadura, así como sus contactos con especialistas en hormigón que han participado en obras en centrales nucleares. Para el desarrollo de los sistemas de transporte automático a utilizarse en la nueva unidad, se contrató a Movitec con la asistencia de los ingenieros Zain. La planificación de los trabajos de la nueva obra acordada con la Autoridad Regulatoria Nuclear, ha permitido mantener la operación de la unidad existente al ciento por ciento, respetando así el normal desarrollo y cumplimiento de los tiempos de tratamiento de ionización de nuestros clientes. También durante 2014 se construyeron las nuevas oficinas administrativas, vestuarios e instalaciones sanitarias para el personal de planta. En 2015 comenzaron los trabajos más importantes: La demolición de las viejas oficinas administrativas y la excavación de la nueva pileta de blindaje, que representa una obra de gran envergadura por el movimiento de terreno que implica un foso de siete metros de profundidad. Además, para generar una estructura monolítica de hormigón con paredes de hasta dos metros de espesor, se construyó un encofrado El colado de hormigón del encofrado de las paredes del bunker de la nueva unidad radiante requerirá unos sesenta camiones de hormigón, que asegurarán una estructura monolítica de blindaje óptimo. especial que por su trincado y apuntalamiento da imagen de obra monumental. Unos sesenta camiones de hormigón elaborado se vaciarán en colada continua con vibración para asegurar el blindaje óptimo definitivo. Todo el equipo de Ionics se siente orgulloso por lo que significa esta obra no sólo para la empresa sino también para la industria nacional. Esta importante decisión significa el inicio de una apuesta al crecimiento junto a nuestros clientes con el compromiso de todo el equipo, pero significa sobre todo, aceptar el desafío de un futuro venturoso para la empresa y para el país. n Nuestros anunciantes Un nuevo concepto en soluciones para la industria farmacéutica Historia Quienes somos La historia de IP&QA Solutions se inició en el año 2014 por un grupo de profesionales en diversos áreas relacionadas con la Industria Farmacéutica, con el claro objetivo de brindar al mercado un servicio distinto a lo disponible hasta ese momento, basados en tecnología, y visión de futuro. En nuestro equipo se destacan Pablo Rolando, como Director Comercial y responsable de Desarrollo de Negocios, y Alejandro López Stanley como Project Manager. Ambos con pasado en SVS y Telstar, y casi 20 años de experiencia en la Industria Farmacéutica, en empresas que incluyen a Idenor Ingeniería, Laboratorios Roemmers, Synthon Argentina, y Phoenix (del grupo GSK), y desarrollando diversos proyectos en el exterior en países como Uruguay, Paraguay, Venezuela, España, Colombia, Brasil, Perú y Chile. Lopez Stanley como responsable Operativo de IP&QA Solutions se encuentra actualmente liderando a un grupo de consultores con pasado en SVS/Telstar y con amplia experiencia en la industria farmacéutica, los cuales forman parte del Staff permanente de IP&QA Solutions. n Presente Contamos con el apoyo tecnológico de Hitec, empresa ya asentada desde hace 29 años, tanto en la industria farmacéutica como en “industria no farma”, y a partir de ese background tecnológico e innovador salimos al mercado a brindar servicios de consultoría en GMP, soluciones PAT y QbD, training y soluciones MES entre otras. En definitiva, con IP&QA Solutions venimos a brindar Soluciones para todas las áreas de la Industria Farmacéutica, con alto valor profesional, técnico y humano. junio 2015, revista safybi 65 Nuestros anunciantes Una cuestión de seguridad El test de esterilidad en productos parenterales es un criterio decisivo para la seguridad de los productos farmacéuticos. Ninguna preparación estéril entra en el mercado sin pasar estos test. ¿Cuáles son los factores clave de los test de esterilidad? Marcus Schütte Gotas para los ojos contaminadas con Pseudomonas aeruginosa pueden, como en el año 1968 sucedió en Suecia, provocar una reducción considerable de la visión o incluso la ceguera total. Si los agentes infecciosos (bacterias, virus, hongos, parásitos) logran introducirse y propagarse en la sangre, hablamos de sepsis, se produce una respuesta inmune significativa a los patógenos o sus toxinas. La industria farmacéutica se compromete cada vez más firmemente con la gran cantidad de requerimientos de las directrices BPF (Buenas Prácticas de Fabricación, GMP en inglés), que a través del concepto de aseguramiento de la