
Volumen 36 No 2 Septiembre 2015 - Revista Politécnica
Volumen 36 No 2 Septiembre 2015 Volumen 36 No 2 Septiembre 2015 La Revista Politécnica es una publicación semestral de la Escuela Politécnica Nacional que pone al alcance de los sectores académico y productivo los desarrollos científicos y tecnológicos en las áreas de las ingenierías y ciencias. Está incluida en: - Latindex: Sistema Regional de información en línea para Revistas Científicas de América Latina, el Caribe, España y Portugal Se autoriza la reproducción total o parcial de su contenido siempre y cuando se cite la fuente. Los conceptos expresados son de responsabilidad exclusiva de sus autores. Contactar con la Coordinación de Edición: [email protected] [email protected] Teléfono: (+593) 3976000 ext. 5218 Guía para autores: http://revistapolitecnica.epn.edu.ec/repo_files/autor_manual.pdf COORDINADORA DE EDICIÓN Ing. Iliana Carrera COMITÉ DE APOYO EDITORIAL Ing. Andrés Proaño Srta. Dayana Gavilanes Sr. Luis Estrada Srta. Sandra Rojas Sr. Daniel Cabrera REVISTA POLITÉCNICA Volumen 36, Número 2 Septiembre 2015 CONSEJO EDITORIAL Enio Da Silveira, Ph.D. Universidad Católica de Río, Brasil. Carlos Smith, Ph.D. University of South Florida, Estados Unidos Gyimah-Brempong Kwabena, Ph.D. University of South Florida, Estados Unidos ESCUELA POLITÉCNICA NACIONAL Rector Jaime Calderón, MBA Vicerrector de Investigación y Proyección Social Alberto Celi, Ph.D. Vicerrector de Docencia Tarquino Sánchez, MBA Director de Investigación y Proyección Social Andrés Rosales, Ph.D. Editor Oscar Camacho, Ph.D. Co Editora Florinella Muñoz, Ph.D. Raymundo Forradelas, Ph.D. Universidad Nacional del Cuyo, Argentina Ricardo Carelli, Ph.D. Universidad Nacional de San Juan, Argentina. Vanderlei Bagnato, Ph.D. Universidad de Sao Paulo, Brasil. Rui Pedro Pinto de Carvalho, Ph.D. University of Coimbra, Portugal. Oscar Ortiz, Ph.D. Universidad Nacional de San Juan, Argentina Gustavo Scaglia, Ph.D. Universidad Nacional de San Juan, Argentina Chen Ning, Ph.D. Universidad de Mineralogía y Tecnología de China, China. Alex Ruiz Torres, Ph.D. Universidad de Puerto Rico, Puerto Rico. Lizandro Solano, Ph.D. Universidad de Cuenca, Ecuador Romel Montufar, Ph.D. Pontificia Universidad Católica, Ecuador Marcos Villacís, Ph.D. Escuela Politécnica Nacional, Ecuador Andrés Rosales, Ph.D. Escuela Politécnica Nacional, Ecuador Danilo Chávez, Ph.D. Escuela Politécnica Nacional, Ecuador Oscar Camacho, Ph.D. Universidad de Los Andes, Venezuela Vicenzo Vespri, Ph.D. Università degli studi di Firenze, Italia Carlos Ávila, Ph.D. Escuela Politécnica Nacional, Ecuador COMITÉ EDITORIAL Andrés Rosales, Ph.D. Investigador Escuela Politécnica Nacional José Luis Paz, Ph.D. Prometeo Escuela Politécnica Nacional, Ecuador Ernesto Jiménez, Ph.D. Prometeo Escuela Politécnica Nacional, Ecuador ESCUELA POLITÉCNICA NACIONAL Campus "José Rubén Orellana" Ladrón de Guevara E11 - 253 Quito - Ecuador Luis Rodríguez, Ph.D. Prometeo Escuela Politécnica Nacional, Ecuador Rafael Uribe, Ph.D. Prometeo Escuela Politécnica Nacional, Ecuador Jose Aguilar, Ph.D. Prometeo Universidad Técnica Particular de Loja, Ecuador CONTENIDO 1 Almeida E.; Espín N. Obtención de extractos enzimáticos con actividad ligninolítica en un fermentador de bandejas con el hongo Phanerochaete chrysosporium 9 Cruz A.; Guamán M.; Castillo M.; Glorio P.; Martínez R. Fibra dietaria en subproductos de mango, maracuyá, guayaba y palmito 16 Reyes J.; Ludeña F. Evaluación de las Características Físico-Químicas, Microbiológicas y Sensoriales de un Yogur Elaborado con Sucralosa y Estevia 25 Morales F.; Marín F.; Morales L.; Morales I.; Marín L.; Gastelurrutia M. Atención Farmacéutica en el Fomento del Desayuno Saludable desde la Oficina de Farmacia 29 Martínez S.; Figueroa J. Cuantificación de Ca, Mg, K, Na y Mn en Subproductos de Cítricos procedentes de Ecuador 34 Fernández J.; Tarazona G. Factores que Influyen en la Composición de la Leche en el Sector el Retorno, Parroquia Sabanilla, Cantón Zamora, Provincia de Zamora Chinchipe – Ecuador 42 Gámez S.; De la Torre E. Recuperación de Oro a Partir de Minerales Polisulfurados con Soluciones Amoniacales de Tiosulfato de Sodio 49 Campos C.; De la Torre E. Estudio de la Detoxificación de Efluentes Cianurados por Oxidación con Dióxido de Azufre, Aire y Catalizadores de Cobre 59 Barrera A.; Endara D.; De la Torre E.; Manangón L. Recuperación de Níquel, Vanadio y Molibdeno del Catalizador Agotado de la Unidad de Craqueo Catalítico Fluidizado (FCC) 67 Loyo C.; Arroyo C.; Aldás M.; Montero R. Extracción y Caracterización de la Fracción No Metálica de las Tarjetas de Circuitos Impresos de Computadoras Desechadas 73 Meza L.; De la Torre E. Modelo de Deconvolución para la Cuantificación de los Componentes del Caucho Vulcanizado Presente en los Neumáticos Fuera de Uso 80 Saltos P.; Chango I.; Aldás M.; Quiroz F. Reciclaje de Poliestireno Expandido por el Método de Disolución Precipitación 89 Proaño A.; Bonilla O.; Aldás M. Desarrollo de un Material Compuesto de Matriz de Poliuretano Rígido Reforzado con Fibra de Raquis De Palma Africana 96 Cardozo E.; Contreras RR.; Bellandi F.; Lopez-Rivera A.; Avendaño J.; Araque C.; Vielma J. Synthesis and Characterization of Six Novel Samarium (III) Complexes with Laspartic Acid, L-glutamic Acid, Glycine and O-phenanthroline, Bipiridile as Ligands 101 Villacís W.; Andrade C. Implementación de Medidas de Prevención y Control de Ruido para los Trabajadores del Centro de Generación de Energía de la Empresa Dipor S.A. 109 Albarracín K.; Jaramillo L.; Albuja M. Obtención de Bioetanol Anhidro a Partir de Paja (Stipa ichu) 1 Obtención de extractos enzimáticos con actividad ligninolítica en un fermentador de bandejas con el hongo Phanerochaete chrysosporium _______________________________________________________________________________________________________________________________ Obtención de extractos enzimáticos con actividad ligninolítica en un fermentador de bandejas con el hongo Phanerochaete chrysosporium Almeida E.*; Espín N.* *Escuela Politécnica Nacional, Facultad de Ingeniería Química y Agroindustria, Quito, Ecuador e-mail: [email protected]; [email protected] Resumen: Se estudió la actividad enzimática de lignino peroxidasa (LiP) y manganeso peroxidásica (MnP) en extractos obtenidos de la fermentación de aserrín de eucalipto con Phanerochaete chrysosporium a escala de laboratorio en frascos de 250 g y en un fermentador de bandejas, con 3 960 g de aserrín, 72 % de humedad, inóculo del 10 %, 30 ºC, y flujo de aire de 0.19 L/s, durante 4 días. Las actividades de LiP y MnP se incrementan inmediatamente después de la inoculación, son máximas en el primer o segundo día y luego descienden. La actividad enzimática de la LiP es mayor. A escala de laboratorio los valores obtenidos son 4 a 5 veces mayores. Palabras clave: Phanerochaete chrysosporium, lignino peroxidasa (LiP), manganeso peroxidasa (MnP), fermentación en medio sólido, enzimas Abstract: Enzyme activity of Lignin peroxidase (LiP) and Manganese Peroxidase (MnP) obtained from sawdust fermentation with Phanerochaete chrysosporium was studied using lab flasks of 250 g and a try fermentor with 3 960 g sawdust, 72 % humidity, 10 % inoculum, 30 ºC and air flow of 0.19 L/s, for 4 days. LiP and MnP activities increase immediately after inoculation; are greatest at the first or second day, and then decrease. LiP activity is higher. Lab scale produces higher activities than tray fermentation. Keywords: Phanerochaete chrysosporium, lignin peroxidase (LiP), manganese peroxidase (MnP), solid fermentation, enzymes. 1. INTRODUCCIÓN La producción de enzimas lignocelulósicas es una alternativa muy rentable para aprovechar los desechos lignocelulósicos debido a la gran cantidad de aplicaciones industriales de las enzimas. Las ligninasas se emplean en la obtención de bioetanol, porque optimiza el proceso y abarata costos [19]. En la industria papelera, el uso de enzimas es conveniente porque las condiciones de operación son menos severas, no generan productos indeseados y son amigables con el ambiente [10]. En el campo de la biorremediación, se emplean enzimas ligninolíticas debido a que son catalíticamente activas sobre una gran variedad de compuestos orgánicos, como fenoles clorinados, plaguicidas, dioxinas, explosivos aromáticos y colorantes [5]. El tratamiento enzimático de efluentes presenta ventajas sobre los métodos tradicionales como: degradación de materiales recalcitrantes, operación dentro de un amplio rango de concentraciones de los contaminantes, pH, temperaturas, salinidad y fácil control del proceso [9]. La fermentación en sustrato sólido, FSS, de los desechos lignocelulósicos con hongos filamentosos, para países en vías de desarrollo podría resultar una alternativa viable para la producción de enzimas se refiere, ya que se emplean desechos como medio de cultivo, el equipo no requiere una inversión alta, lo que permitiría minimizar costos de producción. Por esta razón, el presente trabajo está orientado, al estudio de la producción de extractos con posible actividad enzimática ligninolítica, a partir de la fermentación en medio sólido con el hongo Phanerochaete chrysosporuym con aserrín de eucalipto como sustrato. Se realizó el estudio a escala de laboratorio en frascos de 250 g y en un fermentador de bandejas con el objetivo de determinar los días de máxima generación de activididad lignino peroxidásica (LiP) y manganeso peroxidásica (MnP) presentes en los extractos, durante el proceso fermentativo. 2. MATERIALES Y MÉTODOS 2.1 Materiales Se utilizó una cepa de Phanerochaete chryosoporium Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 2 Almeida E.*; Espín N.* _______________________________________________________________________________________________________________________________ proporcionada por el Laboratorio de Bioprocesos de la Escuela Politécnica Nacional. El aserrín de eucalipto utilizado como sustrato fue obtenido de un aserradero de la cuidad de Ibarra el aserrín de maderas tropicales fue obtenido de la empresa Endesa Botrosa. Los reactivos grado analítico empleados fueron: Alcohol veratrílico de Aldrich, Tartrato de sodio y Ácido acético de Sigma, Ácido tartárico y Fenol de DBH Laboratories, Tween 80 de Life, Acetato de sodio, Agua Oxigenada e Hidróxido de sodio de Merck, Albúmina de JT Baker, Sulfato manganoso monohidratado de Mallinckrodt, Malta Agar de Difco. 2.2 Preparación del inóculo primario del hongo Phanerochaete chrysosporyum La cepa del hongo Phanerochaete chrysosporuym, se duplicó en erlenmeyers con malta agar. Se incubaron a 30 ºC en una estufa marca Boekel Industries INC Model 132000, hasta que el micelio cubrió en su totalidad la superficie de la caja. Con el hongo se inoculó un medio de cultivo de malta agar enriquecido con un 0.7 % de aserrín en peso, contenido en cajas Petri. Estas se almacenaron dentro de la estufa a 30 °C, durante 21 días, hasta que el micelio del hongo cubriera toda la superficie del agar en la caja. El hongo de estas cajas Petri fue utilizado como inóculo durante toda la experimentación. Los frascos fueron colocados en un agitador New Brunswick, durante 30 min; luego se filtró el contenido de cada frasco con una bomba de vacío. Se obtuvo de 60 a 70 mL de cada extracto enzimático, que se clarificaron por centrifugación a 1509 gravedades, durante 15 min, en una centrífuga marca Thermo IEC HN SII. Se midieron las actividades enzimáticas correspondientes para cada uno de los cuatro extractos elaborados. 2.5 Descripción del fermentador de bandejas En la Fig. 1, se presenta el esquema del fermentador de bandejas empleado en la experimentación. Es un armario de madera de madera de 61 X 60.5 X 101 cm (frente, fondo, ancho), recubierto internamente con pintura para facilitar la limpieza. En su interior hay una estructura desmontable de madera en la que se colocan las seis bandejas que pueden deslizarse hacia afuera o adentro del armario; además de un sistema de deflectores para una distribución homogénea del aire. Las bandejas de 32 X 53 cm tienen un marco de madera y una malla metálica fina en el piso que sirve como soporte para el medio de fermentación. 2.3 Preparación del sustrato e inoculación del aserrín a escala de laboratorio En frascos de vidrio de 500 mL, se colocaron 70 g de aserrín seco y 140 mL de agua (relación 2:1 agua: aserrín seco). Se esterilizaron los frascos en un autoclave marca New Brunswick, modelo AE15-10, a 121 °C durante 15 min. Una vez fríos, de 30 a 40 °C, se realizó la inoculación del sustrato en una cámara de flujo laminar, marca Laminar Cabin flow 85 H. La inoculación se realizó de la siguiente manera: se cortó asépticamente el micelio y el agar desarrollado en las cajas petri en ocho partes iguales. Se tomaron cuatro partes opuestas entre sí y se colocaron en la superficie de cada frasco; el inóculo se colocó con el hongo hacia el aserrín, en la parte superior del frasco, y se cubrió con una capa del aserrín. Se cerraron los frascos no herméticamente para poder permitir la entrada y salida de gases al frasco. Los frascos se incubaron durante cinco semanas a 30 °C. 2.4 Muestreo y obtención de extractos enzimático a escala de laboratorio Se muestrearon cuatro frascos, diariamente desde el día de la siembra durante 5 días. A cada frasco se añadieron 70 mL de la solución buffer correspondiente a cada ensayo de determinación de las actividades enzimáticas, es decir, solución buffer de tartrato de sodio para la ligninoperoxidasa y buffer de acetato de sodio para la manganesoperoxidasa. Se utilizaron dos frascos para cada ensayo. Figura 1. Esquema del equipo utilizado. A: compresor de aire, B: resistencia eléctrica, C: entrada de aire seco, D: salida de aire húmedo y caliente, E: entrada de aire húmedo y caliente, F: fermentador de bandejas, G: deflectores, H: salida de gases Las condiciones del proceso fermentativo fueron: 3 960 g de aserrín de eucalipto, 72 % de humedad del aserrín, 30 ºC de temperatura, y flujo de aire de 0.19 L/s. 2.6 Preparación del inóculo para el fermentador de bandejas Los inóculos empleados en el proceso de fermentación a escala piloto, se prepararon de igual manera que los frascos empleados a escala de laboratorio, según el procedimiento descrito en el acápite 2. 3. Todos los inóculos se prepararon el mismo día, por lo tanto, los inóculos tuvieron distintas edades dentro de la cual se encuentran un periodo de incubación y además un periodo de refrigeración. La duración de estos se puede apreciar en la Fig. 2. A los inóculos que no tuvieron periodo de refrigeración se les denominó inóculos frescos y a los que sí tuvieron un periodo de refrigeración, inóculos refrigerados. Los inóculos frescos se utilizaron para las corridas 1 y 2, y los inóculos refrigerados se utilizaron para las corridas 3 y 4. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 3 Obtención de extractos enzimáticos con actividad ligninolítica en un fermentador de bandejas con el hongo Phanerochaete chrysosporium _______________________________________________________________________________________________________________________________ marca Julabo SW22, a 30 ºC, por 60 min. Se leyó la absorbancia a 310 nm (ε = 9300 M-1cm-1s). La actividad manganeso peroxidásica fue medida, espectrofotométricamente, de acuerdo con Lopes et al. [13]. Figura 2. Periodos de incubación y de refrigeración de los inóculos 2.7 Preparación del sustrato e inoculación en el fermentador de bandejas Se esterilizó aserrín de eucalipto en costales cerrados con vapor de agua a 121 °C durante 30 min. Una vez fríos, se extendió su contenido sobre bandejas metálicas desinfectadas y se colocaron 600 g del aserrín estéril sobre cada una de las seis bandejas del fermentador, procurando un espesor homogéneo de la cama, y se las introdujo en el fermentador para aclimatación. Se vació el contenido de dos frascos con aserrín de maderas tropicales inoculado con el hongo Phanerochaete chrysosporium sobre una bandeja metálica desinfectada. Se homogenizó y dividió el contenido de cada frasco en tres partes iguales de aproximadamente 65 g, con cada una de las cuales se inoculó el aserrín de eucalipto de cada bandeja ya aclimatada. El peso total de aserrín húmedo e inoculado en las bandejas fue de 660 g. 2.8 Muestreo y obtención de extractos enzimático a escala piloto Se tomó una muestra de 10 g de sustrato, de cada una de las seis bandejas hasta tener un total de 60 g de sustrato fermentado. El muestreo se realizó diariamente desde el día de la siembra durante 5 días. Para asegurar que la muestra es representativa para todo el contenido del fermentador, en un mismo día se muestreó de los dos mismos puntos elegidos al azar, en todas las bandejas, y de diferentes puntos cada día. En un tubo de ensayo de 10 mL se colocó: 0.1 mL de buffer acetato de sodio 0.1 M (pH 4.5), 0.05 mL de una solución de MnSO4 2 mM, 0.20 mL de una solución de albúmina al 0.5 % y 0.05 mL de una solución de H2O2 2 mM en buffer de fosfato de sodio 0.2 M (pH 8.0). La mezcla se dejó reaccionar en un baño maría a 30 ºC por 10 min. Se detuvo la reacción con adición de 0.04 mL de una solución de NaOH 2 N. Para cada muestra se preparó un blanco sin la adición del peróxido de hidrógeno. De cada muestra y blanco, correspondiente, se leyó la absorbancia a 610 nm (ε = 22000 M-1cm-1). La actividad enzimática se reportó en Unidades Internacionales (UI). Según la Comisión de Enzimas de la Unión Internacional de. Bioquímica y Biología Molecular (UIBBM), una UI se define como “aquella cantidad de enzima que cataliza la transformación de 1 mmol de sustrato o la formación de 1 mmol de producto por minuto, bajo condiciones definidas” [15]. 3. RESULTADOS Y DISCUSIÓN 3.1 Determinación de las actividades enzimáticas de Lignino peroxidasa y manganeso peroxidasa a escala de laboratorio En la Fig. 3 se presentan los resultados de la actividad lignino peroxidásica en función del tiempo de fermentación, de los extractos que corresponden a las muestras tomadas desde el momento de la inoculación del sustrato hasta el cuarto día de fermentación. Las unidades de actividad enzimática (U) se presentan por 1000 g de sustrato seco (gss). La muestra compuesta se homogenizó, se partió en fracciones de 30 g cada una y se colocó dentro de Erlenmeyers de 250 mL de capacidad. Con cada fracción se obtuvo el extracto enzimático de la misma forma que a escala de laboratorio, para determinar la actividad enzimática. 2.9 Determinación de las actividades enzimáticas La actividad lignino peroxidásica fue medida espectrofotométricamente [20, 21, 22]. En un tubo de ensayo de 10 mL se colocaron 0.5 mL de extracto enzimático y se agregaron 0.015 mL de una solución de alcohol veratrílico al 20 % en volumen, 9.4 mL de una solución de tartrato de sodio 0.15 M (pH = 3.0), 0.1 mL de una solución de Tween 80 al 10 % en volumen y se inició la reacción al añadir 0.02 mL de una solución de peróxido de hidrógeno al 10 % en volumen. La mezcla se dejó reaccionar en un baño maría Figura 3. Actividades enzimáticas lignino peroxidásica de los extractos para las tres corridas en frascos La producción de ligninoperoxidasa presenta un máximo al segundo día de fermentación, con valores de actividad que van desde 13 a 17 U/1000 gss. La actividad enzimática desciende a partir de que alcanza el pico de producción, hasta alcanzar valores cercanos a cero hacia el tercero o cuarto día, para luego incrementar nuevamente a un valor tres o cuatro veces menor al alcanzado en el pico de producción. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 4 Almeida E.*; Espín N.* _______________________________________________________________________________________________________________________________ Al comparar el presente estudio con el realizado por Villa [22], la tendencia de producción de lignino peroxidasa es diferente. En el estudio de Villa la producción enzimática baja a partir del octavo día de fermentación, con un valor máximo de 0.17 U/1000 g de sustrato sólido, diez veces menor a la obtenida en el presente estudio. Esta diferencia puede deberse a los sustratos utilizados, en el trabajo de Villa se empleó aserrín de maderas tropicales como sustrato, mientras que en este trabajo se utilizó aserrín de eucalipto. primario, sino que también transcurre durante el desarrollo del hongo hasta, incluso, durante el metabolismo secundario. En la Fig. 4 se presentan los resultados de la actividad Manganeso peroxidásica en función del tiempo de fermentación, de los extractos que corresponden a las muestras tomadas desde el momento de la inoculación del sustrato hasta el cuarto día de fermentación. Es importante mencionar, que sustratos pobres en nitrógeno, como es el caso del aserrín de eucalipto, podría generar otro tipo de enzimas como celulasas, hemicelulasas y proteasas; enzimas ligninolíticas como lacasas, peroxidasas y oxidasas [18]. Según Datta [4], la generación de proteasas por parte del hongo, cuando hay carencia de nitrógeno, puede deberse al intento de liberar enzimas de las paredes celulares de la madera, las mismas que estarían en la capacidad de degradar los polímeros de las paredes para, a su vez, liberar nutrientes, como el nitrógeno y azúcares simples. 3.2 Determinación de las actividades enzimáticas de Lignino peroxidasa en el fermentador de bandejas En la Fig. 5 se presentan los resultados de la actividad enzimática lignino peroxidásica en función del tiempo de fermentación, obtenida para las cuatro corridas realizadas en el fermentador de bandejas. Figura 4. Actividades enzimáticas Manganeso peroxidásica de los extractos para las tres corridas en frascos La producción de actividad manganeso peroxidasa en frascos presenta tendencias diferentes entre sí. En la corrida 1, la producción llega a un pico alto, 3.29 U/1 000 gss, al primer día, a diferencia de las otras dos corridas en las que un máximo de producción se produce al segundo día con valores alrededor de 1.3 U/1 000 gss. Esta diferencia en los resultados obtenidos puede ser consecuencia de la heterogeneidad del sustrato. Del Pilar Castillo et al. [6], comprobaron que la presencia de un 1 % de glucosa en el sustrato aumenta un 30 % la actividad de MnP. La cantidad de carbohidratos simples libres en la madera de eucalipto es muy variable dentro de maderas de una misma especie [7], por lo que, el sustrato utilizado para la corrida pudo haber contenido una mayor concentración de azúcares simples, que motivaron la producción de MnP por parte del hongo. Al analizar el comportamiento de la generación de actividad enzimática manganeso peroxidasa y lignino peroxidasa se puede observar que la actividad enzimática en ambos casos cae a partir de que alcanza su valor máximo. Este comportamiento podría explicarse debido a que existe una producción continua de proteasas por parte del mismo hongo, lo que causaría una inactivación de las enzimas ligninolíticas. Según Cabaleiro et al. [2], las proteasas son producidas por el hongo Phanerochaete chrysosporium como parte de su metabolismo primario, y hay otros autores como Dass et al. [3] y Dosoretz et al. [8], que han demostrado que la producción de proteasas no solo se limita al metabolismo Figura 5. Actividades enzimáticas Lignino peroxidásica de los extractos para las cuatro corridas en el fermentador de bandejas Se puede observar dos tendencias diferentes entre los resultados obtenidos para las cuatro corridas, la tendencia de producción enzimática es similar entre las corridas 1 y 2, y la tendencia también es similar entre las corridas 3 y 4. Sin embargo, hay diferencia entre estos dos tipos de tendencias: en las corridas 3 y 4 la producción alcanza su máximo valor al día uno y luego desciende, más o menos a la misma velocidad con la que subió, hasta casi la mitad de la actividad alcanzada en el máximo, luego la actividad parece que tiende a estabilizarse en el valor alcanzado durante los tres días siguientes. Por otro lado, en las corridas 1 y 2, la producción enzimática se incrementa más lentamente, para alcanzar un máximo al segundo y tercer día, respectivamente para las corridas 1 y 2. Hacia el final de la fermentación, al día cuarto, según la tendencia con la que la producción de lignino peroxidasa llega hasta este punto, la producción enzimática parecería que se estabilizará, en días posteriores. Los valores finales son similares para las corridas 2, 3 y 4, los cuales van de 2.26 a 2.36 U/1 000 gss, estos valores son alrededor de 1 U/1 000 gss mayor que el valor alcanzado al final de la Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 5 Obtención de extractos enzimáticos con actividad ligninolítica en un fermentador de bandejas con el hongo Phanerochaete chrysosporium _______________________________________________________________________________________________________________________________ corrida 1, y a su vez 0.5 U/1 000 gss que el pico de producción alcanzado en la misma corrida 1. Esto lleva a pensar que los tiempos de incubación y almacenamiento en refrigeración de los inóculos influenciarían en el proceso fermentativo y en la generación de actividad enzimática. En la Fig. 6, se presentan las curvas de actividad enzimática ligninoperoxidásica, para las corridas 1 y 2, realizadas con inóculos de 5 y 7 semanas de incubación, respectivamente. Figura 7. Actividades enzimáticas lignino peroxidásicas de los extractos para las corridas 3 y 4 en el fermentador de bandejas Figura 6. Actividades enzimáticas lignino peroxidásicas de los extractos para las corridas 1 y 2 en el fermentador de bandejas La producción de enzimas difiere ligeramente, alcanza un máximo en el segundo o tercer día de fermentación y luego disminuye. El máximo valor obtenido es de 3.24 U/1 000 gss, durante la corrida 2. El máximo alcanzado en la corrida 1 es de 1.83 U/1 000 gss, el cual representa, casi la mitad del valor pico de la corrida 2. Los inóculos utilizados para estas dos corridas tuvieron una diferencia de dos semanas en la incubación; para la corrida 1 se utilizó un inóculo con cinco semanas de incubación y para la corrida 2 se ocupó un inóculo con siete semanas de incubación. Esto podría influir sobre la producción enzimática, en la magnitud y en el día de aparecimiento del pico de producción, porque mientras más tiempo ha estado el hongo en incubación mayor es su biomasa y una gran cantidad de inóculo asegurará una buena producción de metabolitos secundarios en periodos cortos de tiempo [12]. En la Fig. 7, se grafican las curvas de producción enzimática para las corridas 3 y 4, realizadas con inóculos refrigerados por 5 y 7 semanas, respectivamente, luego de una incubación previa de 7 semanas. Se puede notar que las dos curvas son muy similares. Los valores máximos de producción enzimática de 3.79 U/1 000 gss y 3.46 U/1 000 gss, son alcanzados al primer día. Luego, al segundo día la producción cae con una velocidad parecida con la cual subió para luego estabilizarse dentro de los tres días posteriores, en valores de 2.23 U/1 000 gss y 2.36 U/1 000 gss respectivamente. Al analizar los resultados obtenidos en las cuatro corridas, se puede suponer que la producción enzimática podría estar influenciada por el tratamiento sufrido por los inóculos, el mismo que se muestra en la Tabla 1. Tabla 1. Producción pico de LiP según el tratamiento dado a los inóculos utilizados en el fermentador de bandejas Corrida Corrida Corrida Corrida Tratamiento 1 2 3 4 Incubación (semanas) 5 7 7 7 Refrigeración 0 0 5 7 (semanas) Aclimatación (h) 1 1 1 1 Producción pico de 1.83 3.24 3.79 3.46 LiP (U/1 000 gss) Día de aparición del 2 3 1 1 pico Parecería que cuando el inóculo se ha refrigerado, el hongo ha tenido más tiempo para adaptarse al aserrín y penetrar en su estructura a una mayor profundidad debido a que el modo hifático de crecimiento del hongo filamentoso incrementa la capacidad de penetración dentro del sustrato sólido [16]. Según el mismo autor, la penetración incrementa la accesibilidad a todos los nutrientes disponibles dentro de las partículas de sustrato y la estructura de la pared celular sujeta a la punta de la ramificación del micelio asegura una estructura firme; entonces, mientras más tiempo haya tenido el hongo para asegurar esta unión, se esperaría una mejor producción enzimática futura ya que se encuentra más arraigado al sustrato. Al observar la producción y el día de aparición del pico máximo de generación enzimática en función del tiempo de incubación y de refrigeración, parecería que la refrigeración afecta directamente en el día de aparecimiento del pico; mientras que la incubación parecería afectar en la concentración de la enzima. 3.3 Determinación de las actividades enzimáticas de Manganeso peroxidasa en el fermentador de bandejas En la Fig. 8 se presentan los resultados de la actividad enzimática Manganeso peroxidásica en función del tiempo de fermentación, obtenida para las cuatro corridas realizadas en el fermentador de bandejas. Se puede apreciar, al igual que en el caso de la producción de actividad lignino peroxidásica, que existen dos tendencias diferentes de producción enzimática. Las corridas 1 y 2 son similares y las corridas 3 y 4 también son similares entre sí. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 6 Almeida E.*; Espín N.* _______________________________________________________________________________________________________________________________ La tendencia de producción es similar a la ligninoperoxidasa. En las corridas 1 y 2, la producción enzimática sube con una pendiente más reducida, alcanza un máximo al segundo día, y luego baja a una velocidad similar a la observada en la subida; a partir del tercer día hasta el cuarto se observan tendencias diferentes. Figura 9. Actividades enzimáticas Manganeso peroxidásica de los extractos para las corridas 1 y 2 en el fermentador de bandejas En la Fig. 10 se grafican las curvas de producción enzimática para las corridas 3 y 4 realizadas con inóculos de 5 y 7 semanas de refrigeración, respectivamente, luego de una incubación previa de 7 semanas. Figura 8. Actividades enzimáticas Manganeso peroxidásica de los extractos para las cuatro corridas en el fermentador de bandejas Mientras que en las corridas 3 y 4, la producción enzimática se da más rápidamente y alcanza su máximo en el primer día, para luego descender notablemente. Se estima, a partir de los resultados obtenidos por Quintero et al. [14], autores que utilizan aserrín como sustrato, que a partir del cuarto día de fermentación, la actividad manganeso peroxidásica subiría hasta alcanzar otro pico de producción enzimática de la mitad del valor alcanzado durante los primeros días, para después, descender y no volver a subir. En la Fig. 9 se presentan curvas de producción enzimática para las corridas 1 y 2 realizadas con inóculos de 5 y 7 semanas de incubación, respectivamente. Los máximos alcanzados son de 0.72 y 0.76 U/1 000 gss. La actividad enzimática alcanza un máximo al segundo día, igual que para la lignino peroxidasa ya que las dos enzimas, la manganeso peroxidasa y la lignino peroxidasa, son producidas simultáneamente por el hongo [17]. Al final del proceso de fermentación, la actividad en las dos corridas toma direcciones diferentes. En la corrida 1 la tendencia de la producción va en subida, en la corrida 2, en cambio, se ve que la pendiente es negativa y pronunciada, esta diferencia en las tendencias se podría atribuir a que el sustrato utilizado no es completamente homogéneo y la producción de ligninasas se da según los requerimientos de nutrientes que tenga el hongo. Además que la inoculación del sustrato sólido no siempre será homogénea [12], por lo que se podrían esperar desviaciones entre las corridas. En la corrida 1, el hongo pudo haber encontrado dificultad para degradar la lignina durante los primeros días, por lo cual, para el cuarto día renueva su producción enzimática. Esto coincide con las observaciones, de Quintero et al.