calidad y la gestión de calidad, garantizan la seguridad de los productos farmacéuticos. Las directrices BPF no sólo se aplican a los medicamentos en si, sino también a la producción, el empaquetado y los controles, así como al suministro de materias primas incluyendo los equipos y aparatos necesarios. Los puntos clave de las directrices BPF también incluyen al personal, la higiene del personal y su cualificación. La minimización de la contaminación microbiana durante la producción de productos farmacéuticos es absolutamente necesaria. La implementación de controles durante el proceso y el control de productos finales es un componente fundamental para comprobar la contaminación microbiológica o la esterilidad en ciertos casos. Ejemplo de una unidad Sterisart NF versión Septum para pruebas de esterilidad. En la foto, se ve una unidad con doble aguja corta y afilada, ventilación estéril automática y plataforma de seguridad. 66 revista safybi, junio 2015 Pruebas de esterilidad para la liberación de lotes Se aplican requerimientos especialmente elevados, en particular, para las preparaciones estériles (parenterales), que se administran por inyección, infusión o trasplante en el cuerpo humano o de animales. El control de la esterilidad de los productos finales envasados es un criterio crucial y decisivo para la liberación completa de lotes. La prueba de esterilidad de productos finales puede llevarse a cabo por el fabricante del medicamento o por un laboratorio contratado debidamente certificado. De acuerdo a las Farmacopeas internacionales (USP <71>, EP 2.6.1, JP 4.06, WHO [QAS/11.413 final]) se llevan a cabo pruebas de esterilidad no solo en parenterales sino también para la liberación de las gotas para los ojos, pomadas, cremas, aerosoles y formas sólidas como los materiales de sutura quirúrgicos. El método de cultivo de microorganismos para la comprobación de la esterilidad de los productos existe desde alrededor de 1930. Con él se examinan los productos farmacéuticos bien por filtración, que según la USP <71> o la EP 2.6.1 es el método preferible o, cuando no haya otra posibilidad, por inoculación directamente. La prueba de esterilidad se lleva a cabo en un ambiente controlado en condiciones asépticas, bien en aisladores, que corresponden a la sala limpia de clase A o bien en la sala limpia de clase A de un laboratorio, rodeada de una sala limpia de clase B. Las medidas que se toman para evitar la contaminación de las áreas críticas no deben afectar en la prueba de esterilidad al crecimiento de los microorganismos presentes. Esto debe garantizarse por validaciones apropiadas (test bacteriostasis/fungistasis). Se utilizan métodos como la toma de muestras de aire activa con el escáner de aire MD8, para controlar las condiciones ambientales sin interferir en la protección de la zona de sala limpia. En áreas especialmente críticas como las líneas de llenado, aisladores o equipos Blow-Fill-Seal (soplado, llenado y sellado), los clientes se apoyan en el control de aire para preservar las altas tasas de contención y recuperación de virus y microorganismos a través de los filtros de membrana de gelatina (FMG). Pero también los requerimientos sobre el sistema y los materiales, así como en el personal que realiza las pruebas de evaluar los riesgos económicos así como las ventajas y desventajas de un sistema de pruebas de esterilidad. El escáner integrado posibilita la lectura del código de barras del embalaje primario de una unidad Sterisart NF. de esterilidad, son muy altos. Por último, un personal bien entrenado es un componente muy importante para evitar falsos resultados negativos, así como falsos positivos, (U.S. Department of Health and Human Services Food and Drug Administration, (CBER) Office of Regulatory Affairs (ORA), 21 CFR 211.25. Por ello, el personal de laboratorio debe tener en cuenta con anterioridad las aplicaciones respectivas, así como las necesidades, y crear una lista de control o Especificación de Requisitos de Usuarios (ERU). Se han ¿Cuál es la regulación vigente para las pruebas de esterilidad? Antes que nada habría que aclarar en qué país se va a vender el producto farmacéutico, ya que el conocimiento sobre las políticas correspondientes es un requisito fundamental. Por ejemplo, la prueba de esterilidad se realiza en casi todos los países con dos medios y dos recipientes. Para productos que se venderán en China, sin embargo, es necesaria una variante de tres recipientes. En Sudamérica y África, debido al bajo coste del