[14], autores que encuentran un segundo pico de producción de manganeso peroxidasa 5 días después de haber alcanzado el primero. Figura 10. Actividades enzimáticas Manganeso peroxidásica de los extractos para las corridas 3 y 4 en el fermentador de bandejas La producción de la manganeso peroxidasa obedece a la misma tendencia con la que se produce la LiP en las mismas corridas, pero la magnitud de las actividades enzimáticas difiere mucho; las producciones máximas para las corridas 3 y 4, fueron de 0.45 U/1 000 gss y de 0.86 U/1 000 gss, respectivamente. La actividad enzimática, cuando desciende desde el máximo lo hace con tendencias similares en los dos casos, posiblemente debido, también, a la posible producción de proteasas, hasta el día 2, donde, la velocidad del descenso en la corrida 3, empieza a disminuir para luego terminar con una tendencia que predice un leve aumento a partir del día 4. El valor de la actividad enzimática final de las dos corridas son similares, 0.36 y 0.45 U/1 000 gss, respectivamente. En la Tabla 2 se presenta la producción enzimática con respecto a los tiempos de incubación y refrigeración a las que se sometieron los inóculos. Al observar la producción y el día de aparecimiento del pico máximo de generación enzimática en función del tiempo de incubación y de refrigeración, parecería que, a diferencia de lo observado en la producción de lignino peroxidasa, la refrigeración afecta a la concentración de la enzima en el pico Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 7 Obtención de extractos enzimáticos con actividad ligninolítica en un fermentador de bandejas con el hongo Phanerochaete chrysosporium _______________________________________________________________________________________________________________________________ de producción, mientras que la incubación parecería influir en el día del aparecimiento del pico. Tabla 2. Producción pico de MnP según el tratamiento dado a los inóculos utilizados en el fermentador de bandejas Corrida Corrida Corrida Corrida Tratamiento 1 2 3 4 Incubación (semanas) 5 7 7 7 Refrigeración 0 0 5 7 (semanas) Aclimatación (h) 1 1 1 1 Producción pico de 0.72 0.76 0.45 0.86 LiP (U/1 000 gss) Día de aparición del 2 2 1 1 pico Se deduciría que la refrigeración parece anticipar el aparecimiento del pico de producción máxima, ya que cuando los inóculos no fueron refrigerados el pico aparece al día 2 de la fermentación, mientras que en las corridas 3 y 4, en las cuales se utilizaron inóculos refrigerados, el pico aparecen al día 1 de la fermentación. Como lo explica Rimbault [16], el tiempo que haya tenido el hongo para arraigarse dentro del sustrato utilizado para el inóculo facilitará la producción de enzimas durante la fermentación. Al analizar las Tablas 1 y 2 conjuntamente se puede suponer que cuando el inóculo pasa por un periodo de refrigeración el pico de producción aparece al primer día de la fermentación. Este hecho puede deberse a que la refrigeración genera estrés sobre el hongo, y este, al ser retirado de dicho estrés, produce las enzimas LiP y MnP, más rápidamente que cuando no se refrigera. de Bioprocesos por efectos de su almacenamiento y su tratamiento en la preparación del inóculo. Según Lonsane et al. [12], las cepas de los microorganismos son susceptibles a sufrir variaciones en su calidad dependiendo del número de generaciones que esta tenga. Mientras más generaciones se alcanzan, más número de variaciones podrá tener el microorganismo. Cualquier variante producida en una generación, multiplicará su efecto en las generaciones subsiguientes. Este fenómeno puede producir notables efectos sobre fermentaciones en las que el producto de interés no esté asociado con el crecimiento microbiano, es decir si ese trata de un metabolito secundario. Las condiciones en las que se encontraban los inóculos utilizados para cada escala de fermentación no fueron exactamente iguales. Mientras que para inocular los frascos se utilizó un hongo desarrollado en malta agar, para inocular el aserrín de las bandejas se utilizó un hongo desarrollado en madera. En el agar el hongo tendrá siempre mayor cantidad de nutrientes y de humedad que en la madera. Según Lonsane et al. [12], la esterilización del sustrato puede conducir a que este sufra cambios fisicoquímicos, degradación térmica de algunos nutrientes, formación de compuestos tóxicos y daño de los nutrientes. El aserrín utilizado para la fermentación en frascos fue esterilizado durante 15 min, mientras que el utilizado para el fermentador de bandejas se esterilizó a la misma temperatura durante 30 min. Esta diferencia de tiempo podría haber generado una mayor producción de compuestos tóxicos que inhibirían fuertemente la producción de enzimas por parte del hongo. 4. CONCLUSIONES 3.4 Comparación entre las escalas de producción Algunos diseños de bio-reactores han sido probados para una producción eficiente de enzimas fúngicas, algunos reactores han manejado sustratos sumergidos [17] y otros con células inmovilizadas [1]. Poco se ha estudiado sobre la aplicación de bio-reactores en estado sólido para producir enzimas ligninolíticas, y los diseños actuales son escasos. Cuando se desea emplear condiciones sólidas en la producción de enzimas, un estudio sobre el diseño de ingeniería del reactor y el escalado, es absolutamente necesario [17]. En la Tabla 3 se presentan las producciones enzimáticas máximas de cada enzima para cada escala de fermentación. Tabla 3. Producciones enzimáticas máximas a escala de laboratorio (frascos) y a escala piloto (fermentador de bandejas) Escala de producción MnP (U/1 000 gss) LiP (U/1 000 gss) Fermentación en frascos 3.296 17.01 Fermentación en 0.859 3.789 bandejas La producción de lignino peroxidasa es mayor que la de manganeso peroxidasa. Resultados diferentes a los presentados por Kerem et al. [11] y Quintero et al. [14], en donde la producción de Manganeso peroxidasa resulta ser mayor que la ligninoperoxidasa. Esto puede ser el resultado de utilizar una cepa diferente del mismo hongo, o a las variaciones que pudo haber sufrido la cepa en el laboratorio La actividad enzimática máxima de lignino peroxidasa fue de 17.01 U/1 000 gss, en frascos, (a escala de laboratorio), y a escala piloto en el fermentador de bandejas fue de 3.79 U/1 000 gss. La producción enzimática máxima de manganeso peroxidasa en frascos, a escala de laboratorio, fue de 3.296 U/1 000 gss y a escala piloto en el fermentador de bandejas fue de 0.859 U/1 000 gss. La producción de las enzimas ligninolíticas lignino peroxidasa y manganeso peroxidasa, por parte del hongo Phanerochaete chrysosporium, en frascos, a escala de laboratorio, es de 4 a 5 veces mayor, que en el fermentador de bandejas. La tendencia en las producciones enzimáticas de ligninoperoxidasa y manganesoperoxidasa, en las dos escalas de producción analizadas, se comportan de manera similar: inmediatamente después de la inoculación esta sube hasta alcanzar un valor máximo al primer o segundo día y luego desciende a un valor de aproximadamente a mitad del valor máximo hacia el cuarto día. Se puede considerar que en la producción de enzimas ligninolíticas con el hongo Phanerochaete chrysosporium Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 8 Almeida E.*; Espín N.* _______________________________________________________________________________________________________________________________ sobre aserrín de eucalipto, alcanza un máximo de producción un día antes, cuando se utilizan inóculos sometidos a un periodo de refrigeración de 5 a 7 semanas. 5. RECOMENDACIONES Analizar la cinética de producción enzimática de otras enzimas producidas por el hongo Phanerochaete chrysosporium como la lacasa, CMCasa, CBH y BGL, bajo las mismas condiciones de producción con las que se trabajó en el presente estudio dentro del fermentador de bandejas. Emplear mezclas de desechos lignocelulósicos en los que se induzca una mayor producción enzimática por parte del hongo Phanerochaete chrysosporium bajo fermentación en medio sólido en un fermentador de bandejas utilizando las mismas condiciones de cultivo descritas en el presente estudio. Analizar la influencia de la temperatura en la cinética de producción de enzimas Realizar un estudio acerca de la influencia de la edad del inóculo y pretratamientos del mismo sobre la producción enzimática del Phanerochaete chrysosporium bajo fermentación en sustrato sólido. REFERENCIAS [1] [2] [3] [4] [5] [6] [7] [8] [9] M. Asgher, M. Asad, y R. Legge, (2006), “Enhanced Lignin Peroxidase Synthesis by Phaneorchaete chrysosporium in Solid State Bioprocessing of Lignocellulosic Substrate”, World Journal of Microbiology & Biotechnology, Vol. 22, p. 449. D. Cabaleiro, S. Rodríguez, A. Sanromán, A., y M. Longo, (2001), “Characterization of Deactivating Agents and Their Influence on the Stability of Manganese-dependent Peroxidase from Phanerochaete chrysosporium”, Journal of Chemical Technology and Biotechnology, Vol. 76, p. 867. S. Dass, C. Dosoretz, C. Reddy, y H. Grethlein, (1995), “Extracellular Proteases Produced by the Wood-degrading Fungus Phanerochaete chrysosporium Under Ligninolytic and Non-ligninolytic Conditions”, Archives of Microbiology, 163, p. 254 A. Datta, (1991), “Purification and Characterization of a Novel Protease from Solid Substrate Cultures of Phanerochaete chrysosporium”, The Journal of Biological Chemistry, Vol. 267, p. 728. G. Dávila y R. Vázquez-Duhalt, (2006), “Enzimas Ligninolíticas Fúngicas para Fines Ambientales”, Mensaje Bioquímico, Vol. 30, p. 29. M. Del Pilar Castillo, P. Ander, y J. Stenström, (1997), “Lignin and Manganese Peroxidase Activity in Extracts from Straw Solid Substrate Fermentations”, Biotechnology Techniques, Vol. 11, p. 701. J. Del Río, A. Gutiérrez, M. Hernando, P. Landín, J. Romero, y A. Martínez, (2005), “Determining the influence of eucalypt lignin composition in paper pulp yield using Py-GC/MS”, J. Anal. Appl. Pyrolysis, 74, p. 110. C. Dosoretz, H. Chen, y H. Grethlein, (1990), “Effect of Environmental Conditions on Extracellular Protease Activity in Ligninolytic Cultures of Phanerochaete chrysosporium”, Applied and Environmental Microbiology, 56, p. 395. N. Duran, y B. Esposito, (2000), “Potential Applications of Oxidative Enzymes and Phenoloxidase-like Compounds in Wastewater and Soil [11] Z. Kerem, D. Friesem, y Y. Hadar, (1992), “Lignocellulose Degradation during Solid-State Fermentation: Pleurotus ostreatus versus Phanerochaete chrysosporium”, Applied and Environmental Microbiology, Vol. 58, p. 1121. [12] B. Lonsane, G. Saucedo-Castaneda, M. Rimbault, S. Roussos, G. Viniegra- Ganozalez, N. Ghildyal, M. Ramakrishna, y M. Krishnaiah, (1992), “Scale up Strategies for Solid State Fermentation”, Process Biochemistry, 27, p. 259. [13] P. Lopes, M. Teixeira, A. Nunes y L. Durrant (2009), Purification and partial characterization of manganese peroxidase from Bacillus pumilus and Paenibacillus sp, Brazilian Journal of Microbiology, 40, pp. 818826. [14] J. Quintero, G. Feijoo, y J. Lema, (2006), “Producción de Enzimas Ligninolíticas con Hongos Basidiomicetos Cultivas Sobre Materiales Lignocelulósicos”, Vitae, Revista de la Facultad de Química Farmacéutica, Vol. 13, p. 61. [15] E. Racker, (1958), International Unit of Enzyme Activity, National Academy of Sciences-National Research Council, Science, 128, pp. 1920. [16] M. Rimbault, (1998), “General and Microbiological Aspects of Solid Substrate Fermentation”, Electronic Journal of Microbiology, [Online]Vol. 1, Available in www.ejb.org. [17] I. Rivela, S. Rodríguez-Couto, y A. Sanromán, (2000), “Extracellular Ligninolytic Enzyme Production by Phaneorchaete chrysosporium in a New Solid-state Bioreactor”, Biotechnology Letters, Vol. 22, p. 1443. [18] F. Ruíz-Dueñas, y Á. Martínez, (2009), “Microbial degradation of lignin: how a bulky recalcitrant polymer is efficiently recycled in nature and how we can take advantage of this”, Microbial Biotechnology, Vol. 2, p. 164. [19] D. Singh, y S. Chen, (2008), “White-rot Fungus Phanerochaete Chrysosporium: Conditions for the Production of Lignin-degrading Enzymes”, Applications of Microbiology in Biotechnology, Vol. 81, p. 399. [20] M. Tien, y K. Kirk, (1983), “Lignin-degrading Enzyme from Phanerochaete chrysosporium: Purification, Characterization, and Catalytic Properties of a Unique H202-requiring Oxygenase”, Proc. Natl. Acad. Sci., Vol. 81, p. 2280. [21] M. Tien, y K. Kirk, (1988), “Lignin Peroxidase of Phanerochaete chrysosporium”, Methods in Enzymology, Vol. 161, p. 238. [22] Y. Villa, “Determinación de la cinética de crecimiento del Hongo Phanerochaete chrysosporium en residuos lignocelulósicos y determinación de la actividad ligninoperoxidásica en el sustrato a escala de laboratorio”, Tesis Previa a la Obtención del título de Ingeniero Químico, EPN, Quito, Ecuador, 2007. Treatment: A Review”, Appl. Catal. B. Eznym., 28, p.83 . [10] R. Howard, E. Abotsi, E. Jansen van Rensburg, y S. Howsrd, (2003), “Lignocellulose biotechnology: issues of bioconversion and enzyme production”, African Journal of Biotechnology, Vol. 2, p. 602. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Fibra dietaria en subproductos de mango, maracuyá, guayaba y palmito _________________________________________________________________________________________________________________________ Fibra dietaria en subproductos de mango, maracuyá, guayaba y palmito Cruz A.*; Guamán M.*; Castillo M.*; Glorio P.**; Martínez R.* *Universidad Técnica Particular de Loja, Sección de Ciencias y Tecnología de Alimentos, Loja, Ecuador e-mail: [email protected]; [email protected]; [email protected]; [email protected] ** Universidad Nacional Agraria La Molina, Lima, Perú e-mail: [email protected] Resumen: Los subproductos del procesamiento de alimentos vegetales son fuentes importantes de fibra dietaria y especialmente de la fracción insoluble. El objetivo de este estudio fue determinar la fibra dietaria total, sus fracciones y los componentes mayoritarios de la fracción insoluble en los subproductos de mango (Mangifera indica L, var. Tommy Atkins y Haden), maracuyá (Passiflora edulis, var. Flovicarpa), guayaba (Psidium guajava, var. Red) y palmito (Chamaerops humilis, var. Bactrisgasipaes). La fibra dietaria total y sus fracciones se determinaron por el método enzimático gravimétrico al igual que la obtención de la fracción purificada libre de pectina. La determinación de hemicelulosa A y B se realizó por acidificación ligera y precipitación con etanol respectivamente; la celulosa fue extraída de la fracción de lignocelulosa por tratamiento con permanganato de potasio y la lignina por hidrólisis ácida. El subproducto de guayaba presentó los mayores contenidos de fibra dietaria total e insoluble (74.00±2.00 y 69.32±0.96 g/100 g materia seca respectivamente), mientras que la fibra dietaria soluble fue mayor en el de mango (15.10±2.20 g/100 g materia seca). El contenido de hemicelulosa estuvo comprendido entre 2.35±0.26 y 25.92±0.53 g/100 g de materia seca; la hemicelulosa B estuvo entre 2.91±1.05 y 5.89±0.25 g/100 g de materia seca. La celulosa fue el componente mayoritario en los subproductos de guayaba (33.54±1.00 g/100 g de materia seca), mientras que la lignina fue el compuesto mayoritario en la semilla y pulpa de maracuyá (28.17±2.07 g/100 g de materia seca). Todos los subproductos estudiados a excepción de la pulpa de maracuyá y mango se mostraron como buena fuente de fibra dietaria insoluble con un contenido superior a 40 g/100 g. Palabras clave: Subproductos agroindustriales, fibra dietaria, hemicelulosa, celulosa, lignina Abstract: Byproducts of food processing industries are major sources of dietary fiber, especially of the insoluble fraction. The aim of this study was to determine total dietary fiber and it is fractions, and the principal components in insoluble fraction of dietary fiber from the agroindustrial byproducts of guava (Psidium guajava, var. Red), mango (Mangifera indica L, var. Tommy Atkins y Haden), passion fruit (Passiflora edulis, var. Flovicarpa) and heart of palm (Chamaerops humilis, var. Bactrisgasipaes). A gravimetric enzymatic method was used to obtain the total dietary fiber (soluble and insoluble fraction), its fractions and purified insoluble fiber free of pectin. Hemicellulose A and B were determined by soft acidification and hemicellulose B by precipitation with ethanol; cellulose was extracted from lignocellulose fraction by treatment with potassium permanganate and lignin by acid hydrolysis. Total and insoluble dietary fiber were higher in guava (74.00±2.00 and 69.32±0.96 g/100 g dry matter, respectively). The soluble fiber was majority in the byproducts of mango (15.10±2.20 g/100 g dry matter). The hemicellulose A content was between 2.35±0.26 and 25.92±0.53 g/100 g dry matter; hemicellulose B was between 2.91±1.05 and 5.89±0.25 g/100 g of dry matter. Cellulose was the majority component in the byproducts of guava (33.54±1.00 g/100 g dry matter), while lignin was the main component in seeds and pulp of passion fruit (28.17±2.07 g/100 g dry matter). All byproducts analyzed, except passion fruit pulp and mango pulp, show to be good source of insoluble dietary fiber was higher than 40 g /100 g. dry matter. Keywords: Agroindustrial byproducts, insoluble dietary fiber, hemicellulose, cellulose, lignin 1. INTRODUCCIÓN La industria del procesamiento de frutas y vegetales genera una gran cantidad de subproductos infrautilizados tales como cáscara, frutos, pulpa, semillas, tallos entre otros [26]; que se corresponde con todo aquello que no se consume o no es vendible habitualmente, que está estropeado, que no es apto para el consumo humano sin su ulterior procesamiento o que fue cosechado circunstancialmente; pero que puede ser utilizado después de un tratamiento [1, 25, 42]. Los subproductos de maracuyá representan más del 75 % de la Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 9 10 Cruz A.*; Guamán M.*; Castillo M.*; Glorio P.**; Martínez R.* _______________________________________________________________________________________________________________________________ materia prima y los de mango entre el 40 y 60 % [46]; después de la elaboración del jugo de guayaba, el 53 % de la fruta son subproductos [13] y en el proceso de elaboración de conservas de palmito se genera un 85 % de subproductos [47]. Estos constituyen una importante fuente de azúcares, minerales, ácidos orgánicos, fibra dietaria y compuestos fenólicos [12]. Varios de ellos contienen más fibra dietaria que su respectiva porción comestible [17]. En los últimos años la industria alimentaria ha incrementado el uso de componentes naturales como la fibra dietaria y compuestos bioactivos [6], motivando la búsqueda de nuevas fuentes [30], que puedan sustituir a los cereales, recurso de fibra usualmente utilizado [2]. El alto valor nutricional, la capacidad antioxidante y el bajo aporte calórico de los subproductos de frutas tropicales ha generado un creciente interés por su estudio y uso en la elaboración de alimentos, aditivos o suplementos alimentarios; su reutilización puede ser económicamente atractiva y beneficiosa para el ambiente [2, 24]. Los efectos funcionales y tecnológicos de la fibra dietaria dependen del contenido de las fracciones soluble e insoluble, de la relación entre ellas y de la cantidad de los componentes individuales de cada fracción [35], estos parámetros juegan un rol en relación con los beneficios para la salud y su conocimiento es importante para sugerir sus posibles aplicaciones como ingrediente de alimentos funcionales y/o productos nutracéuticos [7, 25]. Se considera como fibra dietaria insoluble (FDI) a una compleja matriz de componentes químicos tales como: celulosa, hemicelulosa y lignina [29]. Las fibras insolubles son escasamente fermentadas, tienen un efecto laxante y regulador intestinal [44], son capaces de retener agua en su matriz estructural y formar mezclas de baja viscosidad; esto produce un aumento de la masa fecal que acelera el tránsito intestinal, influyendo en la prevención y en el tratamiento de la constipación crónica. Por otra parte también contribuyen a disminuir la concentración y el tiempo de contacto de potenciales carcinogénicos con la mucosa del colon [2, 15]; además, la de algunas frutas y verduras podrían promover una disminución significativa en la concentración de colesterol en la sangre [9]. Se considera a la lignina como un potente captador de radicales libres e inhibidor del crecimiento de colonias bacterianas, se cree que actúa como una barrera física para los microbios, limitando la capacidad de fermentación de estos [10]; mientras que la hemicelulosa aumenta el volumen y peso de las heces, incrementa la excreción de ácidos biliares y junto a la pectina (fracción soluble) presenta una notable capacidad de ligar o atrapar metales pesados, también la celulosa y la lignina son capaces de captar metales pesados aunque en menor grado [10, 35, 40, 41]. La fibra dietaria soluble (FDS) constituida por pectinas, gomas, oligosacáridos, β-glucanos y mucílagos [11, 49], es altamente fermentable; está asociada con el desarrollo de la flora intestinal y el fortalecimiento del sistema inmune por la producción de metabolitos [45]; sirve de sustrato para los microorganismos benéficos, por lo tanto actúa como un prebiótico y mejora la salud del huésped, refuerza la protección de la mucosa intestinal disminuyendo el riesgo de enfermedades grastrointestinales [10, 43]; posee una buena capacidad de hidratación e hinchamiento, forma soluciones viscosas, absorbe y retiene sustancias como la glucosa, minerales y moléculas no polares promoviendo la disminución del colesterol y glucosa en la sangre [2, 44]. La adición de fibra dietaria a los alimentos no solamente es importante por su valor nutricional, sino también por sus propiedades tecno – funcionales [21]. La fracción soluble favorece la estabilidad de espumas y emulsiones, actúa como agente gelificante y espesante; mientras que la FDI mejora la textura y densidad, incrementa el volumen y estabiliza la matriz alimentaria; la FDS podría tener un potencial uso en la encapsulación de componentes bioactivos [2, 39]. El objetivo del presente trabajo fue determinar la fibra dietaria total, sus fracciones y los componentes individuales de la fracción insoluble: celulosa, hemicelulosa y lignina de los subproductos de mango, maracuyá, guayaba y palmito. Estos subproductos podrían ser utilizados por la industria alimentaria como fuentes alternativas de fibra dietaria para la elaboración de alimentos. 2. MATERIALES Y MÉTODOS 2.1 Materia prima Fueron utilizados subproductos generados en la elaboración de pulpas o concentrados de mango (Mangifera indica L. var. Tommy Atkins y Haden) y guayaba (Psidium guajava, var. Red), jugos o concentrados de maracuyá (Passiflora edulis, var. Flovicarpa) y conservas de palmito (Chamaerops humilis, var. Bactrisgasipaes); procesados en industrias ecuatorianas. 2.2 Obtención y preparación de muestras El muestreo de cada subproducto se realizó en diferentes momentos del proceso de producción en el cual se procesan materias primas de diferentes proveedores de las industrias. Los subproductos de maracuyá y guayaba fueron principalmente cáscara, pulpa y semilla; los de palmito correspondieron a la capa 2 y 3; y los de mango a la cáscara y pulpa. Los subproductos fueron sometidos a un proceso de deshidratación a temperatura inferior a 60ºC para evitar cambios en sus propiedades y contenidos de los compuestos bioactivos. Se molieron hasta un tamaño de partícula comprendido entre 125 y 250 μm. 2.3 Fibra dietaria La fibra dietaria total y sus fracciones soluble e insoluble se determinaron por el método enzimático – gravimétrico de la Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Fibra dietaria en subproductos de mango, maracuyá, guayaba y palmito _________________________________________________________________________________________________________________________ AOAC 991.43 y AACC 32-07, basados en el método de Lee et al. (1994) y Prosky et al. (1994). 2.4 Fraccionamiento de la fibra dietaria insoluble La extracción y fraccionamiento de la fibra dietaria insoluble se realizó por el método de Monte & Maga (1980) con adaptaciones de Claye et al. (1996) que está basado en el procedimiento de Southgate (1976). 2.4.1 Eliminación de hidratos de carbono complejos y proteínas solubles A las muestras previamente desengrasadas con hexano durante 24 horas utilizando extracción soxhlet [4], se les realizó una maceración dinámica con agua destilada (1:20 w/v, pH 7.0-7.5) y el residuo previamente secado (60 °C, 36 horas) se mezcló con una solución de EDTA 0.01 M a 90 °C (25 mL/g residuo) con la finalidad de eliminar hidratos de carbono complejos del residuo. El producto obtenido se denomina fibra insoluble no purificada (FINP). 2.4.2 Remoción de almidón, proteína y sustancias pécticas insolubles La eliminación de almidón de la FINP se realizó por tratamiento enzimático con amiloglucosidasa (10 %.v/v) Se usó tripsina (2.5 % v/v) para la degradación de las proteínas. Por el efecto disolvente del oxalato de amonio (0.5 % p/v) las sustancias pécticas fueron hidrolizadas en sus respectivas sales. El producto obtenido se denomina fibra insoluble despectinada (FID) 2.4.3 Extracción de Hemicelulosa A y B, celulosa y lignina La FID fue tratada con hidróxido de potasio (5 % p/v) en atmósfera de nitrógeno para extraer la lignocelulosa (residuo). El sobrenadante fue ligeramente acidificado con ácido acético (50 % v/v) para precipitar la hemicelulosa A; mientras que la hemicelulosa B fue precipitada con etanol (95 % v/v). De la lignocelusosa se obtuvieron dos fracciones: la celulosa mediante desmineralización completa y extracción con solución saturada combinada de permanganato de potasio (KMnO4+ buffer lignina 2:1) y la lignina por hidrólisis ácida con H2S04 (72 % v/v), para destruir la fracción de celulosa y luego determinarla por pérdida de peso. 2.5 Análisis Estadístico Los resultados fueron expresados como media ± desviación estándar de tres repeticiones de cada fracción del subproducto, los cuales fueron analizados estadísticamente mediante un ANOVA de un solo factor y la prueba de comparación múltiple de Tukey (p<0.05) para establecer donde se encontraban las diferencias entre los niveles del factor principal (recurso de fibra). El análisis de resultados se llevó a cabo usando el paquete estadístico Minitab® Statistical Software, versión 16. 11 3. RESULTADOS Y DISCUCIÓN El contenido de fibra dietaria total (FDT), fibra dietaria insoluble (FDI), fibra dietaria soluble (FDS) y la relación entre las fracciones FDI y FDS se muestran en la Tabla 1. Respecto a la FDT los subproductos de guayaba, mango y maracuyá mostraron diferencia (p<0.05), con un rango de 23.52 a 74.95 g/100 g MS para pulpa de maracuyá y subproductos de guayaba respectivamente. Todos los subproductos presentaron valores menores a los reportados por Martínez et al. (2012a) para subproductos de piña (75.8 g/100 g) y mezcla de subproductos de maracuyá (81.5 g/100 g) y a los de pulpa de limón fino (81.7 g/100 g) reportados por Marín et al. (2007). La cáscara de maracuyá presentó valores similares a los de cáscara y cascarilla de cacao (51.88 – 56.70 g/100) [27]. A excepción de la pulpa de maracuyá, en todos los subproductos la FDT fue mayor a la de pulpa de melocotón (36.10 g/100 g) [18] y pulpa de café [34]. Según el criterio de Fuentes-Alventosa et al. (2009), los subproductos de guayaba pueden ser considerados como fuente alta de FDT; los de palmito, cáscara, pulpa y semilla de maracuyá como fuentes con contenido medio de FDT, mientras que el mango y la pulpa de maracuyá como recursos con bajo contenido de FDT. La capa 2 y 3 de palmito fueron similares entre sí, tanto para FDT como para sus fracciones. Los subproductos de guayaba, pulpa de maracuyá y capa 2 de palmito mostraron mayor diferencia entre la FDI y FDS. La elevada proporción de fibra dietaria insoluble en las muestras se relaciona con la considerable cantidad de celulosa, hemicelulosa y lignina presentes; esto puede ser considerado una ventaja para su uso en la industria alimentaria, que puede enriquecer productos como jugos y bebidas de baja viscosidad sin modificar notablemente esta característica. Por su capacidad de retener agua, la FDI es recomendada para mejorar el volumen y mantener la frescura en productos de panificación [49], disminuir la sinéresis en yogurt. En el caso de los subproductos de frutas, aportarían además color y sabor para modificar las características sensoriales de los alimentos y disminuir el uso de aditivos químicos como saborizantes y esencias; especialmente en alimentos elaborados a base de quinua, amaranto, chocho, cebada y otros alimentos ancestrales; estos son ideales para incrementar el contenido de componentes insolubles de la fibra dietaria en alimentos asociados con efectos benéficos para la salud intestinal, excreción de ácidos biliares y disminución de la concentración de colesterol en la sangre, como son las granolas, barras para desayuno, galletas integrales y concentrados de fibra [9, 26, 44]. El contenido de hemicelulosa A fue diferente entre los subproductos (p<0.05) y varió de 2.35 a 25.92 g/100 g MS; en la capa 3 de palmito fue significativamente mayor a los demás, como se visualiza en la Tabla 2. Los valores de hemicelulosa A de las capas de palmito fueron mayores a los reportados por Claye et al. (1996) en salvado de trigo (15.18 g/100 g), salvado de arroz (3.91 g/100 g), fibra de Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 12 Cruz A.*; Guamán M.*; Castillo M.*; Glorio P.**; Martínez R.* _______________________________________________________________________________________________________________________________ tomate (15.35 g/100g) y al obtenido por Monte & Maga (1980) en frijoles pintos (3.19 g/100 g); mientras que la cantidad de hemicelulosa A de los subproductos de guayaba fue similar a la determinada en fibra de avena (16.00 g/100 g) y menor a la de fibra de manzana (17.78 g/100 g) [7]. Las capas 2 y 3 de palmito no mostraron diferencia significativa en el contenido de hemicelulosa B, de igual manera los subproductos de mango y guayaba. Los valores de hemicelulosa B determinados en este estudio fueron inferiores a los reportados por Claye et al. (1996) en salvado de trigo (8.67 g/100 g), fibra de avena (12.77 g/100 g), salvado de arroz (12.34 g/100 g), fibra de manzana (6.26 g/100 g) y fibra de tomate (8.70 g/100 g) y mayores al obtenido por Monte & Maga (1980) en frijoles pintos (0.66 g/100 g). El contenido de hemicelulosa A en relación al de hemicelulosa B fue más alto, a excepción de la cáscara de maracuyá; la misma que contiene aproximadamente 90 % de xiloglucanos que son los componentes mayoritarios de hemicelulosa B [16]. De los resultados obtenidos se evidenció que la hemicelulosa se encuentra presente principalmente en las semillas y tallos; por lo que una aplicación importante de los subproductos de palmito como fuente de hemicelulosa, sería su utilización para enriquecer en fibra productos libres de gluten, como harinas, pastas y otros; la alta capacidad de hinchamiento de la fracción insoluble y la afinidad de la hemicelulosa por el agua [19], facilitaría la cocción de las pastas y la resistencia de la masa a la deformación. Así mismo, estos subproductos podrían mezclarse con la harina de trigo para aumentar el volumen del pan y mejorar la estructura de la miga, por la habilidad de las hemicelulosas para liberar arabinoxilanos solubles en agua que participan conjuntamente con el gluten de las proteínas en la red de compuestos de superficie hidratada [19]. Los subproductos de palmito por su peculiar característica de tener color, olor y sabor neutro, pueden ser usados sin afectar las características sensoriales e indistintamente en productos salados o dulces. Pueden ser ideales para la elaboración de la masa de alimentos tradicionales fritos como empanadas de harina, plátano, yuca y morocho; por su baja capacidad de retención de grasa (1.13-1.15 g/g) [28] proporcionará a los productos una sensación menos grasosa. La celulosa (ver Tabla 2) presentó valores entre 2.13 en la pulpa de maracuyá y 33.54 g/100 g MS en los subproductos de guayaba. Todos los subproductos a excepción de los de guayaba exhibieron valores de celulosa inferiores a los subproductos de frutilla (25.86 g/100 g) y grosella negra (21.0 g/100 g), pero similares a los presentes en subproductos de col morada (15.21 g/100 g), manzana (16.10 g/100 g), zanahoria (12.65 g/100 g) [35], cáscara de naranja (13.61 g/100 g) y cáscara de limón (12.72 g/100 g) [50]. La celulosa presente en los subproductos de guayaba fue similar a la de subproductos de cereza de aronia (34.56 g/100 g) [35]. Comparando con sus similares, los subproductos de maracuyá, guayaba y palmito presentaron valores de celulosa inferiores a los de mezcla de subproductos de guayaba (37.20 g/100 g), mezcla de subproductos de maracuyá (39.20 g/100 g) (Lousada et al., 2005), pared celular de la cáscara de maracuyá (33.60 g/100 g) [51] y residuos de la cosecha de palmito (29.03 g/100 g) [47]. El contenido de celulosa en los subproductos de guayaba fue superior al de fuentes tradicionales de fibra dietaria, como salvado de trigo (17.45 g/100 g), salvado de avena (19.98 g/100 g) y salvado de maíz (22.23 g/100 g) [7]; por lo que podrían ser usados en sustitución de estas en la elaboración de cereales para desayuno, galletas, mezclas para papillas, al aportar sabor y dulzor, y disminuir de esta forma el nivel de azúcar añadida en estos productos. Por esta misma razón, se podría disminuir el aporte calórico de postres smothies y snacks. El contenido de lignina entre los subproductos analizados presentó diferencia estadística (p<0.05) a excepción de las dos capas de palmito. En todos los subproductos, a excepción de mango, la lignina cuantificada fue superior a la de alimentos tales como: col (0.83 g/100 g), zanahoria (1.01 g/100 g), lechuga (2.02 g/100 g), papa (0.90 g/100 g), tomate (1.69 g/100 g) y salvado de maíz (2.32 g/100 g), reportados por Anderson & Bridges (1988); y salvado de trigo (2.82 g/100 g) (Claye et al., 1996). Los subproductos de palmito mostraron valores de lignina inferiores a los reportados por Fuentes-Alventosa et al. (2009) para subproductos de espárragos (15.01 g/100 g). La lignina en la semilla y pulpa, y cáscara de maracuyá fue similar a la presente en subproductos de frutilla (20.84 g/100 g), cereza de aronia (22.68 g/100 g) y grosella negra (26.66 g/100 g) [36]. Los subproductos de guayaba mostraron menor contenido de lignina que el reportado por Lousada et al. (2005) para mezcla de subproductos de guayaba (18.59 g/100 g). En el caso de la maracuyá Lousada et al. (2005) reportan 9.50 g/100 g para mezcla de subproductos y Astuti et al. (2011) reportan 31.79 g/100 g para la cáscara. Los subproductos estudiados constituyen un potencial recurso de lignina, componente apreciado por sus efectos benéficos para la salud humana y muy poco consumido [10, 32]. Su uso como ingrediente en la elaboración de productos cárnicos funcionales, que a más de incrementar el rendimiento durante la cocción podría ser interesante para disminuir los niveles de nitrito residual [21], función derivada de su alta capacidad para captar radicales libres [10]. La principal ventaja de la fibra de estos subproductos comparada con las fibras provenientes de cereales es la presencia de compuestos bioactivos asociados a su matriz [21, 26]. En la guayaba se apreció una distribución de los componentes, mientas que en el mano la celulosa fue el compuesto predominante, Los subproductos derivados de la maracuyá presentaron diferencias entre sí en la cáscara y la pulpa más semilla, la ligninia fue el componente preponderante seguido por la celulosa en el primer caso y la hemicelulosa en la pulpa más semilla. La pulpa de maracuyá presentó los valores más bajos de celulosa, mientras que en el mango el contenido de lignina fue el más bajo. Las capas 2 y 3 de palmito mostraron mayor similitud entre los contenidos de los componentes de FDI. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Fibra dietaria en subproductos de mango, maracuyá, guayaba y palmito _________________________________________________________________________________________________________________________ La sumatoria de los componentes individuales de la fracción insoluble cuantificados en este estudio no fue igual al valor de FDI determinado por el método AOAC 991.43, pues este método cuantifica además otros compuestos tales como ceras, fenoles, taninos, productos de reacción de Maillard y cutinas que están unidas a la celulosa [8, 14, 31]. Subproductos El uso de estos subproductos constituye una oportunidad para incrementar la disponibilidad de alimentos procesados saludables sin incrementar los costos económicos y ambientales, añadir nuevos recursos para la producción de alimentos, crear un sistema alimentario más sostenible para garantizar la seguridad alimentaria y disminuir el hambre en la región [37]. Tabla 1. Contenido de fibra dietaria en subproductos FDT FDI FDS (g/100 g) (g/100 g) (g/100 g) 74.95 ± 1.94a 69.32 ± 0.96a 4.19 ± 1.42a FDI/FDS 18.37 ± 7.30a 2.19b 1.59 ± 0.13b ± 1.63c 4.62 ± 0.58c 1.00 ± 0.56d 21.27 ± 9.80d 3.58e 12.05 ± 1.59e 4.63 ± 0.64c ± 1.81e 4.37 ± 0.83a 13.20 ± 3.08a, c, ± 4.12e 5.96 ± 0.43a 9.63 ± 1.18a, b, Guayaba Piel, pulpa y semilla Mango Cáscara y pulpa 42.60 ± 2.22b 23.79 ± 1.36b 15.06 ± Maracuyá Cáscara 52.42 ± 1.16c 41.19 ± 2.75c 9.04 Pulpa 23.52 ± 1.00d 17.81 ± 1.83d Semilla y pulpa 60.05 ± 7.70e 55.22 ± Capa 2 63.10 ± 1.93e 55.99 Capa 3 64.50 ± 5.04e 57.16 Palmito 13 d c Los valores corresponden a media ± desviación estándar; n = 3. Letras iguales en columnas no hay diferencia significativa Tabla 2. Contenido de componentes de fibra dietaria insoluble Hemicelulosa A Hemicelulosa B Celulosa (g/100 g) (g/100 g) (g/100 g) Piel, pulpa y semilla 16.45 ± 0.92a 2.91 ± 1.05a 33.54 ± 1.00a Subproductos Guayaba Mango Cáscara y pulpa 5.67 ± 0.52b 3.32 ± 0.43a Lignina (g/100 g) 16.51 ± 0.58a 15.80 ± 1.06b 0.74 ± 0.08b 15.00 ± 2.46b 21.49 ± 0.21c b Maracuyá Cáscara 2.35 ± 0.26c 4.47 ± 0.66b c Pulpa 4.15 ± 0.34b 3.02 ± 0.09a 2.13 ± 0.57c 5.95 ± 0.86d c Palmito Semilla y pulpa 9.47 ± 0.81d 5.39 ± 0.45c 10.27 ± 0.82d 28.17 ± 2.07e Capa 2 22.05 ± 1.16e 5.89 ± 0.25c 16.36 ± 0.70b 13.01 ± 0.93f Capa 3 25.92 ± 0.53f 5.47 ± 0.23c 10.93 ± 0.47d 12.07 ± 0.96f Los valores corresponden a media ± desviación estándar; n = 3. Letras iguales en columnas no hay diferencia significativa 1 AGRADECIMIENTOS 4. CONCLUSIONES En todos los subproductos analizados la fibra dietaria está constituida mayoritariamente por la fracción insoluble. Los subproductos mostraron distintas peculiaridades en lo referente a la composición de la fracción insoluble. La celulosa fue el componente mayoritario en los subproductos de guayaba y mango, mientras que la lignina lo fue en la cáscara de maracuyá, pulpa y semilla de maracuyá; y la hemicelulosa en los subproductos de palmito Los autores quisieran agradecer a las empresas Grupo Fadesa y Exofrut (Ecuador) por la provisión de la materia prima. REFERENCIAS [1] Ajila, C. M., Bhat, S. G., & Prasada, U. J. S. (2007). Valuable components of raw and ripe peels from two Indian mango varieties. Food Chemistry, 102(4), 1006-1011. doi: http://dx.doi.org/10.1016/j.foodchem.2006.06.036 Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 14 Cruz A.*; Guamán M.*; Castillo M.*; Glorio P.**; Martínez R.* _______________________________________________________________________________________________________________________________ [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] Al-Sheraji, S. H., Ismail, A., Manap, M. Y., Mustafa, S., Yusof, R. M., & Hassan, F. A. (2011). Functional properties and characterization of dietary fiber from mangifera pajang kort. Fruit Pulp. Journal of Agricultural and Food Chemistry, 59 (8), 3980-3985. doi: 10.1021/jf103956g Anderson, J. W., & Bridges, S. R. (1988). Dietary fiber content of selected foods. The American Journal of Clinical Nutrition, 47(3), 440-447. AOAC (1997). Official Methods of Analysis of AOAC International (16th ed.). Washington, DC: Association of Official Analytical Chemists. Astuti, T., Warly, L., Jamarun, N., & Evitayani. (2011). The effect of incubation time and level of urea on dry matter, organic matter and crude protein digestibility of passion fruit (Passiflora edulis var. flavicarpa) hulls. Journal of the Indonesian Tropical Animal Agriculture, 36(3), 180-184. doi: http://ejournal.undip.ac.id/index.php/ jitaa/article/view/7496 Bensadón, S., Hervert-Hernández, D., Sáyago-Ayerdi, S., & Goñi, I. (2010). By-Products of Opuntia ficus-indica as a source of antioxidant dietary fiber. Plant Foods for Human Nutrition, 65(3), 210-216. doi: 10.1007/s11130-010-0176-2 Claye, S. S., Idouraine, A., & Weber, C. W. (1996). Extraction and fractionation of insoluble fiber from five fiber sources. Food Chemistry, 57(2), 305-310. doi: http://dx.doi.org/10.1016/03088146(95)00250-2 Champ, M., Langkilde, A.-M., Brouns, F., Kettlitz, B., & Le Bail, Y. (2003). Advances in dietary fibre characterisation. Definition of dietary fibre, physiological relevance, health benefits and analytical aspects. Nutrition Research Reviews, 16(1), 71-82. doi: 10.1079/NRR200254 Chau, C.-F., & Huang, Y.-L. (2005). Effects of the insoluble fiber derived from Passiflora edulis seed on plasma and hepatic lipids and fecal output. Molecular Nutrition & Food Research, 49(8), 786-790. doi: 10.1002/mnfr.200500060 Chawla, R., & Patil, G. R. (2010). Soluble dietary fiber. Comprehensive Reviews in Food Science and Food Safety, 9(2), 178196. doi: 10.1111/j.1541-4337.2009.00099.x Dhingra, D., Michael, M., Rajput, H., & Patil, R. T. (2012). Dietary fibre in foods: a review, Journal of Food Science and Technology, 49(3), 255–266. doi: 10.1007/s13197-011-0365-5 Djilas, S., Čanadanović-Brunet, J., & Ćetković, G. (2009). Byproducts of fruits processing as a source of phytochemicals. Chemical Industry & Chemical Engineering Quarterly, 15(4), 191-202. El-Deek, A., Hamdy, S., Attia, Y., & El-Shahat, A. (2009). Guava byproduct meal processed in various ways and fed in differing amounts as a component in laying hen diets. International Journal of Poultry Science, 9(8), 866-874. Elleuch, M., Bedigian, D., Roiseux, O., Besbes, S., Blecker, C., & Attia, H. (2011). Dietary fibre and fibre-rich by-products of food processing: Characterisation, technological functionality and commercial applications: A review. Food Chemistry, 124(2), 411-421. doi: 10.1016/j.foodchem.2010.06.077 Escudero, E., & Gonzáles, P. (2006). La fibra dietética. Nutrición Hospitalaria, 2(21), 61-72. Fuentes-Alventosa, J. M., Rodríguez-Gutiérrez, G., JaramilloCarmona, S., Espejo-Calvo, J. A., Rodríguez-Arcos, R., FernándezBolaños, J., & Jiménez-Araujo, A. (2009). Effect of extraction method on chemical composition and functional characteristics of high dietary fibre powders obtained from asparagus by-products. Food Chemistry, 113(2), 665-671. doi: http://dx.doi.org/10.1016/j.foodchem .2008.07.075 Goñi, I., & Hervert-Hernández, D. (2011). By-Products from plant foods are sources of dietary fibre and antioxidants. Phytochemicals Bioactivities and Impact on Health (pp. 95-116). Madrid: Universidad Complutense de Madrid. Grigelmo-Miguel, N., Gorinstein, S., & Martı́n-Belloso, O. (1999). Characterisation of peach dietary fibre concentrate as a food ingredient. Food Chemistry, 65(2), 175-181. doi: http://dx.doi.org/10.1016/S0308-8146(98)00190-3 Guillon, F., & Champ, M. (2000). Structural and physical properties of dietary fibres, and consequences of processing on human physiology. Food Research International, 33(3), 233-245. doi: http://dx.doi.org/10.1016/S0963-9969(00)00038-7 Lee, S., Prosky, L., & DeVries, J. (1994). Determination of total, soluble, and insoluble dietary fiber in foods - enzymatic - gravimetric [21] [22] [23] [24] [25] [26] [27] [28] [29] [30] [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] method, MES-TRIS buffer: Collaborative study. Association Official Analytical Chemistry. 75, 395-416. López, J. F., & Pérez-Alvarez, J. A. (2008). Overview of meat products as functional foods. Technological strategies for functional meat products development, 1-17. Lousada, J., Miranda, J., Rodríguez, N., Machado, C., & Braga, R. (2005). Consumo e digestibilidade de subprodutos do processamento de frutas em ovinos. Revista Brasileira de Zootecnia, 34(2), 659-669. Marín, F. R., Soler-Rivas, C., Benavente-García, O., Castillo, J., & Pérez-Alvarez, J. A. (2007). By-products from different citrus processes as a source of customized functional fibres. Food Chemistry, 100(2), 736-741. doi: http://dx.doi.org/10.1016/ j.foodchem.2005.04.040 Martín-Sánchez, A. M., Cherif, S., Ben-Abda, J., Barber-Vallés, X., Pérez-Álvarez, J. A., & Sayas-Barberá, E. (2014). Phytochemicals in date co-products and their antioxidant activity. Fodd Chemistry, 158, 513-520. doi: 10.1016/j.foodchem.2014.02.172 Martínez, R. (2013). Caracterización de la fracción insoluble de fibra dietaria en residuos de café (Coffea arábica L. var. Typica) y cacao (Teobroma cacao L. var. Complejo Nacional por trinitario). M.S. tesis, Univ. La Molina, Lima-Perú. Martínez, R., Torres, P., Meneses, M. A., Figueroa, J. G., PérezÁlvarez, J. A., & Viuda-Martos, M. (2012a). Chemical, technological and in vitro antioxidant properties of mango, guava, pineapple and passion fruit dietary fibre concentrate. Food Chemistry, 135(3), 15201526. doi: http://dx.doi.org/10.1016/j.foodchem.2012.05.057 Martínez, R., Torres, P., Meneses, M. A., Figueroa, J. G., PérezÁlvarez, J. A., & Viuda-Martos, M. (2012b). Chemical, technological and in vitro antioxidant properties of cocoa (Theobroma cacao L.) coproducts. Food Research International, 49(1), 39-45. doi: http://dx.doi.org/10.1016/j.foodres.2012.08.005 Martínez, R., Torres, P., Meneses, M. A., Figueroa, J. G., PérezÁlvarez, J. A., & Viuda-Martos, M. (2012c). Coproductos de la industrialización del palmito: Composición química y propiedades tecnológicas. Alimentación, equipos y tecnología, 268, 32-33. Mertens, D. R. (2003). Challenges in measuring insoluble dietary fiber. Journal of Animal Science, 81(12), 3233-3249. doi:/2003.81123233x Mildner-Szkudlarz, S., Zawirska-Wojtasiak, R., Szwengiel, A., & Pacyński, M. (2011). Use of grape by-product as a source of dietary fibre and phenolic compounds in sourdough mixed rye bread. International Journal of Food Science & Technology, 46(7), 14851493. doi: 10.1111/j.1365-2621.2011.02643.x Molina, M. E., & Martín, Á. (2007). La fibra dietética procesada como alimento funcional. Offarm, 26(1), 70-77. Mongeau, R., & Brooks, S. P. J. (2001). "Chemistry and analysis of lignin" en Dietary Fiber. New York: Marcel Dekker, Inc. pp. 321-374. Monte, W. C., & Maga, J. A. (1980). Extraction and isolation of soluble and insoluble fiber fractions from the pinto bean (Phaseolus vulgaris). Journal of Agricultural and Food Chemistry, 28(6), 11691174. doi: 10.1021/jf60232a065 Murthy, P., & Naidu, M. M. (2010). Recovery of Phenolic Antioxidants and Functional Compounds from Coffee Industry ByProducts. Food and Bioprocess Technology, 5(3), 897-903. doi: 10.1007/s11947-010-0363-z Nawirska, A., & Kwaśniewska, M. (2005). Dietary fibre fractions from fruit and vegetable processing waste. Food Chemistry, 91(2), 221-225. doi: http://dx.doi.org/10.1016/j.foodchem.2003.10.005 Nawirska, A., & Uklanska, C. (2008). Waste products from fruit and vegetable processing as potential sources for food enrichment in dietary fibre. Acta Scientlarum Polonorum, 7(2), 35 -42. O´Donnell, T. H., Deutsch, J., Yungmann, C., Zeitz, A., & Katz, S. H. (2015). New sustainable market opportunities for surplus food: A food system-sensitive methodology (FSSM). Food and Nutrition Sciences, 6 (10), 883-892. doi: 10.4236/fns.2015.610093 Prosky, L., Asp, N., Schweizer, T., DeVries, J., Furda, I., & Lee, S. (1994). Determination of soluble dietary fiber in foods and food products: collaborative study. Europe PubMed Central, 3(77), 690694. Raghavendra, S. N., Rastogi, N. K., Raghavarao, K. S. M. S., & Tharanathan, R. N. (2004). Dietary fiber from coconut residue: effects of different treatments and particle size on the hydration properties. European Food Research and Technology, 218(6), 563-567. doi: 0.1007/s00217-004-0889-2 Redondo-Márquez, L. (2002). La fibra terapéutica (2da. ed.). Barcelona: Editorial Glosa. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Fibra dietaria en subproductos de mango, maracuyá, guayaba y palmito _________________________________________________________________________________________________________________________ [41] Rodríguez, K. (2007). Análisis de los polisacáridos y la textura de la pared celular en cultivo en suspensión de Beta vulgaris L., Instituto Politécnico Nacional, Yautepec, Morelos. [42] Rustad, T., Storrø, I., & Slizyte, R. (2011). Possibilities for the utilisation of marine by-products. International Journal of Food Science & Technology, 46(10), 2001-2014. doi: 10.1111/j.13652621.2011.02736.x [43] Sangeetha, P. T., Ramesh, M. N., & Prapulla, S. G. (2005). Recent trends in the microbial production, analysis and application of Frucooligosaccharides. Trends in Food Science & Technology, 16(10), 442–457. doi:10.1016/j.tifs.2005.05.003 [44] Saura-Calixto, F. (2010a). Dietary Fiber as a Carrier of Dietary Antioxidants: An Essential Physiological Function. Journal of Agricultural and Food Chemistry, 59(1), 43-49. doi: 10.1021/jf1036596 [45] Saura-Calixto, F. (2010b). "Fibra dietética en la dieta y en alimentos funcionales. Prebióticos" en Alimentos saludables y de diseño específico. Alimentos funcionales. Madrid: Instituto Tomás Pascual Sanz. pp. 97-106. [46] Schieber, A., Stintzing, F. C., & Carle, R. (2001). By-products of plant food processing as a source of functional compounds — recent developments. Trends in Food Science & Technology, 12(11), 401413. doi: http://dx.doi.org/10.1016/S0924-2244(02)00012-2 [47] Soto, G., Luna, P., Wagger, M., & Carpio, S. (2002). Descomposición de residuos de cosecha y liberación de nutrimientos en plantaciones de palmito en Costa Rica. Agronomía Costarricense, 26(2), 43-51. [48] Southgate, D. A. T. (1976). "Determination of Food Carbohydrates". London: Applied Science. [49] Tosch, S. M., & Yada, S. (2010). Dietary fibres in pulse seeds and fractions: Characterization, functional attributes, and applications. Food Research International, 43(2), 450-460. doi: 10.1016/j.foodres.2009.09.005 [50] Ververis, C., Georghiou, K., Danielidis, D., Hatzinikolaou, D. G., Santas, P., Santas, R., & Corleti, V. (2007). Cellulose, hemicelluloses, lignin and ash content of some organic materials and their suitability for use as paper pulp supplements. Bioresource Technology, 98(2), 296-301. doi: http://dx.doi.org/10.1016/j.biortech.2006.01.007 [51] Yapo, B. M., & Koffi, K. L. (2008). The polysaccharide composition of yellow passion fruit rind cell wall: chemical and macromolecular features of extracted pectins and hemicellulosic polysaccharides. Journal of the Science of Food and Agriculture, 88(12), 2125-2133. doi: 10.1002/jsfa.3323 Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 15 Evaluación de las Características Físico-Químicas, Microbiológicas y Sensoriales de un Yogur Elaborado con Sucralosa y Estevia _________________________________________________________________________________________________________________________ 16 Evaluación de las Características Físico-Químicas, Microbiológicas y Sensoriales de un Yogur Elaborado con Sucralosa y Estevia Reyes J.*; Ludeña F.** * Universidad Técnica Particular de Loja, Departamento de Ciencias Agropecuarias y Alimentos, Loja, Ecuador. e-mail: [email protected] ** Universidad Nacional Agraria La Molina, Facultad de Industrias Alimentarias, Lima, Perú. e-mail: [email protected] Resumen: Los productos bajos en calorías han ganado una atención especial en el mercado y son un nicho atractivo para las empresas procesadoras. El propósito del presente trabajo fue desarrollar un yogur bajo en calorías. Se evaluó el efecto del empleo de sucralosa y estevia como edulcorantes, el efecto del porcentaje de sólidos totales, ST (12 y 13 %) y el lugar de proveniencia de la leche (Loja y Zamora), sobre las características sensoriales y fisicoquímicas del yogur. Una prueba sensorial descriptiva determinó la aceptabilidad de cada formulación, mientras que la estabilidad se evaluó estudiando la vida útil hasta 35 días en almacenamiento a 4 °C. Se estableció el uso de 0.015 % de sucralosa y 0.090 % de estevia, respecto de la leche. El tiempo requerido para alcanzar el pH del yogur no fue afectado por los factores de estudio y se mantuvo entre 180 a 193 min. El lugar de proveniencia de la leche no tuvo influencia sobre las propiedades fisicoquímicas y sensoriales del producto. La estandarización al 12 % ST presentó los valores más bajos de acidez (91 a 93 ºD), viscosidad (7300 a 10500 cP) y aporte energético (93.5 Kcal/100 g yogur), mientras que al 13 % ST la sinéresis fue más baja e influyó negativamente la percepción de la viscosidad en la boca. El panel sensorial encontró que la sucralosa mejoró notablemente el dulzor comparado con el producto edulcorado con estevia. Por tanto, para la elaboración de este tipo de yogur puede usarse leche de Loja o Zamora Chinchipe, con 12 % ST y edulcorado con sucralosa; con vida útil de 28 días en refrigeración a 4 ºC, período en el que no hubo presencia de microorganismos patógenos (E.coli y S. aureus), mohos y levaduras. Palabras clave: Yogur, sucralosa, estevia, calorías, edulcorantes. Abstract: Currently different low-calorie foods and no-calorie sweeteners have gained special attention in the society as they have become an attractive niche market for the food industry. The main objective of this work was to develop a yogurt with low calories content. The effects of the following factors were studied: “type of sweetener” (sucralose 0.015 % and stevia 0.090 % (w/v of milk)), “total solids content” (12 and 13 % TSC) and “place of origin of milk” (Loja and Zamora cities), and their influence on the physical-chemical and sensory characteristics of yogurt. The product was stored for 35 days at 4 °C. The acceptability of the product was evaluated using a descriptive sensory analysis and the shelf life study was done by evaluating the microbiological and physicalchemical characteristics. The time required to reach pH was not affected by the study variables and was kept between 180 to 193 min. The place of origin of milk did not affect the physical-chemical and sensorial characteristics of the final product. When the product was standardized at 12 % of TSC the lower values of acidity (91 to 93 °D), viscosity (7300 to 10500 cP) and energy content (93.5 Kcal/100 g yogurt) were shown, whereas at 13 % of TSC the syneresis was lower and negatively affected the perception of viscosity in mouth. The sensory panel found that sucralose sweetness significantly improved compared to product made with stevia. Therefore, the best yogurt formulation was obtained using milk coming from Loja or Zamora Chinchipe, with 12 % of TSC, sweetened with sucralose and its shelf life was 28 days under refrigeration conditions (4ºC), during which there was no presence of pathogens (E. coli and S. aureus), molds and yeasts. Keywords: Yogurt, sucralose, stevia, calories, sweeteners. 1. INTRODUCCIÓN A nivel mundial se ha incrementado notablemente el consumo de yogur, en el período del 2004 al 2011 pasó de 12 a 16 millones de toneladas [32]. Por su parte en el Ecuador entre los años 2006 y 2007 el consumo de este producto creció en un 25 %. La producción de yogur pasó de 120 000 Kg por día en el 2006 a 157 000 Kg en el 2008, que corresponde al 9 % de la producción de leche total que se destina a la Industria [34] . El consumo per cápita de yogur es de aproximadamente 4 Kg/habitante/año, cifra baja si se compara con países como Argentina, Brasil y Uruguay que bordean los 20 Kg [3], esta situación representa para el sector lácteo ecuatoriano una oportunidad de crecimiento, Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 17 Reyes J. *; Ludeña F.** _______________________________________________________________________________________________________________________________ considerando que el Gobierno del Ecuador ha implementado programas para promover el aumento del consumo de lácteos [14]. En el mercado de yogur hay una creciente preferencia por los productos bajos en calorías, debido a la cada vez mayor preocupación de los consumidores por la salud [8], estos productos se introdujeron en el mercado para atender dietas y necesidades específicas, hoy en día su precio competitivo y buenas características sensoriales han logrado expandirse en el mercado con muy buenas expectativas de crecimiento [38]. En el Ecuador entre el 2008 y 2009 se incrementó la compra de los ítems light del 10 % a 15 % [7]. El crecimiento de la demanda de este tipo de productos ha obligado a aumentar su diversificación y disponibilidad en los supermercados [8, 11, 16]. Las sustancias que hacen posible esto son algunos edulcorantes no calóricos, que se caracterizan por proporcionar un sabor dulce muy intenso y se usan en pequeñas cantidades [27]. La sucralosa presenta un alto poder edulcorante (600 veces más que la sacarosa), gran estabilidad a altas temperaturas y al pH [2, 12, 31], dulzor percibido rápidamente, ausencia de sabor amargo o metálico, no cariogénico, puede ser ingerido por diabéticos y su ingesta diaria admisible (IDA) es de 5 mg/Kg peso corporal por día. Pinheiro et al. (2005) indica que es el único edulcorante que no tiene ninguna desventaja. La estevia por su parte, es el primer edulcorante de origen natural sin calorías, proviene de la Stevia rebaudiana (Bertoni), planta sudamericana originaria del Paraguay [29]. Posee un alto poder edulcorante (250 a 300 veces más que la sacarosa), es estable a altas temperaturas y pH ácido, no cariogénico, no afecta los niveles de glucosa en sangre y su ingesta diaria admisible es de 4 mg/Kg de peso corporal/día [29]. La normativa ecuatoriana y la FAO clasifican un alimento como ligero, al que se le ha reducido al menos el 25 por ciento de su contenido de energía o del contenido de nutrientes del producto estándar [9, 21]. Esta investigación buscó desarrollar un yogur bajo en calorías, incorporando como edulcorantes no calóricos sucralosa y estevia, además de estudiar la incidencia de la calidad de la leche por su sitio de proveniencia, siendo la provincia de Zamora Chinchipe y la hoya de Loja, los lugares estudiados. 2. METODOLOGÍA acopio de la provincia de Zamora Chinchipe y de la ciudad de Loja. 2.3 Insumos Se usó sucralosa comercial de la empresa J.K. Sucralose Inc y estevia Granosweet sweta [15], cultivo probiótico FD-DVS Yo-Fast 88 que contiene Streptococcus thermophilus, Lactobacillus delbrueckii ssp. bulgaricus, Lactobacillus acidophilus y Bifidobacterium, leche en polvo descremada de la empresa “El Ordeño” con 34 % de proteínas, 0.50 % de grasa, 4.22 % de humedad y 1.35 % de acidez titulable, estabilizante para yogur G36400 AD de la empresa Aditmaq y sorbato de potasio granulado de la empresa Silperal. 2.4 Métodos de análisis Determinación de pH El valor de pH fue medido en el yogurt a 8 °C usando un pHmetro digital Orión 2-Star, calibrado usando buffers comerciales de pH 4 y 7. El procedimiento se realizó por triplicado [5]. Acidez titulable Se tomó 10 g de muestra a 20 °C; se adicionó unas gotas de solución indicadora de fenolftaleína y se tituló con una solución de hidróxido de sodio 1/9 N hasta cambio de coloración a rosa. La determinación se hizo por duplicado sobre la misma muestra y los resultados se expresaron como ºDornic [40]. Este método es el más difundido y de uso general de las Industrias lácteas ecuatorianas. El factor de corrección para expresar en porcentaje de ácido láctico se da al dividir para 100 los grados Dornic [42]. Viscosidad aparente La viscosidad aparente se determinó siguiendo el procedimiento descrito por Duarte y Hickmann (2007) [8] con modificación, brevemente, las muestras fueron medidas a 4 °C y se agitaron a 4 r.p.m. por 3 minutos con un viscosímetro digital Brookfield DV-I (Brookfield Engineering Laboratories, Inc., USA), equipado con un husillo S63. Sinéresis 2.1 Lugar de ejecución Esta investigación se desarrolló en las instalaciones de la Planta de Lácteos ECOLAC y en el Laboratorio de Alimentos del Departamento de Ciencias Agropecuarias y Alimentos de la Universidad Técnica Particular de Loja. 2.2 Materias Primas La sinéresis se determinó por el método de centrifugación descrito por Jicheng et al. (2010) [24], con modificaciones. Para lo cual se usó una centrifuga a 640 g por 10 minutos y 20 g de muestra (4ºC). El sobrenadante fue recolectado, pesado y la sinéresis se calculó de acuerdo a la siguiente ecuación: 𝑆𝑖𝑛é𝑟𝑒𝑠𝑖𝑠 (%) = Se utilizó leche fresca cruda descremada proporcionada por la Planta de Lácteos Ecolac, proveniente de sus centros de Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 𝑃𝑒𝑠𝑜 𝑑𝑒𝑙 𝑠𝑜𝑏𝑟𝑒𝑛𝑎𝑑𝑎𝑛𝑡𝑒(𝑔) 𝑃𝑒𝑠𝑜 𝑑𝑒 𝑙𝑎 𝑚𝑢𝑒𝑠𝑡𝑟𝑎 (𝑔) 𝑥 100 (1) Evaluación de las Características Físico-Químicas, Microbiológicas y Sensoriales de un Yogur Elaborado con Sucralosa y Estevia _________________________________________________________________________________________________________________________ Tiempo requerido para alcanzar el pH (tpH) Se determinó midiendo la cantidad de tiempo que requiere el yogur para alcanzar pH 4.7 desde el momento de la inoculación con el cultivo láctico [30]. Análisis de los principales ingredientes La cantidad de grasa, proteína, sólidos no grasos y lactosa se determinó por medición de ultrasonido, usando el equipo Lactoscan MCC (Milkotronic Ltd, Nova Zagora, Bulgaria) calibrado previamente. Todas las mediciones se realizaron por triplicado. Calorías El contenido de calorías de yogur se calculó en base a 8.79 Kcal/g de la grasa, 4 Kcal/g de los carbohidratos y 4 Kcal/g del contenido de proteínas [10, 25]. Análisis microbiológicos El recuento de microorganismos se realizó siguiendo los métodos rápidos Petrifilm (3M), para lo cual se hizo una sola dilución de cada muestra tomando 10 g de yogur y adicionándolos a 90 ml de solución salina de peptona. En todos los casos se sembró por duplicado 1 ml de la dilución y se incubó a: 25 °C por 5 días para el caso de mohos y levaduras (Método AOAC 997.02) y 35 °C por 24 horas para E. coli / coliformes totales (Método AOAC 991.14) y S. aureus (Método AOAC 2001.05) [19]. Al término de la incubación se seleccionaron las placas que presentaron entre 15 y 150 colonias, reportando el resultado como unidades formadoras de colonia por gramo (UFC/g). Para la evaluación de la calidad microbiológica se compararon los resultados con los requisitos establecidos en la Norma Técnica Ecuatoriana INEN 2395 para Leches Fermentadas [22]. 18 (dulzura, color y apariencia, cuerpo y textura, sensación olfato gustativa y aceptación general), por diez jueces, usando una escala hedónica de nueve puntos. Los niveles de sucralosa y estevia probados fueron: 0.012, 0.013, 0.014, 0.015 %, y 0.075, 0.080, 0.085 y 0.090 %, respectivamente, usando como control yogur comercial preparado con leche de Loja, estandarizado al 12 % de sólidos totales y edulcorado con sacarosa al 10.6 % [4]. 2.6 Análisis sensorial Selección del panel y entrenamiento De entre un grupo de docentes y estudiantes de la Titulación de Industrias Agropecuarias de la Universidad Técnica Particular de Loja, se seleccionó nueve panelistas (seis mujeres y tres hombres, de entre 22 a 35 años de edad), en base a su habilidad para reconocer sabores básicos, a su experiencia y capacitación previas en evaluación sensorial. El entrenamiento para este estudio consistió en cinco sesiones [26, 36], en las que se establecieron 13 atributos (Tabla 1) [36, 39]. Tabla 1. Lista de atributos para el análisis descriptivo. Descriptores Aspecto Olor Sensación olfato gustativa durante degustación Sensación olfato gustativa después de la degustación Textura oral Atributos Viscosidad Tonalidad Brillo Sinéresis A establo Acidez Dulzor Astringencia Residual a edulcorante Amargo Leche guardada Viscosidad Harinosidad Preparación y evaluación de la muestra 2.5 Metodología Experimental Elaboración de yogur Los yogures fueron fabricados mezclando leche desnatada con leche en polvo para regular a 12 y 13 % sus ST (rango en el que trabaja la empresa Ecolac Cía. Ltda.) y edulcorante. Seguidamente se aplica un tratamiento térmico a 90° C x 10 min y posteriormente se enfría. Las mezclas se inocularon con el cultivo (1 % p/v) y se incubaron a 43 ºC hasta llegar a un pH de 4.7, luego de lo cual se enfriaron (22 °C), se agitaron manualmente, se envasaron en bolsas plásticas de 1 Kg y se almacenaron a 4 °C. Elección de los niveles de sucralosa y Estevia Se aplicó un diseño completamente aleatorio para establecer los niveles de edulcorante (sucralosa y estevia) para el estudio. La variable respuesta que se consideró para el efecto estuvo basada en una prueba de aceptación del yogur Todas las muestras se sacaron de refrigeración media hora antes de empezar la sesión de evaluación. Se sirvió en un rango de temperatura entre 4 a 10ºC. Cada yogur fue presentado en un vaso de plástico de 50 g identificado con código de tres dígitos. El orden de presentación de las muestras fue aleatorio. La evaluación se llevó a cabo en el Laboratorio de evaluación sensorial, los panelistas permanecieron separados y sentados, cada yogur lo evaluaron por triplicado, se facilitó agua para que se enjuaguen la boca entre muestras. Se usó una escala bipolar lineal de 10 cm y se estableció con el panel la valoración óptima de cinco (mitad de la escala) para todos estos atributos. 2.7 Vida útil Se almacenó producto a 4 ºC y se tomó las muestras para su análisis después de 1, 7, 15, 21, 28 y 35 días después de Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 19 Reyes J. *; Ludeña F.** _______________________________________________________________________________________________________________________________ elaborado. Se realizaron los análisis fisicoquímicos: viscosidad, sinéresis, acidez y pH; sensoriales: apariencia, color, olor, consistencia y aceptabilidad general. Las pruebas microbiológicas de coliformes totales y, mohos y levaduras se aplicaron a las muestras en todas las fechas de control y las correspondientes a E. coli y Staphylococcus aureus únicamente el día uno. 2.8 Análisis estadístico Se aplicó un diseño factorial 23 para investigar el efecto de la estandarización de sólidos totales (12 y 13 %), el tipo de edulcorante no calórico (sucralosa y estevia) y el lugar de proveniencia de la leche (ciudad de Loja y provincia de Zamora Chinchipe), sobre las propiedades físicas, químicas y sensoriales del yogur bajo en calorías. Para determinar diferencias significativas en tiempo de vida útil y nivel de edulcorante, se usó un análisis de varianza (ANOVA) de una y dos vías, la prueba de comparaciones múltiples de Tukey (p < 0.05) y el software Minitab versión 16. 3.2 Determinación de los niveles de edulcorante 3.2.1 Sucralosa El edulcorante sucralosa en el nivel 0.015 % resultó significativamente mayor en términos de percepción de dulzor (Tabla 3), que la puntuación obtenida por la muestra control. En cuanto a la sensación olfato gustativa, los niveles 0.013, 0.014, 0.015 % y el control no presentan diferencia estadística entre sí, aunque el nivel 0.015 % obtuvo en promedio una mayor puntuación sensorial que el resto de muestras. En cuanto al nivel 0.012 % presenta puntuaciones estadísticamente más bajas que 0.015 % para este atributo. Para los atributos, color y apariencia, cuerpo y textura, no se presentó diferencia estadística, aunque las puntuaciones más altas las tuvo el tratamiento control, esto puede deberse a que el azúcar (sacarosa) contribuye no solamente a edulcorar, sino que es responsable de las características de color a través de reacciones de Maillard y caramelización [4]. Por tanto, se elige el nivel de sucralosa 0.015 % para la preparación del yogur. 3.2.2 Estevia 3. RESULTADOS Y DISCUSIÓN 3.1 Características fisicoquímicas de la leche Como se puede apreciar en la Tabla 2; la composición de sales minerales es estadísticamente mayor para la leche proveniente de Zamora, esto según lo explica Yildiz (2010) puede deberse a la presencia en la leche mastítica que genera un incremento de los niveles de sodio y potasio en la leche. Saa (2010) realizó un estudio epidemiológico en las fincas proveedoras de la empresa Ecolac, Cía. Ltda. y concluyó que la prevalencia de mastitis clínica es 14 % y subclínica 86 % [35]. Por otro lado, la leche proveniente de los sectores seleccionados para el estudio cumplió con la norma Técnica Ecuatoriana NTE INEN 9:2008, “Leche cruda. Requisitos” [20], excepto la cantidad de sólidos no grasos presentes, encontrándose por debajo del mínimo exigido por dicha norma (8.2 %). Tabla 2. Características físico químicas de la leche cruda descremada de Loja y Zamora Chinchipe Proveniencia de la leche Característica Loja Zamora Grasa (%) 0.1 ± 0.10a 0.1 ± 0.03a Densidad* (g/ml) 1.0309 ± 0.36a 1.0309 ± 0.29a Sólidos no grasos (%) 7.9 ± 0.07a 7.9 ± 0.06a a Proteína (%) 2.90 ± 0.03 2.90 ± 0.02a a Lactosa (%) 4.35 ± 0.04 4.38 ± 0.05a a Sales minerales (%) 0.65 ± 0.01 0.66 ± 0.01b a Acidez (ºD) 13.3 ± 0.49 13.6 ± 0.51a Aporte energético 61.23 ± 2.36a 62.25 ± 1.22a Kcal/200 ml *Densidad corregida a 20ºC Letras diferentes indican diferencias significativas (p < 0.05) Los datos se presentan como media ± desviación estándar (n=12) De la misma manera que para el caso del edulcorante sucralosa, en la Tabla 4 se puede observar los resultados de la prueba de evaluación sensorial para diferentes niveles de edulcorante estevia. Los resultados revelaron que la percepción de dulzor en el yogur con edulcorante estevia fue estadísticamente igual (p > 0.05) para todas las concentraciones estudiadas (0.075, 0.080, 0.085, 0.090 por ciento) incluyendo el tratamiento control. En cuanto a color y apariencia, cuerpo y textura, éste no presentó diferencia significativa (p>0.05) entre los niveles probados, pero si la manifestó respecto al tratamiento control el cual obtuvo los puntajes más altos. Corresponde a 0.09 % la valoración más alta entre los niveles de estevia de este estudio. Para los atributos, sensación olfato gustativa y aceptabilidad general, todos los niveles de estevia probados fueron iguales entre sí, pero estadísticamente menores (p<0.05) que el control. Por lo tanto, se decidió seleccionar para el estudio al nivel 0.09 % de estevia. 3.3 Elección de las mejores condiciones de elaboración de yogur bajo en calorías Para elegir la combinación de factores que se usará en la elaboración de yogur se consideró su efecto sobre las características físicas, químicas y sensoriales, a continuación se presenta los resultados de la investigación. 3.3.1 Efecto de los factores de estudio sobre las características fisicoquímicas del yogur En la Tabla 5 se presentan los resultados del efecto de los tres factores de estudio (edulcorante, proveniencia de la leche y porcentaje de sólidos totales) en las características físicas y químicas de yogur. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Evaluación de las Características Físico-Químicas, Microbiológicas y Sensoriales de un Yogur Elaborado con Sucralosa y Estevia _________________________________________________________________________________________________________________________ a. Tiempo requerido para alcanzar el pH (tpH) El tpH del yogur no se ve afectado significativamente (p>0.05) por los factores estudiados (ver Tabla 5), por lo que puede afirmarse que el crecimiento de los microorganismos del cultivo iniciador fue normal. Weber y Sharareh (2013) [41], estudiaron el efecto de varios edulcorantes sobre el crecimiento y la supervivencia de L. rhamnosus GR-1, comprobando que la adición de los glucósidos de esteviol son apropiados para incorporar en un yogur probiótico, ya que no afectan el crecimiento de estos microorganismos y además se mantiene en un nivel funcional a los 28 días de almacenamiento. Pinheiro et al.(2002), citados por Pinheiro et al. (2005) [33], concluyeron que al estudiar el efecto de algunos edulcorantes (sacarosa, sacarosa + dextrosa, aspartame, aspartame + sacarina y sucralosa) sobre el proceso de elaboración de yogur, es posible obtenerlo exitosamente sin afectar la viabilidad de los microorganismos probióticos del cultivo. Tabla 3. Optimización del nivel de sucralosa en yogur Nivel de Sucralosa (%) Características 0.012% 0.013% 0.014% 0.015% Dulzura 6.3 ± 1.36ab 6.5 ± 0.96ab 6.4 ± 0.88ab 7.2 ± 0.82a Color y apariencia 6.9 ± 1.51a 7.0 ± 1.48a 7.0 ± 1.00a 7.0 ± 1.15a a a a Cuerpo y textura 6.1 ± 1.52 6.2 ± 1.94 6.2 ± 1.40 6.3 ± 1.67a Sensación olfato gustativa 5.8 ± 1.60a 6.4 ± 1.31ab 6.3 ± 1.11ab 6.9 ± 1.18b Aceptación general 6.1 ± 1.37a 6.3 ± 1.36a 6.1 ± 1.27a 6.8 ± 0.92a Letras diferentes indican diferencias significativas (p < 0.05) entre los niveles de edulcorante Los datos se presentan como media ± desviación estándar (n = 10) Características Dulzura 20 6.0 7.1 6.5 6.4 6.3 Tabla 4. Optimización del nivel de estevia en yogur Nivel de Estevia (%) 0.075% 0.080% 0.085% 0.090% 5.9 ± 1.18a 6.0 ± 1.35a 6.0 ± 1.38a 6.3 ± 1.44a Color y apariencia 7.0 ± 1.47ab 6.7 ± 1.58b 6.9 ± 1.78ab ab b Cuerpo y textura 6.6 ± 1.52 6.1 ± 1.68 6.4 ± 1.68ab Sensación olfato gustativa 6.0 ± 1.35a 5.7 ± 1.73a 5.9 ± 1.56a Aceptabilidad general 5.8 ± 1.36a 5.7 ± 1.43a 5.8 ± 1.53a Letras diferentes indican diferencias significativas (p < 0.05) entre los niveles de edulcorante Los datos se presentan como media ± desviación estándar (n = 10) 7.1 6.8 6.1 6.0 ± ± ± ± Control ± 1.19b ± 1.05a ± 1.45a ± 1.06ab ± 1.00a Control 6.7 ± 0.92a 1.17ab 1.14ab 1.58a 1.46a 7.3 6.9 6.9 6.8 ± ± ± ± 0.89a 1.13a 0.91b 0.95b Tabla 5. Características fisicoquímicas de los yogures elaborados con la combinación de los factores de estudio Características Tiempo requerido para alcanzar el pH (minutos) Acidez titulable (ºD) Viscosidad aparente (Cp) 193.3 ± 11.55ª 93.3 ± 6.11ª 13% ST 186.7 ± 11.55ª 96.7 ± 1.53 b 12% ST 186.7 ± 11.55ª 92.7 ± 2.89ª 13% ST 193.3 ± 11.55ª 94.3 ± 3.51 b 12% ST 188.3 ± 10.41ª 91.7 ± 2.08ª Factores de estudio 12% ST Loja Sinéresis (%) Aporte energético Kcal/200 g) 9750 ± 4685ª 41.9 ± 8.09ª 93.9 ± 0.45ª b b 100.9 ± 1.65b 10171 ± 3035 31.7 ± 5.55 Estevia Zamora Loja a 13% ST 180.0 ± 0.00 12% ST 180.0 ± 0.00a Sucralosa Zamora 13% ST 186.7 ± 11.55 97.3 ± 2.08 b 92.7 ± 2.52ª a 98.0 ± 5.29 b 7369 ± 639ª 9299 ± 1843 40.1 ± 11.29ª b 7953 ± 591ª 15061 ± 3473 b 93.6 ± 1.94ª 98.9 ± 1.39b 41.9 ± 5.53ª b 10481 ± 622ª 11058 ± 2340 25.4 ± 2.46 b 93.0 ± 1.03ª b 100.6 ± 0.56b 34.4 ± 11.31ª 93.5 ± 1.86ª 22.8 ± 10.90 31.3 ± 3.18 b 98.9 ± 1.46b Letras diferentes indican diferencias significativas (p < 0.05) entre las medias Los datos se presentan como media ± desviación estándar (n = 10) b. Acidez titulable El factor “sólidos totales” tuvo efecto sobre la acidez titulable del yogur, los tratamientos estandarizados al 12 % ST presentan valores significativamente menores (p < 0.05) (92 a 94ºD) comparados con los estandarizados al 13 % (94 a 98ºD), esto puede deberse a que con el incremento del contenido de sólidos en leche para yogur como resultado de una fortificación, también se crea un mayor efecto buffer debido a la acción amortiguadora de las proteínas adicionales, fosfatos, nitratos y lactatos, que requiere un desarrollo adicional de ácido por los cultivos iniciadores para lograr un pH normal en este tipo de productos [28, 42] . Salvador y Fiszman (2004) [37] reportaron una acidez de entre 0.82 a 1.15 % de ácido láctico para yogures elaborados con leche desnatada. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Evaluación de las Características Físico-Químicas, Microbiológicas y Sensoriales de un Yogur Elaborado con Sucralosa y Estevia _________________________________________________________________________________________________________________________ c. Viscosidad aparente El “tipo de edulcorante y la proveniencia de la leche”, no afectaron significativamente las medidas de viscosidad (p > 0.05), pero si lo hizo el factor sólidos totales (ST). Yogures estandarizados a 13 % tienen una viscosidad aparente significativamente más alta (9000 a 15000 cP) que los regulados al 12 % (7300 a 10500 cP), este efecto puede deberse al aumento de proteínas de la que hace más densa la red del gel [1], según lo explica Lee y Lucey (2010) [28] y Yildiz (2010) [42]. Haque y Ji (2003) [17], por su parte, reportaron viscosidades aparentes de 2000 a 5000 cP para yogur ajustado con proteínas de suero obtenidas del procesamiento de queso cheddar. Por otro lado, Isleten y Karagul-Yuceer (2006) [23] encontraron que al agregar caseinato de sodio y un mejorador de textura sobre yogur, se obtuvo viscosidades de 7363 y 7120 cP, respectivamente, superiores a las reportadas para el tratamiento control (leche en polvo desnatada) 5243 cP, además, estos ingredientes secos mejoraron las propiedades sensoriales de yogur. d. Sinéresis La estandarización de los sólidos no grasos afectó significativamente la medida de sinéresis (p<0.05) del yogur, correspondiendo al regulado al 13 % ST los valores más bajos (Tabla 5). Lee y Lucey (2010) [28], indican que la sinéresis es un defecto del yogur que se previene usando estabilizantes o incrementando el contenido de sólidos totales de la leche que se destina para su producción. e. Aporte energético Los factores “tipo de edulcorante y proveniencia de la leche”, no afectaron significativamente las medidas de aporte energético (p>0.05), sin embargo, se encontró que la regulación de sólidos totales de la leche de partida (12 y 13 %) tuvo efecto sobre esta variable. Yogures estandarizados al 12 % ST tienen un aporte energético significativamente menor (93.5 Kcal/200g) que los llevados al 13 % (99.83 Kcal/200 g). Esto se debe a la relación directamente proporcional existente entre cantidad de leche en polvo desnatada usada para la regulación y los constituyentes principales que la acompañan y que aportan energía (lactosa y proteína). En el presente estudio, los tratamientos con 12 % ST presentaron la mayor disminución del aporte energético (> 50 %), considerando como referencia comercial un yogur entero (3 % de grasa), edulcorado con sacarosa (13 %) que aporta en promedio 190 kilocalorías por porción de 200 gramos. 21 3.3.2 Efecto de los factores de estudio sobre las características sensoriales del yogur Únicamente el atributo dulzor, se vio afectado significativamente (p<0.05) por el “tipo de edulcorante”, resultando la sucralosa como el que mejoraba esta característica en el yogur (ver Tabla 6a), los jueces calificaron con un promedio muy cercano al óptimo establecido (5.08). Un resultado similar obtuvieron Weber y Sharareh (2013) [41], los cuales evaluaron el efecto de diversos agentes edulcorantes (0.12 % estevia, 0.12 % estevia- inulina-cromo (SIC), 0.1 % sucralosa y 5.0 % de sacarosa), sobre las propiedades sensoriales de yogur probiótico (sabor, apariencia, dulzura, textura y apariencia), en ésta se encontró que la muestra con sucralosa se prefirió de manera significativa para las cinco características estudiadas, seguida por la muestra de sacarosa que gustó significativamente más que estevia y SIC, para todas las características excepto sabor. El edulcorante estevia puede impartir algunas características de amargura y un sabor residual indeseable a los productos [13], lo que evidentemente limita las cantidades a dosificar y su correspondiente grado de dulzor, sin embargo, esta desventaja ha impulsado la investigación en laboratorios de desarrollo e innovación de las industrias que buscan encontrar un edulcorante con mejor equilibrio de dulzura y buen gusto [18]. Pinheiro et al. (2005) [33] manifiestan que entre todos los edulcorantes, la sucralosa es el único que no presenta ninguna desventaja, tiene un perfil tiempointensidad de alta calidad, bastante similar a los de sacarosa y aspartame. La dulzura percibida es rápida, persiste durante un período ligeramente más largo que el de la sacarosa, y no presenta ningún sabor amargo o retrogusto metálico. “El contenido de sólidos totales (% ST)” también afecta significativamente a la textura oral (viscosidad percibida en la boca), resultando el yogur estandarizado al 12 %, como el que se acercaba más al óptimo (5.2). 3.3.3 Elección del mejor tratamiento Considerando la relación entre la viscosidad aparente y la sensorial, el nivel de dulzor alcanzado, el menor aporte calórico por porción y la facilidad para la provisión de leche, el tratamiento elegido correspondió al yogur elaborado con leche de Loja o Zamora Chinchipe, estandarizarlo al 12 % ST y edulcorado con sucralosa. Revista Politécnica - Septiembre 2015, Vol. 36, No. 3 Evaluación de las Características Físico-Químicas, Microbiológicas y Sensoriales de un Yogur Elaborado con Sucralosa y Estevia _________________________________________________________________________________________________________________________ 22 Tabla 6a. Características sensoriales de los yogures elaborados con la combinación de los factores de estudio Descriptor Atributo Loja Estevia Zamora Loja Sucralosa Zamora Olfato gustativa durante de la degustación Aspecto 12 % ST 13 % ST 12 % ST 13 % ST 12 % ST 13 % ST 12 % ST 13 % ST Viscosid ad 5.1±0.86 a 5.0±0.28 a 4.2±0.66 a 4.8±0.45 a 4.7±0.37 a 5.6±0.27 a 5.1±0.64 a 4.9±0.48 a Tonalidad Brillo Sinéresis Acidez Dulzor Astringencia 4.2±0.41a 4.6±0.30a 4.1±0.54a 4.8±0.57a 3.9±0.21a 4.8±0.21a 4.9±0.31a 4.5±0.35a 4.1±0.22a 5.1±0.75a 4.0±0.24a 5.3±0.11a 4.2±0.27a 4.8±0.19a 4.1±0.51a 4.7±0.40a 3.8±0.67a 4.6±0.37a 4.1±0.20a 4.5±0.19a 4.1±0.37a 4.6±0.32a 3.9±0.10a 4.6±0.62a 4.3±0.27a 4.5±0.57a 4.1±0.22a 4.5±0.49a 5.2±0.66a 4.5±0.35a 4.3±0.41a 4.5±0.23a 4.1±0.76a 4.9±0.26a 5.5±1.12a 4.5±0.62a 4.5±0.28a 4.4±0.14a 5.1±0.32a 4.9±0.74a 5.2±1.01a 4.6±0.16a 4.3±0.50a 5.0±0.33a 4.1±0.31a 5.0±0.85a 4.4±0.56a 4.5±0.57a Letras diferentes indican diferencias significativas (p < 0.05) entre las medias Tabla 6b. Características sensoriales de los yogures elaborados con la combinación de los factores de estudio Descriptor Olfato gustativa después de la degustación Residual a Leche Amargo edulcorante guardada Atributo Loja Estevia Zamora Loja Sucralosa Zamora 12 % ST 13 % ST 12 % ST 13 % ST 12 % ST 13 % ST 12 % ST 12 % ST Olor Textura Oral A establo Viscosidad Harinosidad 5.0±0.48a 4.8±0.21a 4.9±0.30a 4.1±0.36a 5.5±0.59a 5.1±0.08a 5.3±0.73a 4.8±0.36a 4.6±0.35a 4.1±0.14a 5.9±0.07a 5.1±0.47a 5.0±0.56a 4.9±0.31a 4.5±0.19a 4.1±0.36a 4.8±0.64a 4.9±0.17a 5.4±0.14a 4.8±0.30a 4.7±0.19a 4.1±0.11a 5.1±0.58a 5.2±0.29a 5.3±0.45a 4.5±0.26a 4.8±0.57a 4.1±0.77a 4.7±0.41a 4.8±0.63a 4.8±0.67a 4.9±0.06a 4.8±0.23a 4.1±0.09a 6.1±0.43a 5.0±0.47a 5.1±0.59a 4.3±0.21a 4.5±0.14a 5.1±0.42a 5.7±0.47a 4.8±0.54a 4.6±0.28a 4.9±0.30a 4.8±0.33a 4.1±0.71a 5.5±0.30a 5.1±0.18a Las letras a y b indican que existe diferencia significativa entre las medias a p<0.05 3.3.4 Determinación de la vida útil del producto a. Análisis fisicoquímicos Como puede observarse en la Fig. 1, el tiempo de almacenamiento afectó significativamente (p<0.05) la viscosidad aparente del yogur. Esta característica decreció durante los primeros siete días de almacenamiento, no manifestó diferencia estadística entre los días 14 al 28 y se verificó una disminución al día 35. Por su parte, la sinéresis del yogur (ver Fig. 2) aumenta significativamente (p<0.05) durante todo el período de almacenamiento. Entre los días 21 y 35 presentaron los valores más altos, Jicheng et al. (2010) [24] y Farooq y Haque (1992) [11], encontraron que la cantidad de sinéresis de yogur aumentó durante 14 días de almacenamiento. Salvador y Fiszman, (2004) [37], reportaron que los valores de sinéresis para yogures almacenados en tres temperaturas diferentes (10, 20, y 30 °C) presentan un aumento significativo, que fue más evidente durante los primeros días de almacenamiento. Además, encontraron que las muestras que se almacenaron a 30 °C, tienen un grado mucho mayor de sinéresis en los dos tipos de yogur estudiados (enteros y desnatados). Aryana y McGrew (2007) [5], reportaron un aumento constante en la sinéresis en todos los tratamientos de yogur estudiados a medida que transcurre el tiempo de almacenamiento. La acidez se mantuvo entre 86 y 95 ºD para los días 1 y 35 respectivamente (Fig. 3), fue afectada significativamente (p<0.05) por el tiempo de almacenamiento, observándose los valores más altos a partir del día 28 (94.5 ºD o 0.945 % de ácido láctico). Tales diferencias relacionadas con el almacenamiento implican actividad del cultivo vivo y es un fenómeno esperado [17]. El pH, por su parte, durante el período de estudio alcanzó valores que se ubicaron entre 4.6 a 5.0. Al observar la Fig. 4 se puede notar que esta característica decreció con el tiempo Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 23 Reyes J. *; Ludeña F.** _______________________________________________________________________________________________________________________________ en almacenamiento encontrándose que la primera semana presenta valores significativamente más altos (p<0.05) que la segunda y ésta a su vez mayores (p< 0.05) que el resto de semanas en las que se llevó a cabo el estudio. 5,1 5,0 4,9 pH Al igual que lo reportado por Farooq y Haque (1992) [10], en el presente estudio el decrecimiento del pH no afectó marcadamente las características sensoriales del yogur. 4,8 4,7 a b 4,6 c c c 21 28 Centipoises 4,5 4000 3500 3000 2500 2000 1500 1000 500 0 c 4,4 1 7 14 35 Días Figura 4. pH de yogur durante el almacenamiento a b. b 1 7 bc bc bc 14 21 28 c 35 Días Evaluación sensorial Para todas las características sensoriales estudiadas del yogur, los jueces no percibieron cambios significativos hasta el día que duró el estudio de almacenamiento y los puntajes correspondieron a las características sensoriales “me gusta mucho” a “me gusta poco”. Figura 1. Viscosidad de yogur durante el almacenamiento Porcentaje (%) c. 70,0 68,0 66,0 64,0 62,0 60,0 58,0 56,0 54,0 52,0 En lo concerniente a la calidad y desarrollo microbiológico del yogur evaluado, no hubo presencia de microorganismos patógenos (E.coli y S. aureus), mohos y levaduras durante todo el período de evaluación del producto, demostrando que en el proceso de elaboración se cumplieron las normas básicas de higiene [35]. bc abc ab a 1 7 14 Días 21 c bc 28 4. CONCLUSIONES 35 Figura 2. Sinéresis de yogur durante el almacenamiento Grados Dornic (ºD) 100,0 95,0 90,0 85,0 80,0 Análisis microbiológico. b a a a 1 7 14 b ab En este estudio se ha demostrado que leche proveniente de Loja o Zamora Chinchipe son adecuadas para producir yogur bajo en calorías y no afectan sus características fisicoquímicas y sensoriales. Al mismo tiempo, una mayor proporción de sólidos totales en la formulación presentó yogures con menor sinéresis, mayor acidez y viscosidad, pero con un mayor aporte energético por porción y negativamente afectada la viscosidad percibida en la boca. La sucralosa fue el edulcorante que mejoró las características sensoriales del yogur, sobre todo en lo relacionado con el atributo dulzor. Así mismo, el producto presenta una estabilidad normal almacenado en condiciones de refrigeración a 4 ºC. AGRADECIMIENTO Los autores agradecen a ECOLAC Cía. Ltda. (Ecuador) por el aporte financiero y la provisión de la materia prima. 75,0 Días 21 28 35 Figura 3. Acidez titulable de yogur durante almacenamiento Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Evaluación de las Características Físico-Químicas, Microbiológicas y Sensoriales de un Yogur Elaborado con Sucralosa y Estevia _________________________________________________________________________________________________________________________ REFERENCIAS [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] Abd, E.-K. (2009). Production and Evaluation of a High Protein Version of Non-fat Yogurt. Research Journal of Agriculture and Biological Sciences, 5(4), 310-316. ADA. (2006). Facts About Sucralose. In, vol. 2010. Alvear, G. (2010). Estudio de factibilidad para el procesamiento y comercialización de yogurt en Pedro Vicente MaldonadoPichincha. San Francisco de Quito, Quito. Arora, S., Gawande, H., Sharma, V., Wadhwa, B. K., George, V., Sharma, G., & Singh, A. K. (2009). The development of burfi sweetened with aspartame. International Journal of Dairy Technology, 63(1), 127-135. Aryana, K. J., & McGrew, P. (2007). Quality attributes of yogurt with Lactobacillus casei and various prebiotics. LWT - Food Science and Technology, 40(10), 1808-1814. Diario Hoy, E. (2009). Alimentos light ganan terreno luego de seis meses. In). Quito, EC: Diario Hoy. Duarte, C., & Hickmann, F. (2007). The Influence of additives on the rheological and sensory properties of nonfat yogurt. International Journal of Dairy Technology, 60(4), 270-276. FAO. (1997). Guidelines for use of nutrition and health claims. Suiza: Codex Alimentarius. Farooq, K., & Haque, Z. (1992). Effect of Sugar Esters on the Textural Properties of Nonfat Low Calorie Yogurt. Dairy science, 75, 2676-2680. Farooq, K., & Haque, Z. (1997). Effect of Acylated Proteins on Textural Properties of Nonfat Low Calorie Set Yogurt and Lowfat Ice Cream. Food Science and Technology, 3(3), 274-278. FDA. (1999). Food Additives Permitted for Direct Addition to Food for Human Consumption (Vol. 2010). USA: Federal Register. Fitch, C., Morgantown, W., & Keim, K. (2012). Use of Nutritive and Nonnutritive Sweeteners. Academy of Nutrition and Dietetics, 112(5), 739-758. Fresco, E. (2009). Análisis del contexto y proyección de la lechería a nivel mundial y de la región. In Foro del Sector Lechero Ecuatoriano, (pp. 33). Quito. Granotec. (2010). Ficha Técnica Granosweet sweta (estevia: esteviosidos naturales). In Granotec (Ed.)). Guayaquil. Haque, Z., & Aryana, K. (2002). Effect of Sweeteners on the Microestructure of Yogurt. Food Science and Technology, 8(1), 21-23. Haque, Z., & Ji, T. (2003). Chedar whey processing and source: II. Effect on non-fat ice cream and yoghurt. International Journal of Food Science and Technology, 38, 463-473. Harnish, D. (2012). Leveraging Sweetening Expertise to Realize Stevia’s Full Potential on Taste. In): Dr. Harnish. Horwitz, W., & Latimer, G. (2005). Official Methods of Analysis. Microbiological Methods. WH Andrews;TS Hammack. . In 18 ed. ed., (pp. Ch. 17. p. 11-244). Maryland, USA. AOAC International. INEN. (2008). Norma Técnica Ecuatoriana NTE INEN 9:2008. Leche cruda. Requisitos. In 4. ed ed., (pp. 5 p). Quito-Ecuador. INEN. (2011a). Norma Técnica Ecuatoriana NTE INEN 1334-3. Rotulado de Productos Alimenticios Para Consumo Humano. Parte 3. Requisitos Para Declaraciones Nutricionales y Declaraciones Saludables. In, (pp. 17 p). Quito-Ecuador. INEN. (2011b). Norma Técnica Ecuatoriana NTE INEN 2395:2011. Leches Fermentadas. Requisitos. In 2. ed ed., (pp. 10 p). Quito-Ecuador. Isleten, M., & Karagul-Yuceer, Y. (2006). Effects of Dried Dairy Ingredients on Physical and Sensory Properties of Nonfat Yogurt. Dairy science, 89(8), 2865-2872. Jicheng, W., Zhuang, G. U. O., Qing, Z., Liya, Y. A. N., Yongfu, C., Xia, C., Xiao-Ming, L. I. U., Wei, C., & He-Ping, Z. (2010). Effect of probiotic Lactobacillus casei Zhang on fermentation characteristics of set yogurt. International Journal of Dairy Technology, 63(1), 105-112. Keating, K., & White, C. (1990). Effect of Alternative Sweeteners in Plain and Fruit-Flavored Yogurts. Dairy science, 73, 54-62. King, S. C., Lawler, P. J., & Adams, J. K. (2000). Effect of Aspartame and Fat on Sweetness Perception in Yogurt. Journal of Food Science, 65(6), 1056-1059. 24 [26] Kroger, M., Meister, K., & Kava, R. (2006). Low-calorie Sweeteners and Other Sugar Substitutes: A Review of the Safety Issues. Comprehensive Reviews in Food Science and Food Safety, 5(2), 35-47. [27] Lee, W. J., & Lucey, J. A. (2010). Formation and Physical Properties of Yogurt. Asian-Australasian Journal of Animal Science, 23(9), 1127-1136. [28] Lemus-Mondaca, R., Vega-Gálvez, A., Zura-Bravo, L., & AhHen, K. (2012). Stevia rebaudiana Bertoni, source of a highpotency natural sweetener: A comprehensive review on the biochemical, nutritional and functional aspects. Food Chemistry, 132(3), 1121-1132. [29] Mcgregor, J., & White, C. (1986). Effect of Sweeteners on the Quality and Acceptability of Yogurt. Dairy science, 69, 698-703. [30] Owen, F. (2000). Química de los Alimentos (Segunda ed.). Zaragoza: Acribia. [31] Padilla, A. (2011). El rol de los lácteos en las nuevas tendencias del retail. In). Valdivia-Chile: Zenith International Ltd. [32] Pinheiro, M., Oliveira, M., Penna, A., & Tamime, A. (2005). The effect of different sweeteners in low-calorie yogurts-a review. International Journal of Dairy Technology, 58(4), 193-199. [33] Purtschert, N. (2009). La Industria Láctea en el Ecuador y sus Perspectivas de Inclusión Económica y Sustentabilidad. In C. d. I. L. (CIL) (Ed.), Foro del sector lechero ecuatoriano). Quito. [34] Ray, B., & Bhunia, A. (2008). Fundamentos de microbiología de los alimentos (4ta ed.). México: McGraw-Hill Interamericana Editores, S.A. de C.V. [35] Saa, Luis. (2010). Estudio de las principales infecciosas en ganado bovino en la Provincia de Zamora Chinchipe en las fincas proveedoras a la planta de Lácteos Ecolac, durante el periodo Noviembre 2009-Marzo 2010 (pp. 15). Loja: Universidad Técnica Particular de Loja [36] Saint-Eve, A., Lévy, C., Le Moigne, M., Ducruet, V., & Souchon, I. (2008). Quality changes in yogurt during storage in different packaging materials. Food Chemistry, 110(2), 285-293. [37] Salvador, A., & Fiszman, S. M. (2004). Textural and Sensory Characteristics of Whole and Skimmed Flavored Set-Type Yogurt During Long Storage. Journal of Dairy Science, 87(12), 4033-4041. [38] Sandrou, D. K., & Arvanitoyannis, I. S. (2000). Low-Fat/Calorie Foods: Current State and Perspectives. Critical Reviews in Food Science and Nutrition, 40(5), 427-447. [39] Santana, L., Santos, L., Natalicio, M., Mondragon-Bernal, O., Elias, E., Silva, C., Zepka, L., Isabela, M., Vernaza, M., CastilloPizarro, C., & Bolini, H. (2006). Perfil Sensorial de Iogurte Light, Sabor Pêssego. Ciencia y Tecnología de Alimentos, 26(3), 619625. [40] Tamime, A., & Robinson, R. (1991). Yogurt: Cencia y Tecnología. Zaragoza: Acribia. [41] Weber, A., & Sharareh, H. (2013). The effect of Stevia rebaudiana on the growth and survival of Lactobacillus rhamnosus GR-1 and Sensory Properties of Probiotic Yogurt. Food Research, 2(2), 136-143. [42] Yildiz, F. (2010). Development and manufacture of yogurt and other fuctional dairy products. Florida: CRC Press. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 25 Atención Farmacéutica en el Fomento del Desayuno Saludable desde la Oficina de Farmacia _______________________________________________________________________________________________________________________________ Atención Farmacéutica en el Fomento del Desayuno Saludable desde la Oficina de Farmacia Morales F.*; Marín F.; Morales L.; Morales I.; Marín L.; Gastelurrutia M. *Universidad Nacional del Chimborazo, Riobamba Ecuador e-mail: *[email protected]; [email protected]; [email protected]; [email protected]; [email protected]; [email protected] Resumen: Se considera Desayuno Saludable aquél que contiene una ración equilibrada de todos los nutrientes, tanto de forma cualitativa como cuantitativa. Pese a que debería proporcionar entre el 20-25 % del aporte diario, suele ser la ingesta que con mayor frecuencia es insuficiente o nula. En este trabajo se presenta un estudio de los hábitos alimentarios de escolares, así como una intervención educativa teórica y práctica dentro del programa de Educación para la Salud en Atención Farmacéutica. Los resultados muestran que sólo el 36.6 % de los alumnos encuestados realiza un desayuno completo. La actividad educativa teórico-práctica consigue introducir cambios en los hábitos del desayuno y en las opiniones de los escolares intervenidos. Palabras clave: Atención Farmacéutica, Desayuno Saludable, Educación para la Salud, Escolares, Intervención. Abstract: A Healthy Breakfast is the one that includes a balanced portion of every nutrient qualitatively and quantitatively. Although it should supply the 20-25 %calories of the day, it is usually insufficient or even absent. In this paper, we study the food habits of schoolchildren and we also participate in a health educational intervention of a Pharmaceutical Care Program with them. Only the 36.6 % of students have a healthy breakfast every day. The health education achieves favorable changes in the behavior and opinions of the schoolchildren participating. Keywords: Pharmaceutical Care, Healthy Breakfast, Health Education, Schoolchildren, Intervention 1. INTRODUCCIÓN La Educación para la Salud comprende las oportunidades de aprendizaje conscientemente destinadas a mejorar la alfabetización sanitaria. Esto incluye la mejora del conocimiento de la población y el desarrollo de habilidades personales que conduzcan a la mejora de la salud. Supone, por tanto, una función importante de los profesionales sanitarios, sociales y de la educación. Asimismo, constituye una pieza fundamental en el proceso asistencial, que incluye Prevención, Tratamiento y Rehabilitación [1]. La alimentación durante la edad pediátrica tiene una gran transcendencia en la proyección de la calidad de vida del adulto, ya que una alimentación inadecuada por si sola puede ser un factor de riesgo [2]. Dentro de ésta, el desayuno es la comida más importante para el desarrollo físico e intelectual de los niños [3]. El desayuno debería proporcionar el 2025 % del aporte energético diario, sin embargo, suele ser la ingesta que con mayor frecuencia es insuficiente o nula [4]. Un desayuno puede incluir toda clase de alimentos, aunque la cultura alimentaria de una población marca las costumbres y selecciona las propias de esa hora de la mañana [2]. Se considera Desayuno Saludable aquél que contiene una ración equilibrada de todos los nutrientes, tanto de forma cualitativa como cuantitativa [5]. Puede ser muy diverso, pero debe asegurar la ingesta de alimentos energéticos: hidratos de carbono y lípidos, estructurales: proteínas, y protectores: vitaminas y minerales. Se aconseja preferentemente la tríada formada por lácteos, cereales y frutas o zumos de fruta fresca, que se pueden complementar con otros alimentos proteicos como huevos, jamón, etc., hasta llegar al 20-25 % de las necesidades energéticas diarias [6]. Según la Organización Mundial de la Salud (OMS), la adolescencia comprende desde los 10 a los 19 años de edad, separando un primer periodo de preadolescencia (10-14 años de edad), que resulta ser una etapa crucial para el crecimiento de los niños [7]. Un desayuno bien equilibrado permite evitar los fallos energéticos que describen los profesores a lo largo de la mañana escolar. Un niño con déficit en el desayuno disminuye su atención y se dispersa. La falta de alimentación puede provocar cansancio, dolor de cabeza, desgana y somnolencia, con lo que disminuye el rendimiento intelectual [4]. La Educación para la Salud constituye una herramienta básica en la mejora sanitaria de las personas. El entorno escolar constituye un escenario muy importante durante la Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 26 *Morales F.; Marín F.; Morales L.; Morales I.; Marín L.; Gastelurrutia M. _______________________________________________________________________________________________________________________________ edad infantil para dicha Educación [8]. En las últimas décadas se han acumulado experiencias sobre la importancia que tiene una buena alimentación, especialmente durante las primeras etapas de la vida [9]. Por ello, en este trabajo se presenta un estudio de los hábitos en la alimentación de escolares de 11 a 13 años de edad, así como una intervención educativa teórica y práctica dentro del programa de Educación para la Salud. El objetivo del estudio es conocer el patrón de desayuno de los escolares de Quinto y Sexto Curso de Educación Primaria (11-13 años de edad), así como los factores que influyen en su conducta alimentaria y fomentar el Desayuno Saludable para adecuar su dieta. 