personal, no se utilizan unidades preesterilizadas cerradas, sino componentes individuales, que se ensamblan gradualmente a un sistema. Esto se conoce en Alemania con el nombre de „SchillerSystem“, una metodología conocida que fue común también en Europa hasta aproximadamente 1975. Sin embargo, no ha prevalecido en Europa, USA y Japón debido al tiempo necesario para ensamblar y por los riesgos de resultados con falsos positivos. El procedimiento para la prueba de esterilidad está claramente regulado: lo que se necesita es una fuente de presión adecuada para salas limpias, por ejemplo una bomba peristáltica que transporte el líquido a analizar a las unidades de ensayo estériles, filtrándolo a través de una membrana. Los gérmenes son junio 2015, revista safybi 67 Nuestros anunciantes retenidos por la membrana (tamaño de poro nominal de 0,45 μm), los antibióticos y otras sustancias inhibidoras del crecimiento se deben aclarar por las propiedades de baja absorción de la membrana. En el caso de que el producto sea en polvo, debe ser solubilizado con las medidas adecuadas. Al final del proceso, los dos recipientes estarán cerrados. Posteriormente, se lleva a cabo el llenado de recipientes con el medio nutriente apropiado: El digerido de caseína de soja (CASO, por sus siglas en inglés) para la detección de bacterias aerobias y medio fluido de tioglicolato para la detección de bacterias anaerobias/aerobias. Las soluciones de enjuague permitidas pueden leerse en la USP<71>. Debido a que generalmente los septos de las soluciones de enjuague o de productos se perforan más de una vez, se debe tener en cuenta que las agujas han de ser afiladas y suaves para una introducción múltiple. Para posibilitar un trabajo adecuado según las BPF, ha de ser posible la trazabilidad por lotes de todos los elementos utilizados incluyendo los sistemas de test de esterilidad o mejor: en cada envase ha de ser guardable en pdf un código de barras escaneable y los datos de acuerdo con el “Code of Federal Regulation“ (21 CFR part 11). Seguridad en las preparaciones estériles Para el control de los productos farmacéuticos estériles Sartorius ofrece las unidades desechables Sterisart NF para el control de calidad del producto final. Estos sistemas están concebidos como soluciones desechables cerradas para poner a prueba bolsas, jeringas, viales, ampollas y polvos. El principio básico se basa en el método de filtración por membrana. El objetivo de las unidades Sterisart NF es una prueba de esterilidad reproducible y fiable, sin cabida a resultados falsos, positivos o negativos. Durante los 14 días de incubación para la identificación de contaminación (crecimiento) o para la validación del producto, puede ser necesario extraer muestras de los envases asépticos, o aplicar líquidos, como la adición de penicilinasa para la inactivación de los antibióticos. Esta extracción se lleva a cabo hoy en día sobre todo con una punción con una aguja a través del tubo o cortando el tubo y posteriormente introduciendo la solución con una pipeta. De esta manera el riesgo de contaminación y el riesgo de lesiones del usuario (aguja) aumentan considerablemente. Con las versiones septum de las unidades Sterisart NF, las extracciones son significativamente más fáciles y evitan los riesgos. La tecnología septum está reconocida en el campo de la medicina y se ha probado con éxito. El septum está sellado con una tapa y se puede limpiar, si fuera necesario, con un paño humedecido en isopropanol. Si el propio producto farmacéutico enturbia el medio, la USP <71> recomienda lo siguiente: …”14 días posteriores al comienzo de la incubación transferir porciones del medio (cada una no menor a 1 mL) a vasos frescos del mismo medio ...”. Esta transferencia de líquidos también se puede llevar a cabo de forma segura y fiable con las versiones septum. El concepto de las transferencias de líquidos y la ventilación simultánea del recipiente se puede lograr con una aguja doble patentada. Las agujas de acero inoxidable son muy afiladas, lo que, en primer lugar, facilita considerablemente la perforación en los septos y, en se- 68 revista safybi, junio 2015 gundo lugar, el manejo es más fácil en comparación con las agujas individuales, ya que no se requiere ningún filtro de ventilación por separado. Bomba universal para transferencia de líquidos La bomba universal Sterisart está disponible para la transferencia del líquido del producto en las unidades. Un escáner de código de barras integrado permite, de forma sencilla, la