2. METODOLOGÍA bollos, mermelada, pan, bizcocho, huevo, galletas y embutidos. El grupo C está formado por las frutas, tanto en piezas como en zumos naturales. La información de esta variable dependiente se obtiene a través de la entrevista mediante cuestionario. Las variables independientes que se han estudiado en función del Desayuno Saludable son: sexo, influencia del fin de semana, patrón socio-familiar, postura corporal, horario, patrón alimentario durante recreo, madre trabajadores, convivencia con abuelos y frecuencia de desayuno con abuelos. Además se evaluaron las siguientes variables durante el segundo cuestionario tras la intervención educativa: actividades realizadas, cambios en hábitos y conocimientos teóricos adquiridos. 2.1 Diseño del Estudio 2.6 Método De tipo transversal, descriptivo y observacional. Se utiliza un cuestionario previamente publicado [10] y adaptado al estudio que incluye preguntas tanto abiertas como cerradas, permitiendo así un análisis cuantitativo y cualitativo de los resultados. 2.2 Características Sociodemográficas Se estudian los escolares de un Municipio de Murcia situado a 18 Km de distancia de la capital. Este Municipio cuenta con una población de aproximadamente 9000 habitantes, que se ha visto incrementada en los últimos años por población de inmigrantes procedentes en su mayoría de países hispanoamericanos y de la antigua Unión Soviética, población que se encuentra en general bien integrada con la población autóctona. 2.3 Población de Estudio Escolares matriculados de Quinto y Sexto Curso de Educación Primaria en un Municipio de la Región de Murcia (11 -13 años de edad, 123 alumnos matriculados), previa visita y autorización por escrito. Se recogen los datos sobre hábitos de consumo en el desayuno tanto a los escolares a través de cuestionarios [10] que se hicieron un día sin previo aviso en el colegio, como a sus padres a través de una carta con información del programa y citación para una reunión para impartir Educación Sanitaria relacionada con el tema. También se citó a los abuelos para el mismo objetivo. La Educación Sanitaria dirigida a escolares se realizó en el Colegio y dentro del horario escolar. La Educación Sanitaria dirigida a padres y abuelos también se realizó en el Colegio, pero en horario accesible para los padres trabajadores, coincidiendo con el fin de la jornada laboral. Se realizó un “Taller de Preparación al Desayuno Saludable” donde se citó a los escolares, padres y abuelos con el fin de elaborar recetas tradicionales de productos típicos de la Región de Murcia. En esta actividad colaboraron los escolares conjuntamente con los padres y abuelos que pudieron acudir. Al día siguiente, se llevó a cabo la actividad de “El día del Desayuno Saludable”, donde los escolares desayunaron en el colegio, junto con los padres y abuelos que pudieron asistir. 2.4 Confidencialidad de los Datos Solamente se hacen públicos los resultados de forma global. Con anterioridad al estudio, se solicita el consentimiento informado del Centro Educativo, padres y escolares incluidos en el estudio tras haberles informado de la finalidad, contenido de las encuestas y protocolo a seguir. 2.5 Definición de Variables Las variables necesarias para el estudio deben contener información relativa a la variedad y composición de los alimentos (variable dependiente). Dicha variable dependiente hace referencia al Desayuno Saludable, que es aquél que contiene alimentos de los grupos A, B y C. El grupo A incluye alimentos de tipo lácteo: queso, yogurt, leche entera, semidesnatada y desnatada. El grupo B incluye cereales, La Intervención Sanitaria se evaluó a través de un cuestionario, cumplimentado 1 mes después de la actividad. 2.7 Criterios de Inclusión y Exclusión Criterios de Inclusión: Estar matriculados en el Centro Educativo en los niveles de Quinto y Sexto Curso de Educación Primaria. Criterios de Exclusión: No acudir a clase el día del trabajo de campo y/o no mostrarse dispuesto a colaborar respondiendo el cuestionario. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 27 Atención Farmacéutica en el Fomento del Desayuno Saludable desde la Oficina de Farmacia _______________________________________________________________________________________________________________________________ 2.8 Análisis Estadístico en el desarrollo de los hábitos saludables, el hecho de disfrutar de la compañía familiar en el acto de comer. Se han utilizado los programas Excel y SPSS para el cálculo de frecuencias simples y tablas de contingencia. 3. RESULTADOS Y DISCUSIÓN Un 90.2 % de los niños encuestados afirma consumir alimentos del Grupo A, siendo la leche (87.8 %) el preferido por la mayoría de ellos. Los varones (95.7 %) se declaran consumidores más habituales de alimentos del Grupo A que las niñas (77.8 %), siendo un dato significativo ya que casi dos de cada diez niñas (16.7 %) encuestadas afirma no desayunar leche. Con respecto a los alimentos del Grupo B, se observa que un 70.7 % de los encuestados consume este tipo de alimentos, cifra superior a la obtenida en anteriores estudios [2, 11]. Este alto porcentaje de alimentos del grupo B permite afirmar que el desayuno de los escolares se aproxima a un desayuno adecuado, ya que la leche y los alimentos del grupo B se complementan con sus aminoácidos esenciales, obteniéndose proteínas de mayor valor biológico. No obstante, sería deseable que el porcentaje de encuestados que no consume alimentos del grupo B (29.3 %) fuese menor. Los alimentos del Grupo C fueron consumidos por el 46.3 % de los encuestados, a expensas de los zumos naturales (36.6 %) y de las piezas de fruta (9.8%). Estos porcentajes, aunque sean insuficientes, son superiores a los obtenidos en otros trabajos anteriores [3, 6], lo que resulta esperanzador ya que la inclusión de fruta y zumos en el desayuno es fundamental para el aporte de vitaminas, antioxidantes y fibras, ayudando a prevenir diversos problemas de Salud. Se podría considerar que el hecho de que el Colegio se encuentre ubicado en un Municipio de la huerta murciana influya en el saludable hábito del consumo de fruta. Sin embargo, sólo el 36.6 % de los alumnos encuestados realiza un “desayuno completo” (Fig. 1), entendiendo como tal, aquél que incluye alimentos de los grupos A, B y C. Cabe destacar que aunque el dato es bajo, resulta bastante superior al que presentan otros estudios, como el denominado enKid [12]. El 73.2 % de los encuestados responde desayunar más cantidad los fines de semana, lo que puede atribuirse a una mayor disponibilidad de tiempo. Este porcentaje es superior al encontrado en otros estudios, cuyos autores observan una correlación directa entre el número de alimentos consumidos y el tiempo empleado en desayunar [13]. Al analizar cómo influye el desayuno en familia en los hábitos de consumo, se observa que el 63.4 % de los escolares desayuna habitualmente en soledad, mientras que se destaca que el 40 % de los niños que desayunan en familia realizan un desayuno completo. Estos resultados ayudan a remarcar la importancia que tiene para los preadolescentes, Figura 1. Alimentos consumidos durante el desayuno en los escolares. Significación de la χ2: (**): p <0.05; (***) p <0.01. Además, se encuentra que el 22 % de los alumnos declara desayunar de pie, mientras que el 39 % que desayuna sentado realiza un desayuno completo. Este hecho confirma que la falta de tiempo para desayunar va en detrimento de los hábitos saludables. En cuanto a la hora del desayuno y el tiempo dedicado al mismo, un 9.7 % de los estudiantes desayunan después de las 8.30 am (el Colegio comienza a las 9.00 am), no teniendo el tiempo necesario para un desayuno tranquilo y saludable. Aproximadamente la mitad de los encuestados (53.7 %) respondieron que su madre trabajaba fuera de casa de forma remunerada. Cabe mencionar que el 40.9% de los escolares con madre trabajadora consume un desayuno completo. Al analizar los cambios experimentados en los desayunos de los escolares tras la intervención educativa (Fig. 2), se aprecia que el 50 % han introducido en su dieta algún alimento del grupo B. El 25 % declara haber introducido alimentos de los grupos A y B, y el 17 % alimentos de los grupos B y C. Cuando se investiga sus pensamientos acerca de la conveniencia de desayunar equilibradamente mediante preguntas abiertas, una amplia mayoría (63.6 %) responde obtener así un mayor rendimiento escolar (Tabla 1). También se examinaron los aspectos positivos que los alumnos destacaban de la intervención educativa práctica “El día del Desayuno Saludable”, en la que se destaca que la mayoría (44 %) resaltan el placer derivado de desayunar acompañado por sus padres, abuelos y resto de compañeros. El 16 % respondió sentirse satisfechos por un desayuno variado y equilibrado. Al 14 % le gusto poder tomar los dulces tradicionales murcianos. Un 10 % destacó como aspecto positivo el poder desayunar tranquilos y sin prisas. El tomar fruta fue resaltado como aspecto satisfactorio por un 9 % y un 7 % respondió que sencillamente le había gustado todo. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 28 *Morales F.; Marín F.; Morales L.; Morales I.; Marín L.; Gastelurrutia M. _______________________________________________________________________________________________________________________________ REFERENCIAS [1] [2] [3] Figura 2. Alimentos añadidos a la dieta tras la intervención educativa. Tabla 1. Por qué piensan los escolares que el desayuno saludable es conveniente. Conveniencia de Desayunar Adecuadamente % Mayor Rendimiento Escolar 63.6 Cuidado de Nuestra Salud 15.9 Dieta Equilibrada 13.6 Otras 6.9 [4] [5] [6] Por último se investigó si los alumnos habían aprendido algo nuevo tras la intervención (Fig. 3). El 38 % de los escolares responde haber aprendido la importancia de desayunar bien, entendiendo el desayuno como la comida más importante del día. Un 29 % declara haber descubierto que en la composición del desayuno hay una importante variedad de alimentos y el 22 % ha comprendido la importancia de tomar fruta en el desayuno. [7] [8] [9] [10] [11] [12] [13] Figura 3. Conocimientos adquiridos por los escolares tras la intervención educativa. Ministerio de Sanidad y Consumo. Promoción de la Salud. Madrid, OMS-Glosario; 1999. P. Girón Barranco (1998). “El desayuno” es toda una comida. Experiencia con alumnos de educación infantil en un colegio publico de un barrio marginal de Bilbao. Enferm Cient. 192-193, pp. 13-16. P. Agudo Matarán, M. D. Montore Sánchez, A. M. Aranda Marín. (1998). Hábitos alimentarios en el desayuno y recreo de los alumnos de Primaria. Cent Salud. 6(3), pp. 157-160. L. Mur Frenne, J. Fleta Zaragozano. (1991). Importancia del desayuno en los niños. Enferm Cient. 115, pp. 7-10. Ministerio de Sanidad y Consumo. Diseño y planificación de dietas saludables. Nutrición saludable y prevención de los trastornos alimentarios. Madrid, 1999, pp. 55. P. Cervera. (2004). Comer con inteligencia. Rev Enferm. 27 (2), pp. 6-10. J. C. Surís, N. Parera, C. Puig. (2000). Encuesta de Salud als adolescents de la ciutat de Barcelona 1999. Barcelona, Fundación Santiago Dexeus Font. J. Cruzado Quevedo,F. Bravo Vicente,L. V. Marín Rives,M. Gea Navarro, F. A. Martínez García, M. J. Lázaro Gómez. (1994). Consumo de alcohol entre escolares de séptimo de EGB. Atención primaria.13(9),pp. 495-497. E. Romero-Velarde, O. Campollo-Rivas, A. Celis de la Rosa, E. M. Vásquez-Garibay, J. F. Castro-Hernández, R. M. Cruz-Osorio. (2007). Factores de riesgo de dislipidemia en niños y adolescentes con obesidad. Salud Pública Méx. [Online].49(2), pp. 103-108.Available: http://bvs.insp.mx/rsp/articulos/articulo.php?id=001958. Ministerio de Sanidad y Consumo. Propuestas de actividades practices. Nutrición saludable y prevención de los trastornos alimentarios. Madrid, 1999, pp. 41-46. V. Arija Val, J. Salas Salvadó, J. Fernández Ballart, G. Cucó, C. Martí Henneberg. (1996). Consumo, hábitos alimentarios y estado nutricional de la población de Reus : evolución del consumo de alimentos, de su participación en la ingestión de energía y nutrientes y de su relación con el nivel socioeconómico y cultural entre 1983 y 1993. Medicina clínica. 106(5), pp. 174179. J. Aranceta, L. Serra. Desayuno y equilibrio alimentario: Estudio enKid. Barcelona, Masson, 2000. R.M. Ortega, A.M. Requejo, R. Redondo, A.M. López-Sobaler, P. Andrés, A. Ortega, E. Quintas, M. Izquierdo. (1996). Breakfast habits of different groups of Spanish schoolchildren. Journal of Human Nutrition and Dietetics. [Online] 9, pp. 33–41. Available: 10.1046/j.1365-277X.1996.00434.x. 4. CONCLUSIONES Del estudio realizado se derivan las siguientes conclusiones: El desayuno de los escolares de Quinto y Sexto Curso de Educación Primaria presenta desequilibrios cuantitativos y cualitativos. Existe una relación directamente proporcional entre el consumo de un Desayuno Saludable y el tiempo dedicado a su ingesta, su consumo en familia, y el hecho de que la madre trabaje de forma remunerada fuera del hogar. La Educación para la Salud mediante intervención educativa teórica y práctica favorece los cambios positivos en el patrón de desayuno de estos escolares. Es del todo justificado invertir esfuerzos potenciando la Educación para la Salud en los Centros Educativos. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 29 Cuantificación de Ca, Mg, K, Na y Mn en Subproductos de Cítricos procedentes de Ecuador _____________________________________________________________________________________________________________________________ Cuantificación de Ca, Mg, K, Na y Mn en Subproductos de Cítricos procedentes de Ecuador Martínez S.*; Figueroa J. ** *Universidad Técnica Particular de Loja, Carrera de Ingeniería en Alimentos, Loja, Ecuador e-mail: [email protected] **Universidad Técnica Particular de Loja, Sección de Ciencias y Tecnología de Alimentos, Loja, Ecuador e-mail: [email protected] Resumen: Los subproductos de cítricos son fuentes importantes de nutrientes y componentes bioactivos; siendo estas características las que promueven su uso para el enriquecimiento de alimentos procesados. El objetivo de este trabajo fue cuantificar el contenido de calcio (Ca), magnesio (Mg), potasio (K), sodio (Na) y manganeso (Mn) en subproductos frescos de naranja, mandarina y limón; mediante espectrofotometría de absorción atómica. En la naranja se presentó el mayor contenido de Ca y Mg (970.83 ± 118.07 mg/100 g y 115.58 ± 15.97 mg/100 g, respectivamente), mientras que se encontró mayor concentración de K en naranja y mandarina (916.84 ± 15.90 mg/100 g y 1081.99 ± 56.50 mg/100 g, respectivamente). En el limón se evidenció el mayor contenido de Na (41.56 ± 3.80 mg/100 g) y únicamente en la naranja se encontró Mn comprendido entre 0.30 ± 0.04 mg/100 g y 0.68 ± 0.10 mg/100 g. El lugar de cultivo tuvo efecto sobre la concentración de minerales. Palabras clave: Subproductos de cítricos, minerales, compuestos bioactivos. Abstract: Citrus by-products are important sources of nutrients and bioactive compounds that is the reason why these by-products could be used to enrich processed food. The aim of this investigation was quantify the concentration of calcium (Ca), magnesium (Mg), potassium (K), sodium (Na) and manganese (Mn) in fresh byproducts of orange, mandarin and lemon; using an atomic absorption spectrophotometer. The orange showed the highest content of Ca and Mg (970.83 ± 118.07 mg/100 g and 115.58 ± 15.97 mg/100 g, respectively), while the K concentration was higher in orange and mandarin (916.84 ±15.90 mg/100 g and 1081.99 ± 56.50 mg/100 g, respectively). The lemon evidenced the highest content of Na (41.56 ± 3.80 mg/100 g) and only in the orange was founded Mn in a range between 0.30 ± 0.04 mg/100 g and 0.68 ± 0.10 mg/100 g. The concentration of mineral depends for the place. Keywords: Citrus by-products, minerals, bioactive compounds. _____________________________________________________________________________________________ 1. INTRODUCCIÓN compuestos valiosos que proceden de desechos del procesamiento de alimentos [6]. Durante el proceso de elaboración de jugos de cítricos, se genera una alta cantidad de subproductos [1]; considerando que aproximadamente el 60 % del peso del fruto corresponde a piel, semillas y pulpa, se producen aproximadamente 20 millones de toneladas de subproductos a nivel mundial por año [2]. La eliminación de residuos constituye un problema cada vez mayor en la industria y una posible fuente de contaminación del medio ambiente; estos al ser materia vegetal son propensos a deterioro microbiano; además, los costos de secado, almacenamiento y transporte son limitantes económicos [3, 4], por lo que hasta hace poco su uso se limitaba para alimentación de animales, producción de pectinas y combustibles [5]. En los últimos años se han desarrollado métodos para el manejo y tratamiento de subproductos; recuperando, biotransformando y utilizando Investigaciones previas indican que los subproductos de cítricos contienen cantidades importantes de azúcares solubles, fibra, ácidos orgánicos, aminoácidos, aceites esenciales, flavonoides, compuestos fenólicos, vitaminas y algunos minerales esenciales en la dieta humana [2, 5, 7], convirtiéndolos en fuentes promisorias de componentes que pueden ser reutilizados en la elaboración de alimentos por sus valiosas propiedades tecnológicas y nutricionales [8]; además proveen sustancias químicas y materiales de alto interés para la industria química, textil, cosmética moderna y farmacéutica [9]. Actualmente se están desarrollando investigaciones del uso de subproductos para la obtención de aceites esenciales, limoneno, terpenos, líquidos aromáticos, pellets de pulpa de Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 30 Martínez S.*; Figueroa J.* _______________________________________________________________________________________________________________________________ cítricos, etanol, sabores y fragancias, sustancias para la industria de pinturas y cosméticos y suplementos de alimentación animal [10]. Entre los beneficios de los subproductos de cítricos, encontramos que estos tienen mayor calidad que otras fuentes de extracción de fibra dietaria, debido a la presencia de compuestos como flavonoides, vitamina C y minerales; es por eso que se ha desarrollado investigaciones para la aplicación de estos subproductos en la producción de fibra dietaria que puede ser utilizada en la formulación de productos de panadería, lácteos y cárnicos, dándoles un valor agregado no solo por sus beneficios en cuanto a minerales, vitamina C y compuestos antioxidantes, sino también por sus propiedades tecnológicas, funcionales, nutricionales y su efecto benéfico sobre la salud [11]. Los minerales son fundamentales para regular procesos fisiológicos en los seres humanos; más de un tercio de todas las proteínas humanas requieren iones metálicos para su funcionamiento, la carencia de éstos puede representar un impacto significativo en la salud humana [13]. El Ca, P, Mg, S, K y Na son minerales esenciales requeridos por el hombre en cantidades superiores a 100 mg por día y constituyen 1 % o menos del peso corporal [14]. Los minerales como Zn, Fe, Si, Mn, Cu, I y Cr, que representan menos del 0,01 % del peso corporal y su consumo es menor a 100 mg por día son llamados oligoelementos [14]. Además el Ca, Zn y Mg son elementos que contribuyen en procesos enzimáticos que representan una parte integral de la estructura del sistema de auto reparación del ADN, específicamente, el Ca es necesario para la segregación de cromosomas, el Zn es requerido para la síntesis y reparación de ADN y el Mg para la síntesis de ADN y la segregación cromosómica [17]. El Ca y Mg como minerales esenciales para la nutrición humana tienen un valor diario recomendado establecido. Para adultos la ingesta recomendada es 800 mg de Ca/día, para mujeres embarazadas y lactantes 1200 mg de Ca/día y para niños 60 mg de Ca/kg de peso corporal. De Mg se recomienda 300 mg/día para mujeres, 350 mg/día para hombres, 150 mg/día para mujeres embarazadas y que se encuentren en periodo de lactancia y 50 mg/día para infantes [15, 16, 18]. Para prevenir la carencia de micronutrientes existen alimentos enriquecidos con minerales como el Ca, Mg, K, Na y Mn, estos cuatro últimos poseen un rol importante en el proceso de asimilación del Ca [15, 16]. Muchas frutas y verduras tienen en su composición minerales y compuestos fenólicos, que desempeñan un papel importante en su valor nutritivo [12]. El contenido de minerales en frutos cítricos y sus subproductos han sido reportados por algunas investigaciones [3, 4, 5, 11], sin embargo, no se han encontrado estudios sobre la concentración de minerales en subproductos de cítricos de origen ecuatoriano. Por ello, el objetivo de este estudio fue cuantificar el contenido de minerales en subproductos de limón, naranja y mandarina procedentes de distintas regiones del Ecuador, con el interés de potenciar su uso para el enriquecimiento de alimentos. 2. METODOLOGÍA 2.1 Materia Prima Los subproductos (albedo, flavedo y pulpa agotada) fueron obtenidos del proceso de extracción de jugos de naranja (Citrus sinensis var. Criolla), mandarina (Citrus nobilis var. King) y limón (Citrus aurantifolia var. Sutil). La materia prima procedente de cultivos ecuatorianos, en el caso de la naranja de Quinsaloma Playa, Quinsaloma Cerro y Quevedo pertenecientes a la provincia de Los Ríos; la mandarina de Quinsaloma, Quevedo y Chone, esta última pertenece a la provincia de Manabí y el limón de La Vega, Boquerón y Catamayo de la provincia de Loja. 2.2 Cuantificación de Minerales Las muestras fueron preparadas siguiendo el método AOAC 970.39; cada muestra fue incinerada a una temperatura de 500ºC, las cenizas obtenidas fueron tratadas con una solución ácido clorhídrico: agua (1:9). Para producir digestión de la muestra y disolver todos los sólidos presentes [19]. Se utilizó los métodos oficiales de la AOAC 929.07, 931.10, 965.30, 966.16 y 931.09 para cuantificar Ca, Mg, K, Na y Mn, respectivamente [19]. Los análisis se realizaron empleando el espectrofotómetro de Absorción Atómica (marca Perkin Elmer ®, modelo AAnalyst 400). Las curvas de calibración se prepararon utilizando estándares certificados de la casa comercial MERCK. 2.3 Análisis Estadístico Los datos fueron expresados como medias ± desviación estándar de tres réplicas. Con la finalidad de determinar las diferencias entre las medias se aplicó un Análisis de Varianza (ANOVA) y prueba de Tukey. Se consideró p < 0,05 como estadísticamente significativo. Se utilizó el paquete estadístico Minitab® Statistical Software, versión 16 (Minitab Inc., State College, PA, USA). 3. RESULTADOS Y DISCUSIÓN Se cuantificó cinco minerales, de los cuales cuatro son esenciales (Ca, K, Mg, Na) y uno es oligoelemento (Mn). En el estudio se comparó los subproductos de cítricos procedentes de diferentes regiones de Ecuador, por ello es importante mencionar que la composición química de las frutas varía en función al tipo de suelo, condiciones del tiempo durante el cultivo, fertilización aplicada, estado de madurez en la cosecha, fracción de la fruta y su variedad [5, 20]. El contenido de Ca en los subproductos estuvo comprendido entre 457.85 ± 76.68 mg/100 g y 541.35 ± 33.97 mg/100 g para el limón, 362.20 ± 38.73 mg/100 g y 531.93 ± 23.80 mg/100 g en mandarina y de 656.49 ± 51.54 mg/100 g a 970.83 ± 118.07 mg/100 g en naranja. Al comparar la Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 31 Cuantificación de Ca, Mg, K, Na y Mn en Subproductos de Cítricos procedentes de Ecuador _____________________________________________________________________________________________________________________________ concentración de Ca, se encontró que el subproducto de naranja procedente de Quinsaloma Playa tuvo el mayor contenido (970.83 ± 118.07 mg/100 g), por lo que se puede considerar como “fuente de Calcio” para adultos. Mientras que el contenido de Ca de las muestras de naranja procedentes de Quinsaloma Cerro y Quevedo no presentaron diferencia significativa (Ver Fig. 1). El menor contenido de Ca se encontró en la mandarina de Chone y Quevedo; por lo que se puede considerar que el ácido cítrico presente en estas frutas actúa como agente quelante, aumentando la absorción de Ca [5]. Los subproductos estudiados presentaron un mayor contenido de Ca que fuentes tradicionales de este mineral como la leche (124 mg/100 g) [15], espinaca (90 mg/100 g), garbanzo (145 mg/100 g) [21], y que el jugo de naranja (90 mg/100 g) [20]. Se encontró que los subproductos de naranja procedentes de Quinsaloma Playa y Quinsaloma Cerro tuvieron el mayor contenido de éste mineral y no existe una diferencia significativa entre ellos (115.58 ± 16 mg/100 g y 105.73 ± 14 mg/100 g, respectivamente), considerándolos como “fuente de Magnesio”, debido a que una poción de 100 g aportaría el 40 % del valor requerido en la dieta humana. El contenido de Mg en limón y naranja es mayor al de verduras como la espinaca (54 mg/100 g) pero menor al contenido en frutos secos como almendra (258 mg/100 g) y legumbres como garbanzo (160 mg/100 g) [21]. Según [20] el contenido de Mg en el jugo de naranja es 90 mg/100 g, así que la concentración es mayor en los subproductos. 140 1200 1000 Limón Mandarina Naranja A Limón Mandarina Naranja A 120 A 100 B 800 BC CD 600 DE 80 B DE CDE B 60 B B BC DE E 400 40 C C 20 200 0 0 Figura 1. Contenido de Ca en subproductos de limón, mandarina y naranja (mg/100 g). Valores marcados con diferentes letras tienen diferencia significativa (p < 0.05). El Mg está presente en las mitocondrias y es esencial para la función de enzimas relacionadas con la transferencia de grupos fosfato, todas las reacciones que requieren ATP y cada paso relacionado con la replicación y transcripción del ADN [5, 22]. Los subproductos de limón mostraron un contenido de Mg comprendido entre 58.58 ± 0.25 mg/100 g y 62.08 ± 1.51 mg/100 g, los de naranja entre 55.48 ± 4.45 mg/100 g y 115.58 ± 15.97 mg/100 g y los provenientes de mandarina tuvieron el menor contenido entre 26.32 ± 2.15 mg/100 g y 44.05 ± 4.38 mg/100 g. Figura 2. Contenido de Mg en subproductos de limón, mandarina y naranja (mg/100 g). Valores marcados con diferentes letras tienen diferencia significativa (p < 0.05). El menor contenido se evidenció en los subproductos de mandarina procedentes de Quinsaloma y Chone, no hubo diferencia significativa entre ellos (26.32 ± 2 mg/100 g y 24.21 ± 4 mg/100 g, respectivamente) (Ver Fig. 2). El mayor contenido de K se encontró en los subproductos de naranja (entre 598.76 ± 84.46 mg/100 g y 916.84 ± 15.90 mg/100 g) y en los de mandarina procedentes de Chone (1081.99 ± 56.50 mg/100 g), estos presentaron una diferencia significativa con los demás subproductos (Ver Fig. 3); en los de limón el contenido varió entre 261.95 ± 12.45 mg/100 g y 512.31 ± 29.39 mg/100 g. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 32 Martínez S.*; Figueroa J.* _______________________________________________________________________________________________________________________________ Todos los subproductos tuvieron mayor contenido de K que la leche (150 mg/100 g), en el limón y mandarina el contenido es menor contenido en relación a las almendras (860 mg/100 g) y garbanzo (797 mg/100 g) [21]. Según [4] el contenido de K en la piel de (albedo y flavedo) de mandarina es 6.41 ± 0.02 mg/100 g, es decir, que la mayor concentración de K se encuentra en la pulpa de la mandarina. 1200 1000 ± 14.00 µg/100 g. De acuerdo a las recomendaciones de Mn para la dieta humana, los subproductos de naranja pueden ser considerados “fuentes de Manganeso”. En los subproductos de limón y mandarina no se encontró Mn. 80 70 A Limón Mandarina Naranja B 800 C CD C B 40 BC BCD 30 CD CDE DE 400 A 60 B 50 600 Limón Mandarina Naranja DEF 20 EF F 10 200 EF F 0 0 Figura 4. Contenido de Na en subproductos de limón, naranja y mandarina (mg/100 g). Valores marcados con diferentes letras tienen diferencia significativa (p < 0.05). Figura 3. Contenido de K en subproductos de limón, naranja y mandarina (mg/100 g). Valores marcados con diferentes letras tienen diferencia significativa (p < 0.05). La relación entre K y Na presente en los cítricos juega un papel importante en el equilibrio de los electrolitos de las células del cuerpo humano. Los subproductos de limón poseen un contenido de Na comprendido entre 20.08 ± 2.55 mg/100 g y 41.56 ± 3.80 mg/100 g, en mandarina el contenido estuvo comprendido entre 5.46 ± 1.67 mg/100 g y 57.68 ± 11.02 mg/100 g y en naranja entre 16.98 ± 4.83 mg/100 g y 36.16 ± 3.05 mg/100 g. El mayor contenido de Na estuvo en los subproductos de mandarina procedentes de Quevedo, mostrando diferencia significativa con Quinsaloma y Quevedo, en los subproductos de limón y naranja se evidencia una diferencia significativa entre todos los lugares de procedencia (Ver Fig. 4). Algunos oligoelementos son componentes de enzimas antioxidantes, entre ellos el Mn [5]. Según [20] el contenido de este mineral en jugo de naranja es 0.30 mg/100 g, mientras que en los subproductos se evidenció un mayor contenido comprendido entre 0.30 ± 0.04 mg/100 g y 0.68 ± 0.10 mg/100 g, similar al determinado por [5] que es 339.50 Entre los elementos medidos el que tiene mayor presencia en los subproductos de cítricos es el Ca, seguido de K y Mg. Además el albedo, flavedo y pulpa agotada tuvieron mayor contenido que fuentes tradicionales [9]. De manera general el contenido de minerales fue mayor en los subproductos de naranja, seguidos por mandarina y limón. Como fue mencionado el sitio de procedencia de la fruta, el tipo de suelo, condiciones del tiempo durante el cultivo, fertilización aplicada, estado de madurez en la cosecha y otros factores influyen en la composición química de las frutas, por lo que se puede atribuir a estas condiciones, las diferencias encontradas en la concentración de minerales de los subproductos estudiados, los subproductos procedentes de la provincia de Los Ríos poseen el mayor contenido de minerales. 4. CONCLUSIONES Los subproductos de naranja, mandarina y limón procedentes de distintas provincias del Ecuador son fuentes considerables de minerales esenciales como Ca, Mg, K y Na, y pueden ser aprovechados en la industria para fortificar alimentos y Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 33 Cuantificación de Ca, Mg, K, Na y Mn en Subproductos de Cítricos procedentes de Ecuador _____________________________________________________________________________________________________________________________ bebidas, considerando que no sólo aportan cantidades importantes de minerales sino también otros componentes de interés (flavonoides, vitamina C, fibra, azúcares solubles, compuestos fenólicos, ácidos orgánicos, aminoácidos, entre otros). Debido a sus valiosas propiedades, en la actualidad, también están siendo aprovechados en la industria química, cosmética, farmacéutica, otras. Únicamente los subproductos de naranja presentaron contenido destacable de Mn, su concentración de minerales fue mayor a la de los de mandarina y limón. El lugar de cultivo tuvo efecto sobre la concentración de minerales. REFERENCIAS [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] K. Hayat, X. Zhang, H. Chen, S. Xia, C. Jia, y F. Zhong, "Liberation and separation of phenolic compounds from citrus mandarin peels by microwave heating and its effect on antioxidant activity," Separation and Purification Technology. vol. 73, no. 3. pp. 371-376, 2010. J. Londoño Londoño, J. Sierra, R. Álvarez, A.M. Restrepo Duque, y C.P. Pássaro Carvalho, "Aprovechamiento de los subproductos citrícolas." vol., no. 2012. F.R. Marín, C. Soler-Rivas, O. Benavente-García, J. Castillo, y J.A. Pérez-Alvarez, "By-products from different citrus processes as a source of customized functional fibres," Food Chemistry. vol. 100, no. 2. pp. 736-741, 2007. G. Xu, J. Chen, D. Liu, Y. Zhang, P. Jiang, y X. Ye, "Minerals, phenolic compounds, and antioxidant capacity of citrus peel extract by hot water," Journal of food science. vol. 73, no. 1. pp. C11-C18, 2008. H. Barros, T. Ferreira, y M. Genovese, "Antioxidant capacity and mineral content of pulp and peel from commercial cultivars of citrus from Brazil," Food Chemistry. vol. 134, no. 4. pp. 18921898, 2012. G. Laufenberg, B. Kunz, y M. Nystroem, "Transformation of vegetable waste into value added products: (A) the upgrading concept;(B) practical implementations," Bioresource Technology. vol. 87, no. 2. pp. 167-198, 2003. S. Gorinstein, O. Martı́n-Belloso, Y.-S. Park, R. Haruenkit, A. Lojek, M. Ĉı́ž, A. Caspi, I. Libman, y S. Trakhtenberg, "Comparison of some biochemical characteristics of different citrus fruits," Food Chemistry. vol. 74, no. 3. pp. 309-315, 2001. J. Fernández-López, J. Fernández-Ginés, L. Aleson-Carbonell, E. Sendra, E. Sayas-Barberá, y J. Pérez-Alvarez, "Application of functional citrus by-products to meat products," Trends in Food Science & Technology. vol. 15, no. 3. pp. 176-185, 2004. F. Fava, G. Zanaroli, L. Vannini, E. Guerzoni, A. Bordoni, D. Viaggi, J. Robertson, K. Waldron, C. Bald, y A. Esturo, "New advances in the integrated management of food processing byproducts in Europe: sustainable exploitation of fruit and cereal processing by-products with the production of new food products (NAMASTE EU)," New biotechnology. vol. 30, no. 6. pp. 647655, 2013. R.J. Braddock, "Handbook of citrus by-products processing technology." vol., no. 1999; A. Moure, J.M. Cruz, D. Franco, J.M. Domı́nguez, J. Sineiro, H. Domı́nguez, M.a.J. Núñez, y J.C. Parajó, "Natural antioxidants from residual sources," Food chemistry. vol. 72, no. 2. pp. 145-171, 2001. Y. Lario, E. Sendra, J. Garcı́, C. Fuentes, E. Sayas-Barberá, J. Fernández-López, y J. Perez-Alvarez, "Preparation of high dietary fiber powder from lemon juice by-products," Innovative Food Science & Emerging Technologies. vol. 5, no. 1. pp. 113117, 2004. S.H. Mirdehghan y M. Rahemi, "Seasonal changes of mineral nutrients and phenolics in pomegranate (Punica granatum L.) fruit," Scientia Horticulturae. vol. 111, no. 2. pp. 120-127, 2007. I. Bertini y A. Sigel, Handbook on metalloproteins: CRC Press, 2001, pp. 86-90. [14] M. Özcan, "Mineral contents of some plants used as condiments in Turkey," Food chemistry. vol. 84, no. 3. pp. 437-440, 2004. [15] P. Cervera, J. Clapés, y R. Rigolfas, Alimentación y dietoterapia, ed. edición, España: McGRAW- HILL, 2004, pp. 39-47, 136-139. [16] M.J. Iglesias, A.P. Alejandre, y J.C.E. de Gea, Alimentos saludables y de diseño específico: alimentos funcionales: Instituto Tomás Pascual Sanz, 2010, pp. 119-120. [17] I. Konczak y P. Roulle, "Nutritional properties of commercially grown native Australian fruits: Lipophilic antioxidants and minerals," Food Research International. vol. 44, no. 7. pp. 23392344, 2011. [18] C.M. Weaver y R.L. Chaney, "Intrinsic mineral labeling of edible plants: methods and uses," Critical Reviews in Food Science & Nutrition. vol. 23, no. 1. pp. 75-101, 1985. [19] AOAC, "Fruits and Fruit Products," in Oficial Methods of Analysis of AOAC International, 18 ed. Gaithersburg: AOAC INTERNATIONAL, 2007. [20] P. Ekholm, H. Reinivuo, P. Mattila, H. Pakkala, J. Koponen, A. Happonen, J. Hellström, y M.-L. Ovaskainen, "Changes in the mineral and trace element contents of cereals, fruits and vegetables in Finland," Journal of Food Composition and Analysis. vol. 20, no. 6. pp. 487-495, 2007. [21] O.M. Tuni, Á. Carbajal, L.C. Forneiro, y C.C. Vives, Tablas de composición de alimentos: Ediciones Pirámide, 2013, pp. 56-57, 80-81, 88-89, 92-93, 100-101. [22] J.R. Weisinger y E. Bellorín-Font, "Magnesium and phosphorus," The Lancet. vol. 352, no. 9125. pp. 391-396, 1998. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Factores que Influyen en la Composición de la Leche en el Sector el Retorno, Parroquia Sabanilla, Cantón Zamora, Provincia de Zamora Chinchipe Ecuador 34 ______________________________________________________________________________________________________ Factores que Influyen en la Composición de la Leche en el Sector el Retorno, Parroquia Sabanilla, Cantón Zamora, Provincia de Zamora Chinchipe – Ecuador Fernández J. *; Tarazona G.** * Universidad Técnica Particular de Loja, Titulación de Ingeniería en Industrias Agropecuarias, Loja, Ecuador e-mail: [email protected] ** Universidad Nacional Agraria la Molina, Escuela de Post Grado, Lima, Perú e-mail: [email protected] Resumen: Se evaluó los efectos del mestizaje, ciclo de lactación y suplementos alimenticios en la composición de la leche de vacas del sector El Retorno, las cuales mantenían similares condiciones de manejo y alimentación basada en pasto gramalote (Axonopus scoparius). De acuerdo al mestizaje de las vacas, se agruparon en mínimo mestizaje (criollas), bajo mestizaje y mediano mestizaje con la raza Holstein. Los ciclos de lactación se establecieron hasta 3 meses, desde 4 hasta 6, y mayores a 6 meses. Los suplementos alimenticios consistieron en sal, sal y melaza, y el grupo sal y balanceado. Los análisis de la leche se realizaron mediante un equipo Lactoscanmarca Milkotronic, se determinó el contenido de grasa, proteína y sólidos totales. Los resultados obtenidos indican, que las tres variables influyen en la composición de la leche (p<0.05). El contenido de grasa de la leche proveniente de vacas de mediano mestizaje (4.30 %) fue significativamente mayor que la de vacas criollas (3.63 %), sin mantener diferencia significativa con las de bajo mestizaje (3.99 %), entre las vacas criollas y de bajo mestizaje no existió diferencia significativa en el porcentaje de proteína de la leche (3.07 % y 3.01 % respectivamente), la leche proveniente de vacas de mediano mestizaje obtuvo el menor contenido de proteína (2.89 %). La leche de vacas mayores a seis meses presento el mayor porcentaje de grasa (4.35 %) y mayor contenido de sólidos totales (11.60 %). La leche de vacas alimentadas con sal y melaza contenía un mayor porcentaje de grasa (4.42 %) y sólidos totales (11.90 %). Se concluye que el mestizaje, ciclo de lactación y suplementos alimenticios, influyen en la composición de la leche del hato ganadero del sector El Retorno. Palabras clave: Mestizaje, ciclo de lactación, suplementos alimenticios, grasa, proteína, sólidos totales. Abstract: The effects of crossbred, lactation cycle and food supplements were evaluated in the composition of milk from the cows that belong to El Retorno sector, which had similar handling and feeding conditions from gramalote grass (Axonopus scoparius). According to the crossbreeding of cows, they were classified as creole, crossbred low and crossbred medium. The lactation cycles were established from 0 to 3 months, from 4 to 6 and higher than 6 months. The food supplements consisted of salt, salt and molasses, and the salt and feed. The milk analyses were done through an ultrasonic milk analyzer (LACTOSCAN) Milcotronic brand, the content of milk fat, protein and total solids were determined. The results obtained showed that the three variables influence in the composition of milk (p<0.05). The fat content of the milk that comes from crossbreeding medium cows (4.30 %) was significantly higher than the creole cows (3.63 %); between the creole cows and the ones of crossbreeding low, there was not a significant difference in the percentage of milk protein (3.07 % and 3.01 % respectively); milk that comes from crossbreeding medium cows showed the lowest content of protein (2.89 %). The milk of cows older than six months presented the highest percentage of fat (4.35 %) and the highest of total solids content (11.60 %). The milk of cows fed with salt and molasses had a high percentage of fat (4.42 %) and total solids (11.90 %). It is concluded that crossbreeding, lactation cycle and food supplements influence the composition of milk from the cattle of El Retorno sector. Keywords: Crossbreed, lactation cycle, food supplements, milk fat, milk protein, milk solids. 1. INTRODUCCIÓN Desde la creación de centros de acopio de leche por parte de la planta de lácteos Ecolac de la Universidad Técnica Particular de Loja, se ha promovido el desarrollo de la ganadería del sector, pero hasta la actualidad no se tiene una base de datos de la conformación de la ganadería, de la composición individual, ni del sistema de alimentación que aplican los ganaderos de este sector. Por otro lado, ningún organismo estatal o privado ha promovido el estudio y la investigación referente al mejoramiento de la producción y calidad de la leche que se produce en la provincia de Zamora Chinchipe, lo cual ha dejado sin bases ni sustento para que el sector ganadero pueda implementar nuevos cambios para el mejoramiento. Revista Politécnica-Septiembre 2015, Vol. 36, No. 2 Fernández J. *; Tarazona G.** _______________________________________________________________________________________________________________________________ 35 En función de poder incentivar a quienes se dedican a la actividad ganadera, a realizar acciones que conlleven el mejoramiento de la composición y calidad microbiológica de la leche, el Gobierno Nacional mediante Decreto Ministerial, ha establecido que se incorpore la calidad como base para el pago del litro de leche, es así que con el Acuerdo Ministerial No. 136 del 21 de abril del 2010, se establece el sistema de pago por calidad, el cual se basa además del aspecto microbiológico, en el contenido bromatológico específicamente en los parámetros de proteína y de grasa. Es necesario poder establecer, las condiciones en las cuales se encuentra la composición bromatológica de la leche de la ganadería del sector El Retorno, a fin de que los proveedores puedan acceder al pago mediante las condiciones y parámetros establecidos en el Acuerdo Ministerial y según las tablas de pago, a su vez, Ecolac pueda enmarcarse en el sistema decretado por el Ministerio de Agricultura, Ganadería, Acuacultura y Pesca. Al instituirse el pago por calidad, el ganadero se verá incentivado en cumplir y mejorar los estándares establecidos a fin de poder tener un mejor ingreso económico por la leche producida; sin embargo, el poder acceder a esto implica realizar cambios y mejoras en el manejo ganadero en función de superar los parámetros establecidos para los componentes bromatológicos de grasa y proteína. Actualmente, muchas ganaderías del sector El Retorno están aplicando sistemas de alimentación y cruce de razas sin poder establecer de forma fehaciente un beneficio tanto en la cantidad de la producción, como en la calidad de la leche. El presente trabajo se centra en el estudio sobre la conformación, manejo alimenticio del ganado y la composición de la leche que se produce en El Retorno. Se pretende establecer, bajo la situación de manejo ganadero actual del sector, la influencia de ciertos factores como mestizaje, etapa de lactación y suplementación alimenticia en la composición de la leche, a fin de determinar las mejores opciones que puedan beneficiar en el mejoramiento de los componentes principales de la leche. Se busca establecer los mejores parámetros en el manejo ganadero que permitan cumplir y mejorar los porcentajes de grasa, proteína y sólidos totales, componentes primordiales para el rendimiento en la industrialización de la leche y para el establecimiento de la tasa de pago por calidad. 2. METODOLOGÍA 2.1 Localización La ganadería en la que se enfoca el presente estudio pertenece a los proveedores de Ecolac y está localizado en el sector El Retorno, barrio de la parroquia Sabanilla, en el cantón Zamora, de la provincia de Zamora Chinchipe, Ecuador. La zona de estudio se extiende desde el kilómetro 35 hasta el kilómetro 50 de la carretera Loja a Zamora, ubicada a una altura de 1680 metros sobre el nivel del mar, y sus coordenadas son 3°57’52’’ S, 79°03’19’’ O. El clima de la zona es en general variable, se tiene un régimen climático templado húmedo, la pluviosidad es alta durante el año, existiendo periodos cortos de ausencia de lluvias conocidos como “veranillos”, las precipitaciones anuales van de 1500 a 3000 mm y su temperatura varía entre 18-22 °C, la vegetación consiste en bosque de montaña en las partes altas, matorrales y pastos que cubre gran parte de las pendientes prominentes del área, el relieve orográfico es accidentado 8. Los análisis de las muestras para determinar su composición, se realizaron en el laboratorio de control de calidad de la planta de lácteos Ecolac. 2.2 Materiales - Areteador metálico marca Aret modelo (A-3) - Aretes plásticos marca Dakota - Agitador manual - Frasco plásticos estériles - Caja térmica marca Coleman - Sal - Lactodaily - Lactoweekly 2.3. Equipos - Lactoscan marca Milkotronic, fabricado en Bulgaria modelo MCC 2.4. Levantamiento de información Se realizó el levantamiento de la información correspondiente a las vacas que conforman la ganadería del sector El Retorno, las cuales presentaron similares condiciones de manejo, el ordeño se realizado manualmente y únicamente en la mañana; el sistema de pastoreo era directo y rotativo; la alimentación realizada a base de Gramalote (Axonopus scoparius) y no existía pureza de raza en especial determinada para la ganadería, la mayoría del ganado estaba influenciado por el cruce o tendencia de pureza hacia la raza Holstein. Se identificó a todas las vacas en estado de gestación, con ayuda de un areteador metálico marca Aret modelo (A-3) se perforó una de las orejas de cada vaca y se colocó los aretes plásticos numerados marca Dakota con el fin de evitar confusiones posteriores durante la toma de muestras. Luego de la colación del arete y asignación del número, se consultó información de cada vaca, dicha información fue confirmada por el administrador del centro de acopio y técnico veterinario que presta servicios a los ganaderos del sector, entre la información se enfatizó en lo referente al nivel de mestizaje, ciclo de lactancia y el tipo de suplementos utilizados en la alimentación, dichos elementos conformaron los factores de estudio y son los elementos por los cuales fueron clasificadas las vacas. Se determinó que en total la ganadería de estudio estaba formada por 70 vacas en estado de lactación, con una producción promedio de 11 litros diarios. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Factores que Influyen en la Composición de la Leche en el Sector el Retorno, Parroquia Sabanilla, Cantón Zamora, Provincia de Zamora Chinchipe Ecuador _________________________________________________________________________________________________________________________ 36 2.5 Recolección de muestras 2.8 Análisis estadístico La toma de muestras se realizó basada en las metodologías Luego del levantamiento de datos de las vacas del sector, se procedió a establecer los niveles dentro de cada factor de estudio, en cuanto a mestizaje se clasificó zootécnicamente por sus características fenotípicas de los tipos de raza y el nivel de cruzamiento que se manejaba en el sector 4, 15. Es así que se consideró los siguientes niveles: utilizadas en estudios anteriores 4, 24, para cada vaca se recolecto la totalidad de la leche del ordeño de la mañana, para la toma de la muestra de leche se realizó una agitación previa utilizando un agitador manual durante 30 segundos a fin de obtener homogeneidad en la muestra, luego se trasvasó 200 c.c. de la leche a recipientes plásticos estériles de cierre hermético, los frascos fueron registrados e identificados para cada vaca, luego de tomar las muestras se las colocó en una caja térmica marca Coleman, la cual contenía hielo y sal comercial (cloruro de sodio) con el objetivo de mantener las muestras a una temperatura de 4 oC 1. En estas condiciones, las muestras fueron trasladadas a la ciudad de Loja por un lapso de 30 minutos para el respectivo análisis bromatológico en el laboratorio de calidad de la planta de lácteos Ecolac. El número de muestras analizadas fueron 3 por cada vaca, tomadas en un lapso de 10 días a fin de que las muestras provengan siempre del ciclo de lactación establecido para cada vaca, se evitaba que con el paso del tiempo formen parte de un periodo de lactación diferente al que inicialmente fue tomada la primera muestra. 2.6 Análisis de laboratorio En la determinación de la composición de la leche, los análisis de grasa y proteína requeridos para el presente estudio se realizaron por duplicado para cada muestra y fueron obtenidos según metodología utilizada por autores anteriores 25, 28, quienes realizaron el análisis de leche con la utilización del equipo Lactoscan, este equipo está basado en el principio de ultrasonido lo que permite el análisis inmediato de los valores de todos los componentes de leche, razón por la cual, es utilizado frecuentemente en el pago de leche por calidad, el poco tiempo que requiere para reportar los datos y por su tamaño, se convierte en un instrumento ideal para el control de la leche a nivel de finca, para la limpieza del equipo se utilizó las sustancias Lactodaily (solución ácida de limpieza) y Lactoweekly (solución alcalina de limpieza). Los valores de sólidos totales no son indicados directamente por el equipo, por lo cual se los determino sumando los porcentajes de sólidos no grasos y de grasa que son reportados por el Lactoscan. a) Mínimo (A): ganado local (Criollo) adaptado a la zona, sin cruzamiento o con mínimo aporte de cruzamiento de la raza Holstein (menor al 25 %). b) Bajo mestizaje (B): ganado en el cual participaba la raza Holstein en un 25 %. c) Medio mestizaje (C): ganado con cruzamiento que incluía un aporte del 50 % de la raza Holstein. Para el ciclo de lactación se adoptó la clasificación realizada por otros autores 5, la cual consistió en los siguientes niveles: a) Hasta 3 meses (X): 1- 90 días b) Cuatro a 6 meses (Y): 91-180 días c) Mayor a 6 meses (Z): desde 181 días en adelante En cuanto a la alimentación, se clasificó en función de los tipos de suplementos alimenticios aplicados (2 veces por semana) en la dieta de las vacas por parte de los ganaderos del sector, los cuales consistían en: a) Sal (S) b) Sal y melaza (SM) c) Sal y balanceado (SB) Para determinar el nivel de influencia de las variables independientes, se analizó los resultados de grasa, proteína y sólidos totales, aplicando un análisis de varianza (ANOVA) unifactorial (p<0.05), luego se determinó la significancia de las diferencias entre los niveles de cada factor mediante una prueba Duncan, el análisis estadístico se realizó usando el software R versión 2.15.1.14, 33. 3. RESULTADOS Y DISCUSIÓN 2.7. Análisis de resultados 3.1 Conformación del hato ganadero La influencia de los factores en estudio se determinó mediante el análisis de la composición de la leche (grasa, proteína y sólidos totales) de las 70 vacas en estado de lactación, las cuales fueron previamente clasificadas según el mestizaje, ciclo de lactación, y suplemento alimenticio. Luego se determinó la influencia de éstos factores en estudio mediante el análisis estadístico, para finalmente definir conclusiones y recomendaciones del trabajo realizado. El estudio se realizó con un total de 70 vacas, el mayor porcentaje corresponde al grupo de mínimo mestizaje con Holstein (67.14 %), mientras que las de medio mestizaje con Holstein son las que pertenecen al menor grupo de la totalidad del ganado (8.57 %). Los resultados indican que en el sector no existe predominio de mestizaje con razas productoras de leche, un alto porcentaje (67.14 %) del hato ganadero es conformado por la Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Fernández J. *; Tarazona G.** _______________________________________________________________________________________________________________________________ 37 raza Criolla, cuyas características de volúmenes de producción de leche no son las idóneas para la actividad de producción lechera como sucede con la raza Holstein. Los estudios realizados mencionan valores promedio en la producción de leche en vacas Holstein de 25.86 Kg/día. Se determinan producciones diarias en Holstein de18.40 Kg/día, así como como producciones de 25 kg/día 3, 22, 34. Mientras que en la raza Criolla, la producción diaria puede alcanzar valores de 6.60 kg/día 26. Incluso la producción de leche en las vacas Criollas puede tomar valores menores como 2.80 kg/día 30. - El mestizaje influye sobre el contenido de la grasa, en el contenido de proteína, pero no influye sobre la cantidad de sólidos totales. - El ciclo de lactación influye sobre el contenido de grasa y sólidos totales, pero no causa efecto sobre la proteína. -Los suplementos alimenticios influyen en el contenido de grasa y sólidos totales, sin producir variación significativa sobre la proteína. La comparación de medias y determinación de diferencias significativas entre niveles de cada factor se resumen en la Tabla 2, los datos provienen de la aplicación de la prueba Duncan realizada para los diferentes factores que en el ANOVA se analizó la influencia sobre los componentes de la leche en estudio. De acuerdo al ciclo de lactación, las vacas que correspondían al grupo de hasta 3 meses de lactación lo conformaron el 32.86 %, al grupo desde 4 meses hasta 6 meses el 40.00 % y las que tenían mayor a 6 meses de lactación representaban el 27.14 % del total de las 70 vacas en estudio. En lo que respecta a los suplementos alimenticios, el grupo alimentado con suplemento de sal y el grupo que consumía sal y balanceado, constituyen los mayores porcentajes (44.28 % para cada grupo), siendo el grupo alimentado con sal y melaza, los que representan el menor porcentaje del hato ganadero (11.44 %). Tabla 1. Análisis de varianza para las diferentes variables de estudio Variables independientes La adición de suplementos alimenticios aplicados en el sector no tiene mucha diversidad, son suplementos comunes y de fácil adquisición en el medio (sal, melaza, balanceado), en las fincas no preparan de forma individualizada los suplementos de alimentación, ni tienen un análisis de los requerimientos nutricionales del ganado. La importancia de proporcionar suplementos alimenticios al ganado lechero radica no sólo en mejorar la composición de la leche, sino en aumentar los volúmenes de producción lechera 6, 29, 33. Variable dependiente Mestizaje Grasa Suplemento alimenticio 6.62 e-04* Ciclo de lactación 9.29 e-08* Proteína 3.15 e-06* 0.56 Sólidostotals 0.15 8.93 e-08* 0.23 4.50 e-06* 6.50 e-04* *Diferencia significativa, con p<0.05 3.2.1 Factor mestizaje La respuesta del contenido de grasa de la leche en función del mestizaje mostró variabilidad (p<0.05), la de vacas Criollas tiene significativamente menor contenido de grasa que las de medio mestizaje y sin diferencias significativas con la de bajo mestizaje, se observa un incremento en el contenido de grasa conforme aumenta el cruzamiento de las vacas Criollas con la raza Holstein. 3.2 Influencia del mestizaje, ciclo de lactación y suplementos alimenticios sobre la grasa, proteína y sólidos totales En la Tabla 1, se indican los resultados del ANOVA. Los datos obtenidos demuestran que: Tabla 2. Resultados de la prueba Duncan de las variables en estudio Variable independiente Variable dependiente Mestizaje A (%) Grasa Proteína Sólidostotales 3.63b 3.99ab a a 3.07 10.84 B (%) 3.01 a 11.11 Ciclo de lactación C (%) Y (%) Z (%) P (%) Q (%) R (%) 3.60b 4.30a 3.33c 3.75b 4.35a 3.82b 4.42a b a a a a a 2.89 a X (%) Suplemento alimenticio 11.19 3.07 a 10.48 3.02 b 10.82 3.02 b 11.60 3.07 a 11.06 3.00a 3.06 b 11.90 a 10.58c Valores con letras distintas en la misma fila para los diferentes grupos de cada variable independiente, difieren significativamente (p < 0.05), según prueba Duncan Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Factores que Influyen en la Composición de la Leche en el Sector el Retorno, Parroquia Sabanilla, Cantón Zamora, Provincia de Zamora Chinchipe Ecuador _________________________________________________________________________________________________________________________ El menor contenido de grasa en la leche de vacas de raza Criolla, no concuerda con la información de otros estudios realizados en los cuales el porcentaje de grasa en la leche de vacas Criollas es mayor que en los cruzamientos de la raza Holstein. Se realizan estudios sobre la composición de la leche en la raza Criolla, obteniéndose valores entre 4.73 % y 5.65 % en el contenido graso, y también se determinan resultados con un contenido promedio de grasa en leche de vacas Criollas de 4.25 % [2, 26]. Los estudios realizados en cruzamiento de raza Holstein con Normanda y Holstein con Escandinavas Rojas, indican contenidos de grasa en la leche de 3.74 % y 3.66 % respectivamente [14]. Los datos reportados para raza Holstein, indican porcentajes de grasa entre 3.21 % y 3.49 %, menor al de sus cruces y Criollas [3]. Con dietas especiales (hiervas de ensilado y concentrados) utilizadas en la alimentación se obtiene valores de 4.35 % para grasa en leche de vacas Holstein, aun en esas condiciones la cantidad de grasa en la leche de vacas Holstein no supera los valores reportados para la raza criolla [2, 35]. Respecto a la proteína, los resultados indican diferencias significativas entre los diferentes grupos de mestizaje para el contenido de proteína (p<0.05), la leche de las vacas Criollas presentó significativamente mayor contenido de proteína que las de medio mestizaje, sin embargo no existió diferencias significativas con las de bajo mestizaje. Los valores de proteína disminuyen conforme se incrementa el cruce con la raza Holstein, estudios anteriores indican el mismo comportamiento, comprobándose que el contenido de proteína en la leche de vacas Criollas, es mayor que en vacas con cruzamiento de Holstein. Se determinan valores de proteína en la leche de 3.24 % para el cruce Normanda con Holsteins, 3.20 % para los cruces Montbeliarde con Holstein y 3.20 % para los cruces Escandinavas Rojas con Holstein, mientras que las Holstein puras no sobrepasaron el 3.13 % 14. En otros estudios se determinaron un contenido de 3.12 % para la proteína proveniente de vacas de cruzamiento Jersey con Holstein 15. En los estudios respecto al contenido proteico de la leche en vacas Criollas, se reporta el contenido de 3.52 % y de 4.03 % 2, 26, los cuales son mayores a la de los cruces de la raza Holstein reportada por los autores anteriores. De igual manera otros estudios realizados en vacas Cebú indican un contenido de proteína de 3.83 %, el cual es mayor significativamente al de cruzamientos de la raza Cebú con Holstein (3.42%) y Holstein puras con 3.00 % 21. El mestizaje no intervino en el contenido de sólidos totales (p>0.05), contrario a los determinado en el presente estudio, investigaciones realizadas anteriormente se reportan diferencias significativas entre diferentes razas y sus niveles de cruzamientos. Estudios comprueban que los cruces Angus con Hereford producían leche con menor contenido de sólidos totales (13.58 %) de forma significativa, que los cruces de Jersey con Hereford con 14.34 % 7. Briñez et al. 4 determinan que el cruce de Holstein 50 % con Cebú 50 % produce un contenido de 13.82 % de sólidos totales en la leche, mientras que en el cruce Holstein 75 % con Cebú 25%, era significativamente mayor (15.28 %) [4]. En el 38 presente estudio los sólidos totales aumentan según el incremento de mestizaje con la raza Holstein, sin embargo en estudios anteriores esa tendencia es contraria. Estudios comprueban que la raza Criolla puede poseer mayor cantidad de sólidos totales (14.50 % y 14.16 % respectivamente) que los cruces de raza Holstein con 13.65 % reportados por otros autores. 2, 4, 26. Los resultados obtenidos son de comportamiento contrario a los esperados, el contenido de grasa en la leche de vacas de raza Criolla indica valores muy inferiores a los determinados en investigaciones anteriores, de igual manera la tendencia a aumentar el contenido de sólidos totales con la intensidad del cruce con Holstein (aunque sin diferencia significativa), es contradictorio a lo definido en estudios anteriores, caracterizándose mayor contenido de sólidos totales en la leche de vacas de origen Criollo, el poco número de vacas del grupo medio mestizaje y la falta de uniformidad de suplementos en la alimentación y estados de gestación para cada grupo de mestizaje pudo ocasionar resultados contrarios a los esperados al no proporcionar similitud de condiciones. 3.2.2 Factor ciclo de lactación En lo que respecta a los efectos del ciclo de lactación, los datos obtenidos determinan que existe diferencias estadísticamente significativas (p<0.05) entre los ciclos de lactación para la cantidad de grasa en la leche, los resultados de la prueba Duncan indican que la leche de vacas que se encuentran en el ciclo mayor a seis meses contienen un superior contenido de grasa que la leche de vacas que se encuentran en el ciclo hasta tres meses y que las del ciclo entre el cuarto y el sexto mes. El análisis realizado en producciones de vacas Holstein, confirman la dependencia del contenido graso de la leche del ciclo de lactación, siendo los ciclos inicial y final de la lactación (7-105, 211-315 días respectivamente) los periodos con mayor producción de grasa, obteniéndose valores de 4.46% para ambos ciclos, mientras que en los periodos medios de lactación (106-210) sólo alcanzo un contenido de grasa de 3.7 % [13]. Los resultados obtenidos también concuerdan con el estudio previo realizado en vacas Holstein, en el cual se reporta que existe diferencia significativa entre los diferentes ciclos de la lactación, se presenta menor porcentaje de grasa (3.50 %) al inicio de la lactancia (75-90 días) y la tendencia de mayores valores (3.70 %) en los últimos meses o periodo final de la lactancia (255-270 días) [31]. Resultados obtenidos en el primer periodo de lactancia en vacas Frisian, indican la existencia de diferencias en la composición de grasa, siendo mayor en las lactaciones iniciales y tardías, y disminuyendo en las lactaciones de ciclo intermedio [27]. Respecto al contenido de proteína, el ciclo de lactación no influyó significativamente (p>0.05), lo cual concuerda con los estudios anteriores para ganado mestizo en los mismos periodos y para Holstein Friesianen otros ciclos (7-105, 106– 210, 211–315 días) [4, 13]. Sin embargo otros estudios en lactaciones clasificadas como tempranas (75-90 días), Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 39 Fernández J. *; Tarazona G.** _______________________________________________________________________________________________________________________________ medias (150-165 días) y tardías (255-270 días) indican la variación de forma significativa en el contenido de la proteína, siendo en las lactaciones medias las de mayor contenido de proteína [31]. La composición de la leche presenta un elevado porcentaje de sólidos totales en el ciclo de lactación mayor a seis meses, con diferencias significativas (p<0.05) respecto a los otros ciclos, otros estudios revelan resultados similares en cuanto a la diferencia en la cantidad de sólidos totales para distintos ciclos de lactación, la tendencia en la etapa final o últimos meses de lactación es el incremento de los sólidos totales de la leche. En estudios previos realizados en diferentes meses de lactancia se reportan contenidos de 12.33 % y 12.60 % para el primer y segundo mes, mientras que para el séptimo y octavo mes la cantidad de sólidos totales alcanzó promedios de 13.07 % y 12.95 %, siendo los más altos promedios [12]. En otros periodos de lactancia se determinan menor porcentaje de sólidos totales en el ciclo de 1 a 90 días (13.79 %) y significativamente mayor (14.98 %) para el ciclo de más de 180 días [4]. Por los resultados obtenidos y la información precedente respecto al efecto de la lactación sobre la composición de la leche, se comprueba que depende mucho de la forma de división del tiempo de lactación (meses, días, periodos continuos o separados) y la amplitud de los periodos analizados en el estudio, para definir el tipo de efecto, el tipo de elemento afectado y el periodo de mayor o menor contenido del componente en estudio. 3.2.3 Factor suplemento alimenticio El tipo de suplemento intervino sobre el contenido de grasa (p<0.05), las vacas alimentadas con suplemento de sal y melaza, presentaron mayor contenido con diferencia significativa en relación al grupo alimentado con suplemento de sal y al grupo alimentado con suplemento de sal y balanceado. La diferencia de la composición de la lechepor efecto de la alimentación es comprobada en otras investigaciones. Una alimentación a base de pasto provoca mayor contenido de grasa (5.49 %) en la leche, con diferencias significativas que dietas con suplemento a base de maíz (4.98 %),el pastoreo de vacas Holstein en gramineas provoca una producción de leche con 4.38 % de grasa, mientras que la adición al pastoreo de 2 kg de Gliricidia sepium (leguminosa) disminuía significativamente el contenido de grasa a 3.89 %, de igual manera se comprueba un mayor contenido de grasa en la leche de vacas Holstein alimentadas con un suplemento alimenticio Megalac (sales y ácidos grasos) como fuente de energía alcanzando valores de 4.14 %, mientras que en las vacas alimentadas con linaza se registró contenidos de 3.81 % [17, 24, 20]. La alimentación a base de concentrado de heno, ensilado ryegrass y heno de pastizal natural, provocan diferencias significativas en el contenido graso de la leche, obteniéndose valores de 4.04 %, 3.41 % y 3.71 % respectivamente [11]. 