trazabilidad de los productos utilizados, como los medios y las unidades Sterisart NF. Se prestó una atención especial en la idoneidad de la bomba en salas limpias. La superficie de la bomba está hecha de acero inoxidable tipo 316 L. Puede funcionar ya sea debajo de un banco limpio o integrada en un aislador. Además de la compatibilidad con H2O2, los componentes individuales se caracterizan por su resistencia UV y por ser autoclavables. Estos dos aspectos son particularmente importantes para su funcionamiento debajo de un banco limpio. La bomba universal Sterisart cumple con las GMP, es completamente cerrada y no tiene ventilación. Sartorius Stedim Biotech es un proveedor profesional de productos y servicios para la industria farmacéutica a la vanguardia en la seguridad y la calidad de los productos. La compañía ofrece soluciones fiables y económicas también para el control de calidad en la industria farmacéutica. La oferta de productos abarca desde balanzas analíticas hasta sistemas de agua ultrapura, pipetas o consumibles e incluso productos de microbiología. Para el control microbiológico durante el proceso, incluye en su catálogo los filtros Microsart@ y @media, kits de detección de Mycoplasma y un muestreador de aire. El control del final del proceso (pruebas de esterilidad) de parenterales puede llevarse a cabo con unidades de Sterisart NF. Algunas preguntas para Marcus Schütte, Product Manager de Microbiología en Sartorius Stedim Biotech –Señor Schütte, la ejecución de un test de esterilidad es compleja y por lo tanto propensa a errores. Según su experiencia, ¿cuáles son los errores que se cometen con mayor frecuencia? Bomba universal Sterisart, incluye una pantalla y un escáner de código de barras, para proveer de los medios y líquidos necesarios para el test de esterilidad. –De acuerdo a las regulaciones pertinentes, la ejecución de un test de esterilidad está claramente definida, al igual que las condiciones ambientales y el requisito de que sólo el personal cualificado puede realizar esta prueba. Sin embargo, el error humano nunca se puede descartar completamente. En las cartas de advertencia de la FDA, entre otras investigaciones inadecuadas en una prueba de esterilidad positiva, se critica una desinfección inadecuada y un control ambiental insuficiente. –A menudo, los fabricantes de parenterales optan por tercerizar la prueba de esterilidad a proveedores de servicios. ¿Qué cualidades debe tener un buen laboratorio? –Los laboratorios de terceros deben tener la experiencia necesaria en la aplicación de las pruebas de esterilidad y deben tener el certificado de BPF/BPL respectivamente. Dependiendo del producto y los deseos del cliente, un proveedor de servicios puede realizar la prueba tanto en un aislador como en un banco limpio en una sala limpia de clase B. Desde mi punto de vista, el conocimiento de la validación de productos específicos y la experiencia en el trato con las autoridades (FDA) son requisitos esenciales. n M. Schütte es Product Manager de Microbiología en Sartorius Stedim Biotech GmbH, Goettingen. Contacto: Tel. +49.551.308.3522 revista safybi 69 14o Exposición Internacional del Envase y Embalaje 14th International Packaging Show 9o Exposición Internacional de Maquinaria y Equipamiento para el Procesamiento de Alimentos y Bebidas 9th International Machinery and Equipment of Food and Beverages Processing Show 2o Exposición Internacional de Maquinaria y Equipamiento para la Industria Farmacéutica 2nd International Machinery and Equipment for the Pharmaceutical Industry Show En simultáneo con: August 4 - 7 4 al 7 de agosto 2015 www.envase.org Buenos Aires I Argentina Centro Costa Salguero I Costa Salguero Exhibition Grounds Organiza / Organizer INSTITUTO ARGENTINO DEL ENVASE Av. Jujuy 425 (C1083AAE) Ciudad Autónoma de Buenos Aires, Argentina te: (54-11) 4957-0350 e-mail: [email protected] www.packaging.com.ar Contacto de ventas: e-mail: [email protected] te: (54-11) 4957-0350 ext. 112 Auspicia / Sponsor Nuestros anunciantes Medición de humedad y densidad con tecnología de resonancia de microondas Aplicación del sistema de medición por resonancia de microondas en la determinación de humedad en línea en secadores de lecho fluido El método resonador desarrollado y patentado por Tews Elektronik, Hamburgo, Alemania, se basa en la medición de los cambios de atenuación y de frecuencia de las microondas tomadas como dos parámetros diferentes. Esta alternativa posibilita la medición de humedad independientemente de la