27.2 % producían leche con valores de 3.88 % y 3.98 % de grasa respectivamente, los cuales no eran significativos con la leche de aquellas vacas que no se alimentaron con el ensilaje, en las cuales la leche contenía 3.84 % de grasa 34. Se verifica según el análisis estadístico, de que la alimentación se mantiene sin influir en el contenido de proteína de la leche, similares conclusiones se verifican en estudios anteriores al realizar cambios en la alimentación de las vacas incorporando melaza, minerales, o un suplemento balanceado que contenía proteína, grasa, fibra y minerales [19, 32, 33]. Al estudiar el efecto en la cantidad de proteína en las dietas de vacas alimenticias a base de ensilado de maíz (3.19 %), ensilado de ryegrass (3.07 %), heno de ryegrass (3.20 %) y heno de pastizal natural (3.245) sin existir diferencia significativa entre los resultados obtenidos [11]. Los resultados de otros estudios no confirman lo anterior y se comprueba la influencia de la alimentación sobre la proteína. El contenido de proteína en la leche de vacas Holstein alimentadas con linasa es significativamente mayor (2.98 %) que en las alimentadas con soya (2.87 %) [20]. La adición de suplementos en la alimentación de vacas Holstein utilizando concentrados de cereales y remolacha, proporcionan aumentos significativos, obteniéndose valores 3.01 % y 2.99 % respectivamente, mientras que en vacas en las cuales únicamente se las alimento con pastoreo durante el periodo de ordeño, su contenido de proteína sólo alcanzo el 2.88 % [22]. Otros estudios comprueban que la leche de vacas Holstein alimentadas con diversas concentraciones de ensilaje (soya y melaza) de 13.6 % y 27.2 %, tenía menor contenido (3.24 % y 3.19 % respectivamente) de proteína que la leche producida por vacas que no se incluyó el ensilaje (3.38 %) [34]. El contenido de sólidos totales, fue afectado en su contenido por el tipo de alimentación, el grupo de vacas alimentadas con suplementos de sal y melaza presentaron mayor contenido de sólidos totales (p<0.05) que los grupos alimentados con suplemento de sal y que el grupo con suplementos de sal y balanceado. La adición de 2 kg de concentrado comercial en la alimentación de vacas en pastoreo de Panicum maximun produce mayor cantidad de sólidos totales (12.70 %), el incremento fue significativo a la adición de únicamente 1 kg del concentrado comercial, con el cual sólo se obtuvo un contenido de sólidos de 12.26 % [24]. De acuerdo a los resultados obtenidos y los determinados en estudios anteriores, se puede establecer que el efecto de la alimentación en los componentes lácteos analizados dependerá del tipo de suplemento alimenticio y también de la cantidad suministrada, los comportamientos de mayor o menor contenido de los componentes de la leche pueden ser diferentes en función del tipo de alimento y su dosificación. 3.3 Resultados generales de las variables de estudio en la composición de la leche En la tabla 3, se pueden apreciar los promedios generales de todo el hato ganadero para las variables dependientes. Por el contrario, al sustituir un alimento balanceado por ensilaje (soya con melaza) en proporciones de 13.6 % y Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Factores que Influyen en la Composición de la Leche en el Sector el Retorno, Parroquia Sabanilla, Cantón Zamora, Provincia de Zamora Chinchipe Ecuador _________________________________________________________________________________________________________________________ 40 Tabla 3. Caracterización general de la leche para las variables en estudio Variable Grasa Proteína Sólidostotales n = 210 Promedio (g/100g) 3.79 1.01 3.04 0.17 10.94 1.20 REFERENCIAS [1] [2] Los resultados obtenidos son acordes a los esperados, en estudios anteriores respecto a la composición de la leche se determina valores de 3.72 % y 3.83 % para la grasa, mientras que 3.04 % y 3.08 % para la proteína, guardando mucha similitud a los de la presente investigación [9]. De igual manera se reporta contenidos de 3.07 % para la proteína, muy similar al valor obtenido en el presente estudio [10]. Los sólidos totales indican ser el componente de mayor variabilidad, al comparar los datos reportados en estudios anteriores se indica un valor de 14.30 % (en el presente estudio se obtuvo 10.94 %), mientras que el valor reportado por los mismos autores para la grasa es de 3.89 %, con menos diferencia al comparar con el obtenido en el presente estudio de 3.79 % [4]. De acuerdo a los establecido por el INEN en su norma para leche cruda, los requisitos mínimos en la composición de la leche son de 3.00 % para la grasa, 2.90 % para la proteína y 11.20 % para sólidos totales [16]. Los resultados obtenidos en la presente investigación indican que se cumple con los requisitos establecidos para la grasa y proteína, pero no para los sólidos totales. [3] [4] [5] [6] [7] [8] [9] [10] 4. CONCLUSIONES [11] El mestizaje influye en la composición de la leche (p<0.05), el cruzamiento de la raza Criolla con Holstein produce mayor contenido de grasa en la leche de las vacas con medio mestizaje (4.30 %) que en las criollas (3.63 %). El cruzamiento provocó disminución del contenido de proteína, la leche de vacas de medio mestizaje presentaron menor contenido de proteína (2.89 %) que las criollas (3.07 %). Los sólidos totales no se ven influenciado por el cruzamiento. En los diferentes ciclos de lactación se producen cambios en los componentes de la leche, como grasa y sólidos totales (p<0.05), la grasa (4.35 %) y sólidos totales (11.60 %) son mayores en el ciclo de lactación mayores a 6 meses. La proteína no presenta diferencias significativas durante la lactación. [12] [13] [14] [15] [16] La alimentación puede afectar la composición de la leche (p<0.05), la suplementación con sal y melaza aumenta el contenido de grasa (4.42 %) y de sólidos totales (11.90 %). Los suplementos no ocasionaron cambios en el contenido de proteína. Las mejores opciones para el mejoramiento de la composición de la leche, según las condiciones actuales de manejo del hato ganadero, están centradas en el empleo de vacas de medio mestizaje y proporcionar suplementos alimenticios basados en sal y melaza para producir mejores resultados. [17] [18] [19] [20] Atasoglu, C; Uysal-Pala, C; Karagul-Yuceer, Y. 2009. Changes in milk fatty acid composition of goast during lactation in a semi-intensive production system. Archiv Tierzucht.52(6): pp. 627-636. Bartl, K.; Gómez, C.A.; Aufdermauer, T; Garcia, M; Kreuzer, M; Hess, H.D.; Wettstein, H.-R. 2008. Effect of diet type on performance and metabolic traits of Peruvian local and introduced cow types kept at 200 at 360 m of altitude. Livestock Science.122(1), pp. 30-38. Bobe, G; Lindberg, G; Freeman, A; Beitz, D. 2007. Composition of milk Protein and Milk Fatty Acids Is Stable for Cows Differing in Genetic Merit for Milk Production. Journal of dairy Science. 90(8), pp. 3955-3960. Briñez, W; Valbuena, E; Castro, G; Tovar, A; Ruiz, J; Román, R. 2003. Efectos del mestizaje, época del año, etapa de lactancia y número de partos sobre la composición de leche cruda de vacas mestizas. FCVLUZ. 13(6), pp. 490-498. Briñez, W; Valbuena, E; Castro, G; Tovar, A; Ruiz, J. 2008. Algunos parámetros de composición y calidad en leche cruda de vacas doble propósito en el Municipio Machiques de Perijá. Estado Zulia, Venezuela. FCV-LUZ. 13(6), pp. 607-617. Chaudhary, L; Sahoo, A; Agarwal, N; Kamra, D; Pathak, N. 2001. Effect of Replacing grain with Deoiled Rice Bran and Molasses from the Diet of Lactating Cows. Journal Animal Science. 14(5), pp. 646650. Chenette, C; Frahm, R. 1981. Yield and Composition of Milk from Various Two-Breed Cross. Journal of animal Science. 52(3), pp. 483492. Correa Campués CJ. 2007. Análisis de la susceptibilidad de los fenómenos. Tesis Ing. Geo. Quito- Ecuador, Escuela Politécnica del Ecuador. pp. 20-22. DePeters, E; Cant, J; 1992. Nutritional Factors Influencing the nitrogen Composition of bovine Milk: A Review. Journal of Dairy Science.75(8), pp. 2043-2070. DePeters, E; Ferguson, J; 1992. Nonprotein Nitrogen and Protein Distribution in the Milk of Cows. Journal of Dairy Science. 75(11), pp. 3192-3209. Ferlay, A.; Martin, B.; Pradel, Ph.; Coulon. J.B.; Chillard. Y. 2006. Influence of Grass-Based Diets on Milk Fatty Acid Composition and Milk Lipolytic System in Tarentaise and Montbeliarde Cow Breeds. Journal of Dairy Science. 89(10), pp. 4026-4041. Fuenmayor, C; Chicco, C; Bodisco, V; Capó, E. 1973. Estudio de los componentes de la leche de vacas Holstein y Pardo Suiza durante cuatro lactancias en Venezuela. Agronomía Tropical. 23(6), pp. 541554. Gurmessa, J; Melaku, A. 2012. Effect of Lactation Stage, Pregnancy, Parity and Age on Yield and Major Components of Raw Milk in bred Cross Holstein Friesian Cows. World Journal of Dairy & Food Sciences. 7(2), pp. 146-149. Heins, BJ; Hansen, LB; Seykora, AJ. 2006. Production of Pure Holstein Versus Crossbreds of Holstein with Normande Montbeliarde, and Scandinavian Red. Journal of Dairy Science. 89(7), pp. 2799-2804. Heins, BJ; Hansen, LB; Seykora, AJ; Johnson, DG; Linn, JG; Romano, JE; Hazel, AR. 2008. Croosbreds of Jersey x Holstein Compared with Pure Holsteins for Production, Fertility, and Body and Udder Measurements During First Lactation. Journal of Dairy Science. 91(3), pp. 1270-1278 INEN (Instituto Ecuatoriano de Normalización). 2012. Norma Técnica Ecuatoriana: NTE INEN 9:2012; Leche cruda. Requisitos. 5 rev. Quito. 2p. Mackle, T; Bryant, A; Petch, S; Hooper, R; Auldist, M. 1999. Variation in the composition of milk protein from pasture-fed dairy cows in late lactation and the effect of grain and silage suplementation. New Zealand Journal of agricultural Research.42(2), pp. 147-154. MAGAP (Ministerio de Agricultura, Ganadería, Acuacultura y Pesca). 2010. Acuerdo 136: Reglamento para normar el pago por calidad de la leche y sanidad animal, pp. 2. Mullins, C; Bradford, B. 2010. Effect of a molasses-coated cottonseed product on diet digestibility performance, and milk fatty acid profile of lactating dairy cattle. Journal of Dairy Science. 93(7), pp. 3128-3135. Petit, H.V. 2002. Digestion, Milk Production, Milk Composition, and Blood Composition of Dairy Cows Fed Whole Flaxseed. Journal of Dairy Science.85(6), pp. 1482-1490. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 41 Fernández J. *; Tarazona G.** _______________________________________________________________________________________________________________________________ [21] Ponce, P. 2009. Composición láctea y susinterrelaciones: Expresión genética, nutricional, fisiológica y metabólica de la lactación en las condiciones del trópico. Revista Salud Animal. 31(2), pp. 69-76. [22] Pulido, R; Berndt, S; Orellana, P; Wittwer, F. 2007. Effect of source of carbohydrate in concentrate on the performance of high producing dairy cows during spring grazing. Red de Revistas Científicas de América Latina, el Caribe, España y Portugal.39(1), pp. 19-26. [23] Razz, R; Clavero, T. 1998. Calidad química de la leche en vacas suplementadas con harina de mata ratón (Gliricidia sepium). FCVLUZ.8(4), pp. 312-314. [24] Razz, R; Clavero, T. 2007. Efecto de la suplementación con concentrado sobre la composición química de la leche en vacas doble propósito pastoreando Panicum maximum-Leucaena leucocephala. FCV-LUZ. 17(1), pp. 53-57. [25] Razzaque, M; Mohammed, S; Al-Mutawa, T; Bedair, M. 2009. Growth, Reproduction and Milk Yield of Holstein friesian heifers Born and Adapted in Kuwait. Pakistan Journal of Nutrio. 8(8), pp. 11591163. [26] Rojas, I.; Méndez. J.A.; Portillo, M.; Rincón, X.; Martínez, G.; Contreras, G. 2011. Efecto del polimorfismo genético de las proteínas lácteas sobre la producción y composición de la leche en ganado criollo limonero. FCV-LUZ. 21(6), pp. 517-523. [27] Rook, J.A.F.; Campling, R.C. 2009. Effect of stage and number of lactation on the yield and composition of cow's milk. Journal of Dairy Research.32(1), pp. 45-55. [28] Saad, MSA; El Zubeir, IEM; Fadel Elseed, AMA. 2013: Effect of lactoperoxidase enzyme system and storage temperature on the keeping quality of sheep milk. Livestock Research for Rural Development. 2013:25, 102. [29] Sánchez, N; Ledin, I. 2006. Efecct of feeding different levels of foliage from Cratylia argentea to creole dairy cows on intake, digestibility, milk production and milk composition. Trop Anim Health Prod. 38(1), pp. 343-351. [30] Santellano, E; Becerril, C; Alba, J; Chang, Y; Gianola, D; Torres, G. 2008. Inferring Genetic Parameters of Lactation in Tropical Milking Criollo Cattle with Random Regression Test-Day Models. Journal Dairy Science. 91(11), pp. 4393-4400. [31] Sapru, A; Barbano, D; Yun, J; Klei, L; Oltenacu, P; Bandler, D. 1997. Cheddar Cheese: Influence of Milking Frequency and Stage of Lactation on Composition and Yield. Journal of Dairy Science. 80(3), pp. 437-446. [32] Srinivas, B; Gupta, B. 1997. Urea-Molasses-Mineral Block Licks Suplementation for Milk Production in Crossbred Cows. The Asian Journal of Animal Science. 10(1), pp. 47-53. [33] Strusinska, D; Minakowski, D; Kaliniewicz, J. 2006. Effects of fatprotein supplementation of diets for cows in early lactation on milk yield and composition. Czech J. Anim. Sci. 51(5), pp. 196-204. [34] Tobía, C; Rojas, A; Villalobos, E; Soto, H; Uribe, L. 2004. Sustitución parcial del alimento balanceado por ensilaje de soya y su efecto en la producción y calidad de la leche de vaca, en el trópico húmedo de costa rica. Agronomía Costarricense. 28(2), pp. 27-35. [35] Vance, E.R.; Ferris, C.P.; Elliott, C.T.; McGettrick, S.A., Kilpatrick§, D.J. 2012. Food intake, milk production, and tissue changes of Holstein-Friesian and Jersey × Holstein-Friesian dairy cows within a medium-input grazing system and a high-input total confinement system. Journal of Dairy Science.95(3), pp. 2011-4410. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 42 Recuperación de Oro a Partir de Minerales Polisulfurados con Soluciones Amoniacales de Tiosulfato de Sodio _________________________________________________________________________________________________________________________ Recuperación de Oro a Partir de Minerales Polisulfurados con Soluciones Amoniacales de Tiosulfato de Sodio Gámez S.*; De la Torre E.** *Escuela Politécnica Nacional, Facultad de Ingeniería Química, Quito, Ecuador e-mail: [email protected]; [email protected] Resumen: La extracción de oro a partir de minerales polisulfurados mediante soluciones amoniacales de tiosulfato de sodio y la presencia de iones cúpricos es una alternativa no tóxica a la cianuración. Por esta razón se realizó una comparación entre la cianuración y la lixiviación con soluciones amoniacales de tiosulfato para determinar la efectividad de ambos métodos. En la cianuración se obtuvo una recuperación de oro del 80.6 % después de 24 horas de agitación mientras que en la lixiviación con tiosulfato se consiguió el 80.9 % de recuperación de oro en una hora. Los lixiviados obtenidos fueron sometidos a cinco técnicas con el fin de extraer el oro contenido en dichas soluciones. La técnica más exitosa fue flotación iónica cuya recuperación de oro fue del 84 %. Posteriormente se realizó la electrólisis del concentrado de flotación iónica (espuma) a un voltaje de 1.5 V durante 3 horas lo que permitió alcanzar una recuperación de oro del 82 %. Palabras clave:Lixiviación, recuperación, flotación iónica, electrólisis. Abstract:Gold extraction from polysulphide ores by ammmoniacal thiosulfate solutions in presence of cupric ions is a non-toxic alternative to cyanidation. For that reason, a comparison between cyanidation and ammoniacal thiosulfate leaching was performed to verify the effectiveness of both methods. With cyanidation, a gold recovery of 80.6 % was accomplished at 24 hours, whereas with ammoniacal thiosulfate leaching process a 80.9 % of gold recovery was achieved at just one hour of agitation. The ammoniacal thiosulfate solutions obtained were treated with five different techniques in order to extract gold from the mentioned solutions. The most successful technique was ion flotation which gold recovery ascended to 84 %. Then electrowinning, applied to ionic flotation concentrate, was developed with 1.50 V during three hours obtaining 82 % of gold recovery. Keywords:Leaching, recovery, ion flotation, electrowinning. en el sistema amoníaco-tiosulfato se explican con las siguientes reacciones. 1. INTRODUCCIÓN La lixiviación de oro con tiosulfato de sodio es una técnica no tóxica que permite manejar minerales refractarios [4]. Sin embargo, el tiosulfato no puede disolver el oro tan rápido como lo hace el cianuro, por esta razón se requiere la presencia de amoníaco acuoso y iones cúprico que hacen las veces de catalizador en el proceso de lixiviación [4, 9, 14]. La disolución de oro se desarrolla a través de un conjunto de reacciones redox que permiten transformar el oro metálico en el complejo oro-tiosulfato. El amoníaco acuoso y los iones cúprico forman el complejo cuprotetramina el cual es el responsable de acelerar la disolución del oro [4, 8]. La cuprotetramina oxida el oro metálico contenido dentro del mineral y forma el complejo diaminoauroso, además el ion cúprico es reducido a ion cuproso el cual es re-oxidado con la ayuda del oxígeno disuelto en solución. Mientras tanto el complejo diaminoauroso reacciona con el tiosulfato y produce el complejo oro tiosulfato [4, ]. La lixiviación de oro (1) El compuesto diaminoauroso formado en la reacción anterior, reacciona con el tiosulfato de sodio formando el complejo oro-tiosulfato de la siguiente manera: (2) Por último, el ion cuproso vuelve a oxidarse debido a la presencia del oxígeno disuelto para formar nuevamente la cuprotetramina y de esa forma continuar con la lixiviación: (3) Reacción global: Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 43 Gámez S.*; De la Torre E.* _______________________________________________________________________________________________________________________________ (4) Existen muchos parámetros que afectan el desarrollo de la lixiviación con soluciones amoniacales de tiosulfato. De acuerdo a las características del mineral a tratar, uno o varios factores son decisivos al momento desarrollar un buen proceso de lixiviación [6]. La presencia de amoníaco acuoso está relacionada con el pH de la solución. Para una adecuada lixiviación con tiosulfato es necesario establecer el pH de la solución en un rango comprendido entre 9.6 y 11 [1, 9]. En dicho rango la formación de la cuprotetramina se ve [10]. Si el pH de la solución se encuentra por debajo de 9.6 o arriba de 11, la concentración de la cuprotetramina se ve reducida y otros complejos aminos aparecen en la solución los cuales no intervienen en la disolución del oro [4, 10]. Los iones cúprico permiten formar junto al amoniaco el complejo cuprotetramina. Por esta razón, un incremento en la concentración de iones cúprico permite conseguir una rápida disolución de oro [1, 12]. Sin embargo, un incremento excesivo en la cantidad de iones cúprico provoca la oxidación del tiosulfato en politionatos (compuestos formados al oxidarse el ion tiosulfato). La presencia de politionatos causa una disminución en la concentración de tiosulfato lo que va en detrimento de la disolución del oro [1, 12]. Un incremento en la cantidad del agente lixiviante permite aumentar la recuperación de oro en el lixiviado. No obstante, se debe tener en cuenta que un excesivo aumento en la cantidad de tiosulfato añadida a la solución puede derivar en la oxidación de este en politionatos que perjudican la disolución de oro [1]. Otro aspecto a tener en cuenta es que una excesiva cantidad de la sal de tiosulfato puede favorecer la reacción entre el ion tiosulfato y el oro metálico la cual transcurre más lento que la reacción entre el complejo diaminoauroso y el ion tiosulfato [4]. Una elevada concentración de tiosulfato de sodio puede minimizar el efecto de la cuprotetramina y la disolución de oro progresará más despacio [12]. Es importante recalcar que la eficacia de la lixiviación con tiosulfato está relacionada con las características del mineral. La presencia de ciertos elementos como hierro y cobre que al disolverse, pueden transformar el tiosulfato a politionatos[9]. Más aún, si la cantidad de estos elementos se incrementa debido a un aumento en la cantidad de mineral a tratar, la disolución de oro disminuirá [12]. La baja afinidad del tiosulfato hacia la superficie del carbón activado conduce a una baja recuperación de oro [1, 4]. No obstante, un incremento en el pH de los lixiviados puede mejorar el desenvolvimiento de la adsorción de oro con carbón activado [13]. Si se aumenta la cantidad de amoníaco acuoso, se favorece la formación del complejo diaminoauroso el cual es sí es afín a la superficie del carbón activado [7]. Por lo tanto, para obtener una elevada recuperación de oro, la concentración de carbón activado debe incrementarse pero, la carga de oro sobre carbón activado será muy baja [1]. La precipitación de oro es una técnica aceptable para la recuperación de oro a partir de lixiviados. Los principales precipitantes utilizados en este proceso son el cobre y el zinc cuya diferencia de potencial electroquímico con el oro disuelto permiten su deposición en forma de metal [4-5]. Es importante recalcar que el precipitante metálico puede reducir el tiosulfato en sulfuros que al precipitar disminuyen el área disponible para la cementación del oro [4]. También, una elevada concentración de iones cúprico puede disminuir la recuperación de oro debido a su reducción en forma de óxidos o sulfuros los cuales pasivan la superficie del cobre metálico, especialmente cuando el pH de la solución se encuentra por debajo de 10.0 [3]. En extracción por solventes, el lixiviado está en íntimo contacto con la fase orgánica el cual contiene un adecuado extractante diluido en un solvente orgánico [4]. Muchos extractantes han sido probados en los últimos años los cuales requieren el ajuste del pH para obtener elevadas recuperaciones de oro en la fase orgánica. El extractante más exitoso es el trioctil-metil-amonio cloruro (Aliquat) el cual no necesita un ajuste de pH cuando es usado en el proceso de extracción por solventes [2]. El problema con esta técnica radica en la etapa de re-extracción del oro de la fase orgánica para la posterior recuperación de oro. La electrólisis para la deposición de oro a partir de soluciones amoniacales de tiosulfato por la aplicación de una corriente directa presenta algunos inconvenientes. La presencia de iones cúprico pueden contaminar el producto metálico obtenido por la reducción del complejo oro-tiosulfato en el área catódica [4]. Otro aspecto a tener en cuenta es el elevado consumo de corriente directa debido a las reacciones paralelas y parásitas que se dan en el área catódica [13]. 2. METODOLOGÍA 2.1 Caracterización del mineral La razón por la que la lixiviación con tiosulfato no ha sido considerada como una técnica industrial se debe a la dificultad que se tiene para recuperar el oro contenido en los lixiviados [4]. El proceso convencional de adsorción con carbón activado es inservible para este propósito. En los últimos años, la adsorción con resinas se ha convertido en la técnica más adecuada para recuperar oro de los lixiviados [1]. Sin embargo, su elevado costo de instalación y la necesidad de regenerar las resinas no han permitido que esta alternativa sea un camino rentable para la recuperación de oro [4]. La composición química del mineral fue obtenida mediante disgregación ácida y ensayo al fuego. Las muestras resultantes fueron analizadas por espectrofotometría de absorción atómica en el equipo Perkin AA300. La composición mineralógica fue determinada por difracción de rayos x en el equipo Bruker D8 Advance. Finalmente, el mineral fue reducido a un d100 = 2 mm y luego fue molido a una concentración de sólidos del 62.5 % durante 37 minutos para obtener un tamaño de partícula de 100 µm. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Recuperación de Oro a Partir de Minerales Polisulfurados con Soluciones Amoniacales de Tiosulfato de Sodio _________________________________________________________________________________________________________________________ 2.2 Lixiviación con soluciones amoniacales de tiosulfato de sodio En todos los ensayos de lixiviación se preparó en primera instancia la solución lixiviante estabilizada a un pH de 10.5 antes de añadir el mineral a lixiviar. Se empleó tiosulfato de sodio del 95 % de pureza al igual que amoníaco al 20 % de concentración en volumen. Los iones cúprico fueron introducidos en forma de sulfato cúprico pentahidratado. Durante todos los experimentos se mantuvo una velocidad de agitación de 1500 RPM y a diferentes intervalos de tiempo se extrajeron muestras de la pulpa lixiviada para analizar el contenido de oro mediante espectroscopia de absorción atómica. Con el fin de conocer la evolución de la concentración de tiosulfato durante el experimento, se utilizó el método yodimétrico el cual consiste en utilizar las muestras clarificadas para titular una solución de yodo. Terminada la lixiviación, la pulpa era filtrada en el filtro prensa y los lixiviados obtenidos eran cuantificados y almacenados para la posterior recuperación de oro. Los relaves obtenidos fueron sometidos a ensayo al fuego para determinar la cantidad de oro que no se disolvió. 2.3 Cianuración En el proceso de cianuración, se utilizó una densidad de pulpa del 33 %. Se empleó cianuro de sodio técnico hasta conseguir una concentración de 1 g/L. Inmediatamente, cal fue añadida para controlar el pH de la solución en un valor de 10.5. Se procedió a agitar la pulpa a 1500 RPM durante 24 horas y a diferentes intervalos de tiempo se extraían muestras para analizar la cantidad de oro mediante espectrofotometría de absorción atómica. Con el fin de controlar la concentración de cianuro se empleó la argentometría, la cual consiste en titular las muestras clarificadas con una solución de nitrato de plata de 4.3 g/L. Una vez terminada la agitación, se filtraba la pulpa y el relave conseguido era sometido a ensayo al fuego para determinar la cantidad de oro que no se disolvió. 2.4 Adsorción con carbón activado y carbón activado impregnado con cobre Durante los experimentos de adsorción con carbón activado, se mantuvo una velocidad de agitación de 500 RPM para reducir la excesiva ruptura del carbón. En todos los ensayos se empleó carbón activado cuyo rango de tamaño varió de 2.3 a 4.7 mm con 523 m2/g de área superficial. El mismo tipo de carbón activado fue utilizado para impregnar cobre metálico mediante electrodeposión con la ayuda de electrodos de cobre. 2.5 Cementación con polvo de cobre Todos los experimentos de cementación se realizaron con polvo de cobre de 38 µm de tamaño de partícula. La velocidad de agitación se mantuvo en 1500 RPM durante todos los ensayos. La concentración de cobre metálico se 44 modificó en 8 y 16 g/L. Es importante resaltar que todas las pruebas de cementación se ejecutaron en recipientes cerrados con la finalidad de evitar la disolución de oxígeno en los lixiviados. 2.6 Flotación iónica Para el desarrollo de estos ensayos se utilizó el colector Aliquat a una concentración de 100 %, y el reactivo espumante Flomin F-121 a una concentración del 1 %. Ambos reactivos de flotación se añadieron a una concentración de 0,1 % con respecto al volumen de lixiviado a flotar. Los experimentos de flotación iónica se desarrollaron durante 40 minutos y el concentrado de flotación iónica fue recogido y cuantificado a media que rebosaba de la celda de flotación. 2.7 Electrólisis Los ensayos de electrólisis fueron aplicados tanto a los lixiviados como a los concentrados de flotación iónica con el fin de determinar la factibilidad de dicha técnica de recuperación en ambas corrientes. Además, se evaluó el efecto que tenía la adición de sulfito de sodio como estabilizador del tiosulfato. Este proceso fue desarrollado en una celda de plástico de 200 mL de capacidad. Se utilizaron electrodos de acero inoxidable y el voltaje de varió de 0.4 a 1.5 V. 3. RESULTADOS En la Tabla 1 se indica la concentración de los principales elementos que contiene el mineral de Agroindustrial “El Corazón” [11]. Tabla 1. Caracterización química del mineral de Agrocorazón [11] Elemento Concentración Elemento Concentración Au (g/t) 10.93 Fe (%) 0.89 Ag (g/t) 25.98 Na (%) 2.58 Mg (g/t) 55.00 K (%) 0.95 Co (g/t) 115.00 Cu (%) 0.05 Es importante notar que el mineral posee un alto contenido de oro y plata lo que lo convierte en un mineral apto para la extracción de metales preciosos. Además, se debe tener en cuenta que la concentración de hierro y cobre no es muy elevada por lo que la descomposición del ion tiosulfato en politionatos debido a las reacciones con dichos metales no parece ser un problema tangible en primera instancia. En cuanto a los compuestos que posee el mineral, se puede agrupar a los compuestos que conforman el mineral sulfurado en tres grandes grupos: silicatos, compuestos arcillosos y sulfuros. De acuerdo a la Tabla 2 el mineral de Agroindustrial “El Corazón” posee en su mayoría silicatos como el cuarzo [11]. La pirita se encuentra en una concentración del 1 % dentro del mineral aurífero sulfurado. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 45 Gámez S.*; De la Torre E.* _______________________________________________________________________________________________________________________________ Tabla 2. Caracterización mineralógica del mineral de Agrocorazón [11] Compuesto Contenido (%) Cuarzo 75 Caolinita 12 Poligoskita 5 Muscovita 5 Clinocloro 2 Pirita 1 Cabe resaltar que, al tener una baja concentración de sulfuros en el mineral, la lixiviación con soluciones amoniacales de tiosulfato de sodio se convierte en una técnica más plausible. Como la cantidad de sulfuro en el mineral es muy baja, la descomposición del ion tiosulfato durante la lixiviación no sería un problema grave a tener en cuenta. mayor cantidad de material a lixiviar con una mayor cantidad de los reactivos de lixiviación para el caso de la lixiviación con tiosulfato. 3.2 Evaluación de los procesos empleados para la recuperación de oro a partir de lixiviados de tiosulfato Uno de los principales problemas de la lixiviación de oro mediante el uso de soluciones amoniacales de tiosulfato, radica en la recuperación del oro a partir de los lixiviados obtenidos. En este trabajo, se ha experimentado con cinco alternativas tecnológicas con el fin de determinar cuál de ellas permite obtener la mayor recuperación de oro. Como se puede advertir en la Fig. 2, la técnica de flotación iónica permite obtener la mayor recuperación de oro con respecto a las cuatro técnicas restantes. 