masa o la densidad. Secadores de Lecho Fluido: El secado después de la granulación es uno de las etapas más importantes durante la producción de los productos farmacéuticos. Varias tecnologías son utilizadas para realizar el secado. Para muchas aplicaciones, los Secadores de Lecho Fluido son la primera elección debido a la calidad del producto que se obtiene (Figura 1). Típicamente, el proceso de secado se detiene cuando el contenido de humedad del producto está en un intervalo cuyo máximo es aproximadamente 2%. Esto puede variar dependiendo del producto, pero casi todas las aplicaciones tienen en común que el intervalo buscado de la humedad del producto final es de un 0,4%. Esta es una ventana relativamente pequeña teniendo en cuenta los errores inevitables y las incertidumbres asociados a los métodos de medición de los sistemas de referencia tradicionales. En este momento, el procedimiento más común es detener el proceso cuando se considera estar cerca de la humedad objetivo, se toma una muestra y se mide. Luego se decide si el proceso continúa y por cuánto tiempo. El proceso que se viene utilizando presenta el inconveniente de que el mismo incluye la necesidad de detener y reiniciar el proceso de secado conjuntamente con el retardo generado por la medición en sí misma. Teniendo en cuenta esto, la mejor opción es un sistema que mida la humedad, en tiempo real, dentro del secador Figura 1 de lecho fluido y que de la señal de parada o corte cuando se consigue la humedad deseada. La tecnología de microondas El sistema desarrollado por Tews-Elektronik de medición de humedad que utiliza la tecnología de microondas, tiene la capacidad de medir el contenido de humedad así como la densidad del producto en forma independiente. Dicha propiedad hace posible y altamente confiable la medición en línea de humedad en el lecho fluido, pues los equipos de Tews Elektronik tienen la capacidad de calcular el porcentaje de humedad en forma independiente de la densidad del producto o de la cantidad de producto que pase por delante del sensor en un momento dado, independizándolo de cualquier variabilidad instantánea propia de la dinámica del secador del lecho fluido. Otra ventaja de la medición con microondas es que la penetración del campo de microondas es de varios cm, mientras que por ejemplo las mediciones ópticas tienen una penetración en el producto de apenas algunas longitudes de onda (milímetros). Por lo tanto el sistema de medición de microondas puede medir no sólo en la superficie sino en la totalidad del producto que pasa frente del sensor. Experiencia práctica El punto de instalación típica del sensor de lecho fluido se encuentra en la parte inferior del mismo, según se observa en las Figura 1 y 2. El sensor (Figura 3) tiene características acordes con lo requerido en las GMP. Mide a través de su superficie frontal que genera un campo de microondas cuya potencia máxima utilizada es de no más de 10 mW, lo que no impacta ni en el proceso de secado ni en el produc- Figura 2 Figura 3 junio 2015, revista safybi 71 Nuestros anunciantes Figura 4 rante los últimos años. Un ejemplo típico de la evolución de la humedad y temperatura medidas por el sistema de microondas puede observarse en la Figura 4. Los equipos de medición de resonancia de microondas Tews tienen la habilidad tanto de conectarse a computadoras como de realizar el corte automático del secador de lecho fluido. En el rango bajo de humedad el sistema de microondas presenta especialmente una muy buena correlación con los valores de referencia del laboratorio, teniendo una exactitud del orden del 0,1% al 0,2%. Ventajas de las mediciones en la línea to en sí mismo. Dicha potencia es suficiente para medir la humedad en los polvos y gránulos que pasan delante de él, sin importar el tamaño de las partículas ni los cambios de coloración de las mismas. Un esquema del proceso de medición puede observarse en la Figura 2. Muchas mediciones utilizando diferentes productos y diferentes secadores de lecho fluido se han realizado du- • Medición de la humedad de proceso en tiempo real. • Medición completa (interna y externa) de los gránulos. • Alta exactitud independientemente de las variaciones del tamaño de las partículas, densidad o de color. • No hay interrupciones. • No hay muestreo manual. • Ahorra energía y tiempo de proceso. • Documentación disponible sobre la calificación completa de la instalación y operación (IQ/OQ). • El software de comunicación con el Secador de Lecho Fluido cumple con la CFR (Code Federal Regulation, FDA) parte 11. • Mejora la procesabilidad y la vida útil del producto. n Anuncian en este número 2MS............................................................ AEROFARMA ............................................ AKRIBIS...................................................... Akrimet.................................................... ANALYTICAL.............................................. ANDREANI................................................. APTAR PHARMA........................................ ARGENPACK.............................................. ARTICA....................................................... ASISTHOS.................................................. BLUE SOLUTION....................................... Business Point...................................... CARPE SCHEIDER...................................... CAS INSTRUMENTAL................................ CATLAB...................................................... CIMA-SINOTEK.......................................... COSMOBIO............................................... COSTER GROUP........................................ DROMEX.................................................... EDYAFE...................................................... ESPIRAL DE CALIDAD............................... ESTUDIO ZE-K........................................... ETICOR....................................................... EXPOFYBI 2015......................................... FARMAWALL............................................. FERRETTI VALIDACIONES........................ GENERAL ELECTRIC.................................. 72 revista safybi, junio 2015 41 13 45 54 21 57 9 34 53 33 48 69 19 23 36 1 63 3 37 69 44 20 49 12 Retiración de Contratapa 29 5 GS1............................................................. GUIBA TÉCNICA........................................ HANNA INSTRUMENTS............................ HITEC INGENIERÍA / IP&QA..................... HÖGNER.................................................... IBUTTON DEVICES.................................... IDEAGRAF.................................................. INSTITUTO ARG. DEL ENVASE................. INSTRUMENTACIÓN CIENTÍFICA............ IONICS....................................................... KIMS........................................................... LA POSTAL................................................. LOG-IC....................................................... MEDIGLOVE.............................................. NOVOCAP.................................................. PALL TECHNOLOGIES.............................. PHARMAFOIL............................................ PHARMA PANEL ....................................... ROEMMERS............................................... SARTORIUS................................................ SINAX S.A................................................... SOLUCIONES ANALÍTICAS....................... TESTO ARGENTINA .................................. TEWS.......................................................... VIRGINIA GALLINO - DISEÑO.................. WORLDWIDE SERVICES........................... 31 36 24 59 11 10 67 70 43 Retiración de Tapa 55 46 22 61 51 25 35 15 7 Contratapa 63 39 27 47 63 17 Junio de 2015 | Nº 146 de sus tareas diarias en el laboratorio Volumen 55 Nº 146 Junio de 2015 ISSN: 0558/1265 Uruguay 469, piso 2º B C1015ABI Buenos Aires, Argentina Tel: (54-11) 4373-0462 / 8900 (54-11) 4372-7389 Fax:(54-11) 4374-3630 www.safybi.org Si usted está buscando calidad y precisión en el laboratorio, nosotros lo tenemos. Descúbralo usted mismo y conozca nuestras balanzas, sistemas de agua, productos para el manejo de líquidos, control de calidad microbiológico y soluciones de filtración, respaldados por un equipo de servicio accesible a nivel mundial para ayudar a mantener el alto nivel de desempeño de sus productos Sartorius. Revista Revista SAFYBI | Lo ayudamos a cumplir con los requerimientos Revista SAFYBI SAFYBI: CLAPROMEDIS en Palabras del Presidente ANMAT: Uso racional de medicamentos Sartorius Argentina S.A. Tel. +54.11.4721.0505 [email protected] Visite nuestra revista online en www.sartorius.com/es Pesaje de Laboratorio Control de Calidad Microbiológico Manejo de Líquidos Filtración de Laboratorio Agua para Laboratorio Servicio www.safybi.org CONICET: Entrevista al Director de Vinculación Tecnológica USP: Materiales de ensayo clínico durante la distribución
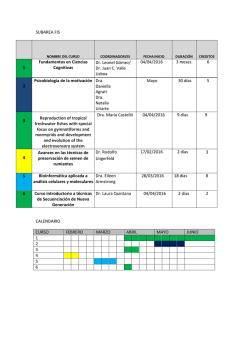

© Copyright 2026