3.1 Comparación entre cianuración y lixiviación con soluciones amoniacales de tiosulfato En la Fig. 1 se muestran las recuperaciones de oro obtenidas aplicando tanto la lixiviación con tiosulfato de sodio y la cianuración con una concentración de sólidos de 33 %. Figura 2. Comparación de las técnicas utilizadas para recuperación de oro de los lixiviados con tiosulfato de sodio Figura 1. Recuperación de oro en cianuración (33 % de sólidos; pH = 10.5; [CNNa] = 1 g/L; d80 = 100 µm) y lixiviación con Na2S2O3 (33 % de sólidos; pH = 10.5; [Na2S2O3] = 1.5 M; [NH3] = 0.1 M; [Cu2+] = 10 mM; d80 = 100 µm) En el caso del proceso de lixiviación con tiosulfato se utilizó una concentración de tiosulfato de sodio de 1.5 My la concentración de iones cúprico fue de 10 mM. De esta forma se evitó una disminución en la recuperación de oro. Cabe resaltar que al disponer de mayor cantidad de mineral para lixiviar, existe mayor cantidad de elementos como cobre y hierro. Estos elementos al disolverse pueden provocar la reducción del tiosulfato formando otros compuestos que no intervienen en la disolución del oro. La recuperación de oro obtenida con el proceso de cianuración fue de 80 % mientras que con la lixiviación con tiosulfato de sodio se consiguió una recuperación de 81 %. En este caso las recuperaciones de oro son similares con la salvedad que la recuperación de oro en la lixiviación con tiosulfato se consigue a la primera hora de iniciado la agitación. Por lo tanto se debe considerar que al incrementar la cantidad de mineral a lixiviar se puede das un descenso en la recuperación de oro debido a que se debe compensar la En la Fig. 2 se exponen los mejores resultados obtenidos en los experimentos correspondientes a cada una de las técnicas utilizadas para recuperar oro de los lixiviados. Claramente se puede observar que la técnica de electrólisis fue la que menor porcentaje de recuperación de oro entregó. Entre los procesos de cementación con polvo de cobre y adsorción con carbón activado se obtuvo una diferencia de 18 % en la recuperación de oro. La presencia de cobre metálico ayuda a la precipitación del oro contenido en los lixiviados debido a la diferencia de potencial electroquímico entre ambos metales [3,5]. La técnica de recuperación de oro por cementación con polvo de cobre permite una mayor recuperación de oro que la técnica de adsorción de oro con carbón activado impregnado con cobre metálico. Por lo tanto, se puede afirmar que la presencia de cobre metálico puro es más beneficiosa que la presencia de cobre en la superficie de carbón activado granular de 523 m2/g de área superficial. Posiblemente, la capa de cobre tapó los poros del carbón activado reduciendo el área superficial del mismo. Al disminuir el área superficial, se redujo la superficie disponible para la precipitación del oro sobre el carbón activado impregnado con cobre. Otra razón puede ser la heterogeneidad del carbón activado con cobre obtenido. En el proceso de impregnación de cobre sobre carbón activado, se obtuvieron muestras de carbón con capas de cobre totalmente definidas sobre su superficie, mientras que otras muestras no mostraban aparentemente Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Recuperación de Oro a Partir de Minerales Polisulfurados con Soluciones Amoniacales de Tiosulfato de Sodio _________________________________________________________________________________________________________________________ cobre en esta. Esto sumado a la poca afinidad del tiosulfato al carbón activado, deriva en una baja recuperación de oro. La técnica de flotación iónica fue la que mayor porcentaje de recuperación de oro entregó debido a que sí existe afinidad entre el complejo oro-tiosulfato y el reactivo colector trioctilmetil-amonio cloruro (Aliquat). La principal ventaja de esta técnica está en la poca cantidad de reactivos de flotación a utilizar (concentración al 0.1 % en lixiviados). Además se obtiene un concentrado de flotación acuoso que puede ser sometido a electrólisis para la posterior recuperación de oro. 46 3.4 Ensayos de electrólisis Los ensayos de electrólisis se aplicaron tanto a los lixiviados como a los concentrados obtenidos en flotación iónica. También se realizaron ensayos con la presencia de sulfito de sodio con el fin de estabilizar el tiosulfato presente en los lixiviados. Por último se modificaron parámetros como el tiempo y el voltaje aplicado a la celda electrolítica. En la Fig. 4 se observa la recuperación de oro en el área catódica obtenida con y sin la adición de sulfito de sodio. 3.3 Flotación iónica de lixiviados En la Fig. 3 se muestra la recuperación de oro acumulada en el concentrado de flotación iónica. Las pruebas de flotación iónica se realizaron con el colector Aliquat al 100 % de concentración y con el espumante Flomin F-121 al 1 % de concentración. Los reactivos de flotación fueron añadidos al 0,1 % de concentración en los lixiviados a flotar. Se obtuvo una recuperación acumulada de oro del 84% en un tiempo de 40 minutos. Otro aspecto importante a recalcar es el volumen acumulado de concentrado obtenido. En el 60 % del volumen inicial de lixiviado, se recuperó el 84 % del oro. En otras palabras, en la mayor parte del lixiviado original, se logró concentrar y recuperar la mayor parte del oro. Figura 3. Recuperación de oro a partir de lixiviados con tiosulfato de sodio mediante flotación iónica (tiempo = 40 min; [Na2S2O3] = 1.0 M; [NH3] = 0.1 M; [Cu2+] = 2 mM; pH = 10.0; colector Aliquat al 100 % y espumante Flomin F-121 al 1 % de concentración) La razón por la que la se recupera la mayor cantidad de oro contenido en el lixiviado, radica en la afinidad que presenta el compuesto Aliquat (amina cuaternaria) por el oro. El colector Aliquat se adhiere con el complejo oro-tiosulfato y la parte hidrofóbica de esta amina cuaternaria se adsorbe con las burbujas de aire estabilizadas por el espumante Flomin F-121. Las burbujas de aire suben junto al oro en forma de espuma y son recuperadas en el concentrado de flotación. Asimismo, es importante tener en cuenta que el concentrado de flotación obtenido es acuoso ya que la cantidad de colector orgánico usado es menor en comparación al lixiviado flotado. Al agregar el Aliquat en una concentración del 0.1 % en el lixiviado, se logra obtener un concentrado de flotación acuoso, lo que evita la aplicación de una etapa de reextracción para separar el oro de la fase orgánica. Por lo tanto, es factible usar el concentrado de flotación iónica en el proceso de electrólisis para la posterior recuperación de oro. Figura 4. Recuperación de oro a partir de lixiviados con tiosulfato de sodio sin sulfito de sodio (0.4 V; 12.6 mA; pH = 10.0) y con sulfito de sodio (0.35 V; 12.6 mA;pH = 10,0; [Na2SO3] = 0.1 M) La concentración de sulfito de sodio en el lixiviado fue de 0.1 M. La razón por la que se agregó sulfito de sodio en uno de los ensayos de electrólisis fue para estabilizar el complejo oro-tiosulfato presente en los lixiviados. De esta manera se evita que el oro precipite en forma de sulfuro durante la reducción del complejo oro-tiosulfato en el área catódica. No obstante, la recuperación de oro obtenida en el ensayo con sulfito de sodio fue de 25 % la cual es menor a la recuperación de oro obtenida en el ensayo sin sulfito de sodio que fue de 28 %. A pesar de que se evitó la formación de sulfuro de oro en forma de precipitado negro con la adición de sulfito de sodio, la recuperación de oro conseguida fue menor. La baja recuperación de oro obtenida se debe a que las reacciones parásitas (como la reducción de agua y oxígeno además de la coprecipitación de otros metales como la plata) sucedidas en el área catódica consumen gran parte de la corriente aplicada para la precipitación del oro. Se aplicó la técnica de electrólisis tanto a lixiviados como a concentrados de flotación iónica para determinar si existía algún impedimento para la aplicación de este proceso en los concentrados de flotación iónica debido a la presencia de componentes orgánicos. Los ensayos se realizaron durante una hora aplicando una corriente de 0.4 V y sin la presencia de sulfito de sodio. En la Fig. 5 se puede observar que las recuperaciones obtenidas para los dos ensayos fueron muy similares. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Recuperación de Oro a Partir de Minerales Polisulfurados con Soluciones Amoniacales de Tiosulfato de Sodio _________________________________________________________________________________________________________________________ 47 4. CONCLUSIONES La lixiviación con soluciones amoniacales de tiosulfato de sodio permite obtener recuperaciones de oro de 81 % al cabo de una hora de iniciado el proceso al 33 % de sólidos. Al incrementar la cantidad de mineral a lixiviar, se requiere de un aumento de los agentes de lixiviación con el fin de compensar la mayor cantidad de oro a disolver. Por lo tanto, al encontrar un adecuado equilibrio entre las concentraciones de los reactivos de lixiviación, se puede superar al proceso de cianuración tanto de recuperación como en velocidad de disolución de oro. Figura 5. Recuperación de oro a partir de lixiviados con tiosulfato de sodio (0.4 V; 12.6 mA; pH = 10.0) y a partir de concentrados de flotación iónica (0.4 V; 12.6 mA;pH = 10.0) En el ensayo de electrólisis aplicado al lixiviado se obtuvo una recuperación de oro de 28 % mientras que en el ensayo realizado con el concentrado de flotación iónica se obtuvo una recuperación de oro de 29 %. Por lo tanto se puede afirmar que la presencia de Aliquat en el concentrado de flotación iónica no impide la aplicación del proceso de electrólisis para la deposición de oro. Debido a la poca cantidad de Aliquat utilizado en la etapa de flotación iónica, el concentrado obtenido se comporta de forma similar a los lixiviados. Por último se varió la corriente inducida con el fin de recuperar una mayor cantidad de oro. En estos ensayos se utilizaron solo concentrados de flotación iónica. Además se incrementó el tiempo de duración del proceso para conseguir la recuperación de oro más alta posible. Se aplicaron voltajes de 0.4 y 1.5 V para determinar si una mayor corriente aplicada permite incrementar la recuperación de oro. En la Fig. 6 se advierten los resultados obtenidos a los dos voltajes aplicados. Al inducir un voltaje de 0.4 V durante 3 horas, se consigue una recuperación de oro de 63 %. En cuanto al experimento desarrollado con 1.5 V, la recuperación de oro obtenida fue de 82 %. Evidentemente, un aumento en el voltaje aplicado permitió obtener una mayor recuperación de oro. Al tener una mayor corriente disponible tanto para la reducción del complejo oro-tiosulfato como para el resto de reacciones parásitas y paralelas, se logra mejorar la recuperación de oro. El pH del proceso de lixiviación con tiosulfato debe mantenerse en un valor superior a 10 pero no mayor a 11 para favorecer la acción de la cuprotetramina, responsable de catalizar la disolución del oro contenido en el mineral. La concentración de iones cúprico permiten acelerar la disolución de oro pero un excesivo incremento de estos puede provocar la oxidación del tiosulfato hacia politionatos lo que va en detrimento de la recuperación de oro en el lixiviado, debido a que estos no intervienen en la disolución del oro. Además un menor tamaño de partícula del mineral favorecerá la recuperación de oro en el lixiviado al darse un contacto más íntimo entre el mineral a lixiviar y los reactivos de lixiviación. Las técnicas convencionales de recuperación de oro a partir de lixiviados como son adsorción con carbón activado y cementación con polvo de cobre, no arrojaron buenos resultados en cuanto a recuperación de oro se refiere. En el caso de la adsorción con carbón activado se obtuvo una recuperación de oro de tan solo el 40 %, mientras que en la cementación con polvo de cobre se consiguió una recuperación de oro de 58 %. A pesar de que se experimentó con carbón activado impregnado con cobre metálico, con el fin de aumentar el área disponible de cementación, solo se consiguió una recuperación de oro del 46 %. Las bajas recuperaciones obtenidas se deben, en el caso de la adsorción con carbón activado, a la poca afinidad del complejo oro-tiosulfato hacia la superficie del carbón activado. En los experimentos de cementación, las bajas recuperaciones obtenidas posiblemente se deban a la pasivación de la superficie disponible debido a la precipitación de óxidos o sulfuros. El ensayo de flotación iónica fue el más exitoso de todos, ya que permitió obtener una recuperación de oro del 84 %. La elevada recuperación de oro se debe a la afinidad del colector Aliquat y el complejo oro-tiosulfato. La poca cantidad de colector aplicado en el lixiviado (0.1 % de concentración en solución) permitió obtener un concentrado de flotación acuoso el cual no presenta inconveniente para ser tratado mediante electrólisis. Al aplicar un voltaje de 1.5 V al concentrado de flotación iónica por tres horas, se consiguió una recuperación de oro de 82 % en el área catódica. Figura 6. Influencia de la corriente aplicada en la recuperación de oro a partir de concentrados de flotación iónica (pH = 10.0) Revista Politécnica-Septiembre 2015, Vol. 36, No. 2 48 Recuperación de Oro a Partir de Minerales Polisulfurados con Soluciones Amoniacales de Tiosulfato de Sodio _________________________________________________________________________________________________________________________ REFERENCIAS [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] M.G. Aylmore y D.M. Muir, (2000). Thiosulfate leaching of gold-a review, Minerals Engineering, 14(2): 135-174. T. Fujita, L. Kejun, W.Tai Yen, A.Shibayama y T. Miyazaki, (2004). Gold extraction from thiosulfate solution using trioctylmethylammonium chloride, Hydrometallurgy 73: 41-53. E, Guerra y D.B. Dreisinger, (1999). A study of the factors affecting copper cementation of gold from ammoniacal thiosulfate solution, Hydrometallurgy 51: 155-172. P. Haddad, A. Grosse, G. Dicinoski, y M. Shaw, (2003). Leaching and recovery of gold using ammoniacal thiosulfate leach liquors, Hydrometallurgy 69: 1-18. J. Hiskey y J. Lee, (2003). Kinetics of gold cementation on copper in ammoniacal thiosulfate solutions, Hydrometallurgy 69: 45-56. D. Michel y E. Delgado (2011). Lixiviación de minerales de oro con el uso de tiosulfato: tecnología alterna a la cianuración de minerales de oro, RIIGEO 13(26): 67-72. P. Navarro, C. Vargas, M. Alonso y F. Alguacil, (2006). The adsorption of gold on activated carbon from thiosulfate-ammoniacal solutions, Gold Bulletin 2006: 93-97. P. Navarro, A. Villarroel y F. Alguacil, (2001). Lixiviación de oro con tiosulfato de amonio catalizado con ion cúprico desde un concentrado polimetálico, Jornadas SAM-CONAMET (pp. 93-97). Santiago de Chile, Chile. G. Senanayake, (2012). Gold leaching by copper (II) in amoniacal thiosulphate solutions in the presence of additives. Part I: A review of the effect of hard-soft and Lewis acid-base properties and interactions of ions, Hydrometallurgy 115-116: 1-20. G. Senanayake y X. Zhang, (2012). Gold leaching by copper (II) in amoniacal thiosulphate solutions in the presence of additives. Part II: Effect of residual Cu (II), pH and redox potentials on reactivity of colloidal gold, Hydrometallurgy 115-116: 21-29. Serrano, J. (2012). Diseño de una planta para la recuperación de oro de minerales sulfurados con ditioxamida como lixiviante de baja toxicidad. (Proyecto de titulación Previa a la Obtención del Título de Ingeniero Químico no publicado). EscuelaPolitécnica Nacional, Quito, Ecuador. M. Tsunekawa, R.K. Rath, N. Hiroyoshi T. y Hirajima, (2003). Ammoniacal thiosulfate leaching of gold ore, ejmp&ep, 3(3): 344-352. S. Ubaldini, C. Abbruzzese, P. Fornari, R.Massida y F. Veglio, (1995). Thiosulfate leaching for gld hydrometallurgy, Hydrometallurgy 39:265-276. A. Zelinsky, O. y Novgorodtseva, (2013). EQCM study of dissolution of gold in thiosulphate solutions, Hydrometallurgy 138: 79-83. Revista Politécnica-Septiembre 2015, Vol. 36, No. 2 Estudio de la Detoxificación de Efluentes Cianurados por Oxidación con Dióxido de Azufre, Aire y Catalizadores de Cobre _________________________________________________________________________________________________________________________ 49 Estudio de la Detoxificación de Efluentes Cianurados por Oxidación con Dióxido de Azufre, Aire y Catalizadores de Cobre Campos C.*; De la Torre E.* *Escuela Politécnica Nacional, Facultad de Ingeniería Química y Agroindustria, Quito, Ecuador e-mail: [email protected]; [email protected] Resumen: Se estudió la detoxificación de efluentes cianurados, mediante oxidación con dióxido de azufre (SO 2), aire y catalizadores de Cu. Se ensayaron diversas especies de Cu tales como: CuSO4 en solución, carbón activado granular (CAG) impregnado con CuSO4, CAG impregnado con Cu0 y Cu0 granular. Este proceso es conocido como INCO, el cual oxida el cianuro WAD a cianato y precipita los metales tales como Cu, Ni, Zn, Cd y Fe. Se realizaron ensayos a escala de laboratorio con soluciones sintéticas de 500 mg/L NaCN y efluentes cianurados industriales. Se midió la variación de CN- con respecto al tiempo por titulación con AgNO3. Se estudió la influencia del pH, la dosificación de Na2S2O5 (fuente de SO2) y las concentraciones de los diferentes catalizadores. Los tiempos de oxidación del 98 % de CN- de las soluciones sintéticas de NaCN, bajo las mejores condiciones de operación del proceso INCO (pH 10.0; 276 N L/h aire; 400 RPM; y 1000 mg/L Na2S2O5) fueron 210 min con 50 mg/L Cu2+ (CuSO4), 40 min con 50 g/L de CAG impregnado con CuSO4, 8 min con 50 g/L de CAG impregnado con Cu0 y 195 min con 10 g/L Cu0 granular. Los efluentes industriales fueron detoxificados utilizando 5 g/L CAG impregnado con Cu0. Estos efluentes, con concentraciones iniciales de 432.50 mg/L y 112.50 mg/L de cianuro total alcanzaron 0.06 mg/L cianuro total en 90 min y 0.05 mg/L cianuro total en 40 min, respectivamente. El contenido de metales en los efluentes (Cu, Cd, Zn, Ni y Fe) luego de la precipitación llegaron a los límites permisibles de descarga a un cuerpo de agua dulce según lo establecido en el TULAS. El precio del tratamiento de efluentes cianurados por el método INCO con CAG impregnado con Cu0 es de 6.70 USD/m3. Palabras clave: efluentes cianurados, proceso INCO, carbón activado impregnado con cobre Abstract: It was studied the detoxification of cyanide effluents by oxidation with sulfur dioxide (SO 2), air and copper catalyzer. The CuSO4 solution, granular activated carbon (GAC) impregnated with CuSO4, GAC impregnated with Cu0 and granular Cu0 were studied. This process is called INCO process and it oxidizes WAD cyanide to cyanate and precipitates metals such as Cu, Ni, Zn, Cd and Fe. Experiments were conducted at laboratory scale using synthetic solutions of 500 mg/L NaCN and industrial cyanide effluents. The variation of the CN- concentration in time was determined titrimetrically with AgNO3 solution. The influence of pH and the concentrations of Na2S2O5 (SO2 source of process) and Cu catalysts on the rate of the removal of CN - were estudied. The oxidation times of the 98 % of cyanide removal from the synthetic solutions of NaCN based on the best operation conditions (pH 10.0; 276 N L/h air; 400 RPM and 1000 mg/L Na2S2O5) were: 210 min (50 mg/L Cu2+ obtained from CuSO4), 40 min (50 g/L GAC impregnated with CuSO4), 80 min (5 g/L GAC impregnated with Cu0) and 195 min (10 g/L granular Cu0). The industrial effluents were detoxified using 5 g/L GAC impregnated with Cu0. These effluents with initial concentrations of 432.50 mg/L and 112.50 of total cyanide reached 0.06 mg/L total cyanide in 90 min and 0.05 mg/L total cyanide in 40 min, respectively. Besides, the metal content (Cu, Cd, Zn, Ni and Fe) of the industrial effluents after precipitation reached the permissible limits for discharges into a fresh body water according to TULAS. The price of cyanide wastewater treatment by the INCO method using GAC impregnated with carbon is 6.70 USD/m3. Keyword: Cyanide effluents, INCO process, copper impregnated activated carbon 1. INTRODUCCIÓN En el Ecuador, a lo largo de la historia la industria minera ha causado grandes impactos ambientales, especialmente en las zonas de Portovelo-Zaruma y Ponce Enríquez debido a las descargas directas o indirectas de relaves contaminados con cianuro y metales pesados a los ríos. Estas descargas han provocado la extinción de todas las formas de vida, ya que el agua no es apta para el consumo de los seres vivos [7, 10]. El cianuro puede encontrarse en forma de cianuro libre, complejos cianurados débiles y moderadamente fuertes (cianuro WAD, formado por Cd, Cu, Zn, Ni y Ag) y complejos cianurados fuertes (cianuro SAD, formado por Au, Hg, Fe y Co). En cualquiera de sus clasificaciones el cianuro constituye un compuesto muy tóxico sobre las especies vivas, razón por la cual la industria minera se ha visto obligada a Revista Politécnica - Septiembre 2015, Vol. 34, No. 2 50 Campos C.*; De la Torre E.* _______________________________________________________________________________________________________________________________ desarrollar procesos de destrucción y recuperación de cianuro. Los procesos de destrucción se basan en convertir el cianuro en compuestos menos tóxicos a través de reacciones de oxidación. Entre estos procesos se encuentran el método INCO, la oxidación con peróxido de hidrógeno, ácido de caro, carbón activado y aire, cloración alcalina y tratamientos biológicos. Por otro lado, los procesos de recuperación tienen como objetivo recuperar el cianuro empleado en la lixiviación del oro y reutilizarlo, tal es el caso de los procesos AVR y SART [6, 2]. Esta investigación se basa en el estudio del proceso INCO, el cual requiere el uso de SO2 y aire en presencia de iones Cu2+ como catalizador bajo condiciones controladas de pH (8-10). El O2 requerido para la oxidación del cianuro es suministrado en el reactor mediante burbujas de aire. El sulfito de sodio (Na2SO3) y el metabisulfito de sodio (Na2S2O5) son los agentes reductores comúnmente usados como fuente de SO2 del proceso y el Cu2+ es obtenido a partir de CuSO4 en solución. El pH de la solución es controlado con cal (CaO) o hidróxido de sodio (NaOH) [8]. El proceso INCO oxida rápidamente el cianuro WAD a cianato y precipita los metales liberados (Cd, Cu, Zn, Ni y Ag) en forma de hidróxidos, a excepción de los complejos cianurados de Fe que precipitan como sales metálicas de ferrocianuro de Cu, Ni o Zn. Las principales variables de este proceso son el pH, las dosificaciones de SO2 y Cu, la velocidad de alimentación de aire y el tiempo de retención. Las dosis de SO2 y Cu empleadas en el proceso dependen de la concentración de cianuro WAD en la solución [7]. El SO2 -- disuelto en la solución forma el ión sulfito de acuerdo a la Ecuación 1. a pH alcalino - El es el reactante del proceso del proceso INCO [3, 4]. La conversión de cianuro WAD a cianato se da de acuerdo a las a reacciones presentadas en las ecuaciones 2 y 3, donde M puede ser Zn2+; Cu2+; Ni2+; Cd2+ and Ag2+ [4]. - - - - → - - (1) → - (2) - - (3) La remoción de cianuro de hierro inicia con la reducción del Fe de estado férrico a estado ferroso, como se muestra en la Ecuación 4. - - - (4) Posteriormente, los complejos de cianuro ferroso precipitan con el Cu, Ni o Zn, de acuerdo a la reacción mostrada en la Ecuación 5. - (5) Los metales remanentes en la solución precipitan en forma de hidróxidos. La precipitación del Cu ocurre de acuerdo a la Ecuación 6, mientras que el Ni y el Zn precipitan conforme la Ecuación 7. - - - - (6) (7) El objetivo principal de este estudio es dar una nueva alternativa al sector minero del Ecuador, para el tratamiento de los efluentes cianurados con base en la innovación del proceso INCO. Para esto se analizará los principales parámetros de operación del proceso (pH, dosificación de Na2S2O5 y CuSO4) y se estudiará el empleo de otros catalizadores como el Cu0 granular, carbón activado granular (CAG) impregnado con CuSO4 y CAG impregnando con Cu0 a fin de mejorar la cinética del proceso INCO y llegar al límite permisible de cianuro total y metales establecidos en el TULAS. Cabe indicar que estos catalizadores han sido escogidos debido a que el CAG en presencia de Cu ha demostrado adsorber aproximadamente 22.4 mg CN-/g CAG. Además, el Cu2+ y Cu1+ cataliza la reacción de oxidación del cianuro e incrementa la capacidad de adsorción del CAG [2, 5, 9]. 2. METODOLOGÍA Se realizaron ensayos con soluciones sintéticas de 500 mg/L NaCN (265 mg/L CN-) y con efluentes cianurados provenientes del proceso de cianuración de dos compañías mineras del Ecuador. Los reactivos empleados para los ensayos fueron soluciones de CuSO4 al 5 %, Na2S2O5 al 20%, NaOH 1N y HCl al 5 %, las mismas que fueron preparadas con reactivos grado analítico y agua destilada. Además se usó Cu0 granular (0.8 × 0.6 mm) y CAG virgen (4.8 × 2.4 µm y No. Iodo 523 mg I2/g CAG) procesado a partir de cuesco de palmiste por carbonización y activado físicamente con vapor de agua. 2.1 Ensayos con soluciones sintéticas de NaCN Se realizaron pruebas con 1 L de solución de 500 mg/L NaCN a temperatura ambiente (18 - 20 °C) y presión atmosférica (0.72 atm). Las variables analizadas fueron: el pH (10.0 – 11.0), la dosificación de Na2S2O5 (500 mg/L - 3000 mg/L) y la concentración de cada uno de los diferentes catalizadores de Cu: CuSO4 en solución (20 mg/L - 50 mg/L), CAG impregnado con CuSO4 (0 g/L - 50 g/L), CAG impregnado con Cu0 (0 g/L - 50 g/L) y Cu0 granular (10 g/L). El pH fue medido con el pHmetro HANNA HI98128 y contralado con soluciones de HCl al 5 % y NaOH 1N. Se alimentó un flujo de aire constante de 276 NL/h, suministrado por el compresor Air America de 1 HP y medido con un rotámetro de gases marca ROTA. Se Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Estudio de la Detoxificación de Efluentes Cianurados por Oxidación con Dióxido de Azufre, Aire y Catalizadores de Cobre _________________________________________________________________________________________________________________________ empleó agitación mecánica a 400 RPM con el agitador BOECO modelo bUSD-20. 51 2.1.5 Obtención de CAG impregnado con Cu0 Se realizaron dos tipos de ensayos, en el primero se adicionó el Na2S2O5 al 20 % solo al comienzo del proceso de oxidación y se determinó la cantidad de N2 S2O5 en la solución cianurada cada 30 min con el fin de analizar la cinética de la reacción con respecto a este agente oxidante. En el segundo tipo de ensayo se mantuvo constante la concentración de Na2S2O5 durante todo el proceso de oxidación, para ello se determinó cada cierto tiempo (10 a 30 min, según la cinética del proceso) la cantidad de Na2S2O5 consumido y se repuso dicho consumo. En los dos casos la cantidad de Na2S2O5 se determinó con el método de Ripper Simple (Análisis basado en titulación yodimétrica, empleado para la determinación del contenido de SO2 en vinos y mostos). El CAG impregnado con Cu0 se obtuvo mediante la electrodeposición de Cu0 sobre el CAG virgen. El ánodo y el cátodo fueron placas de Cu de 15×15 cm con un espesor de 3 mm. El cátodo fue recubierto con 25 g de CAG virgen por cada lado de la placa sostenido por una malla plástica. Los electrodos se colocaron dentro de un recipiente de 6 L de solución electrolítica (120 g/L H2SO4 y 60 g/L CuSO4) con un pH de 1.5. La electrodeposición se llevó a cabo por 4 h con un voltaje de 1 V. Una vez finalizada la electrodeposición, se retiró los cátodos del recipiente y se quitó la malla plástica que cubría el cátodo a fin de liberar el CAG impregnado con Cu0, el cual fue lavado con agua potable para eliminar el sulfato de cobre acumulado en su superficie y secado en la estufa a 110 ºC. Finalmente, se determinó el contenido de Cu0 del CAG mediante la diferencia de pesos del CAG antes y después de la electrodeposición. 2.1.2. Estudio Cinético de la oxidación de cianuro libre 2.1.6 Obtención de CAG impregnado con CuSO4 2.1.1. Dosificación de metabisulfito de sodio - Para el estudio cinético de la oxidación cianuro libre (CN ), se determinó la concentración de CN- con respecto al tiempo por volumetría con una solución de 1.6987 g/L AgNO3 y KI al 10 % como indicador, con base en la norma ASTM D-2036. 2.1.3. Determinación de cianuro WAD y cianuro total Una vez definidas las mejores condiciones de operación del proceso (pH, dosificación de Na2S2O5 y la concentración del catalizador seleccionado), se determinó el contenido de cianuro total y cianuro WAD. Para esto la solución detoxificada por fue sometida al proceso de destilación ácida que consiste en la descomposición de los complejos cianurados, mediante la acidificación de la muestra a fin de producir vapores de ácido cianhídrico (HCN) que a través de la destilación son recogidos en una solución de hidróxido de sodio (NaOH) para formar NaCN, que finalmente se cuantifica por el método colorimétrico piridina-pirazolona con el espectrofotómetro HACH DR 2800 (método que determina de 0.002 a 0.204 mg/L CN-). 2.1.4 Determinación del contenido de Cu en las soluciones sintéticas de NaCN Se determinó la cantidad de Cu (proveniente de los diferentes catalizadores) en la solución de NaCN por absorción atómica antes y después de la precipitación de Cu. Para determinar la cantidad de Cu antes de la precipitación, se tomó una alícuota de 10 mL una vez finalizado el proceso de oxidación y se añadió 0.1 mL de HCl concentrado, a fin de bajar el pH a 4.5 y disolver el Cu precipitado. Mientras que para conocer el contenido de Cu residual en la solución cianurada después de la precipitación se tomó una alícuota de 10 mL y se filtró la solución para separar el Cu precipitado. Para la obtención del CAG impregnado con CuSO4 se colocaron 50 g de CAG virgen y 100 mL una solución de CuSO4 al 15 % en un vaso de precipitación de 600 mL. Se agitó la mezcla a velocidad media en una plancha de agitación magnética (Lab Tech, LMS-3006, 110V) por un lapso de 2 h. Luego, se separó el CAG impregnado con CuSO4 de la solución remanente de CuSO4 con ayuda de una coladera, se lavó con agua potable el CAG impregnado con CuSO4 y se secó en una estufa a 110 ºC. Finalmente, se determinó el contenido de Cu presente en el CAG por absorción atómica (equipo Perkin Elmer AAnalyst 300). 2.2 Ensayos con efluentes cianurados industriales El tratamiento de detoxificación de los efluentes industriales se realizó bajo las mejores condiciones de pH, dosificación de Na2S2O5, y concentración del catalizador de Cu seleccionado, determinadas en este estudio a partir del tratamiento de detoxificación de las soluciones sintéticas de NaCN. El estudio cinético del proceso de oxidación se realizó de la misma manera que en los efluentes sintéticos, es decir con base en la de determinación de CN- a diferentes intervalos de tiempo. 2.2.1 Muestreo de los efluentes industriales Para evaluar el tratamiento de detoxificación de efluentes industriales del proceso de cianuración, se realizó un muestreo simple de los efluentes de las compañías mineras “ r na .A.” y “Paz Borja” ubicadas en el cantón Camilo Ponce Enríquez (Azuay), con base en la norma NTE INEN 2 169:98, la cual describe el muestreo, manejo y conservación de muestras de agua. Revista Politécnica - Septiembre 2015, Vol. 34, No. 2 52 Campos C.*; De la Torre E.* _______________________________________________________________________________________________________________________________ 400 Una vez obtenidas las muestras representativas de los efluentes industriales, se realizó la caracterización química de las mismas mediante la determinación del pH y del contenido de Au, Ag, Cu, Zn, Cd, Ni y Fe por absorción atómica. Además, se analizó el contenido de CN-, CNWAD y CNTOTAL como se explico en la sección 2.1.3. 350 Una vez terminado el proceso de oxidación se realizaron análisis de CNWAD, CNTOTAL, y del contenido de Au, Ag, Cu, Zn, Cd, Ni y Fe de los efluentes cianurados detoxificados, a fin de conocer el porcentaje de remoción de dichos parámetros con respecto a los valores obtenidos en la caracterización química y compararlos con los límites de descarga a un cuerpo de agua dulce, establecidos en el Anexo 1 del Libro VI del TULAS. Los efluentes industriales detoxificados que no cumplieron con la normativa ambiental, fueron sometidos al proceso de oxidación por un mayor tiempo al determinado por el método volumétrico, hasta llegar a valores iguales o inferiores a 0.1 mg/L de CNTOTAL. 3.1 Detoxificación de soluciones sintéticas de NaCN 3.1.1. Influencia del pH en la oxidación de cianuro libre y remoción de Na2S2O5 En la Fig. 1 se muestra el efecto del pH (10.0 y 11.0) en la oxidación de CN- y en la remoción de Na2S2O5 en función del tiempo en una solución sintética de 500 mg/L NaCN (265.4 mg/L CN-). Los ensayos se realizaron con un flujo constante de aire de 276 NL/h, una concentración inicial de 2000 mg/L Na2S2O5 y 50 mg/L Cu2+. A pH 10.0 la remoción de CN- es muy rápida durante los primeros 150 min en los cuales se oxidan alrededor de 160 mg CN-, luego de este tiempo la reacción se vuelve lenta oxidando alrededor de 60 mg CN- en 360 min. Esto se debe a que a pH 10.0 el Na2S2O5 se consume por completo a los 150 min, por ende a partir de este momento la oxidación del CN- se da únicamente debido a la presencia de O2 de acuerdo a la Ecuación 8. - (8) a Por otro lado, a pH 11.0 la oxidación de CN- es lenta desde el inicio del proceso hasta el final del mismo al igual que el consumo de Na2S2O5, ya que luego de 480 min de reacción aún hay una concentración aproximada de 100 mg/L de Na2S2O5. Esto demuestra que a pH 10.0 se ve favorecida la oxidación de CN- a CNO- según la Ecuación 2, en la cual el - obtenido del Na2S2O5 actúa como agente reductor pasando a ión sulfato - . [CN-] (mg/L) 300 1600 250 1200 200 150 800 100 400 50 0 0 0 100 200 300 400 500 Tiempo (min) [CN-]; pH=11 [CN-] pH=11 [CN-] pH=10 pH=11 [Na2S2O5]; [CN-]; pH=10 [Na2S2O 5] pH=11 [Na2S2O5] pH=10 [Na2S2O5]; pH=10 Figura 1. Influencia del pH en la oxidación de CN- y consumo de Na2S2O5 en función del tiempo en soluciones sintéticas de 265 mg/L CN(2000 mg/L Na2S2O5; 50 mg/L Cu2+; 400 RPM y 276 NL/h aire) 3.1.2 Influencia de la dosificación de Na2S2O5 en la oxidación de CN- 3. RESULTADOS Y DISCUSIÓN - 2000 [Na2S2O5] (mg/L) 2.2.2 Caracterización química de los efluentes industriales En la Fig. 2 se muestra la influencia de la dosificación inicial de Na2S2O5 (1000, 2000 y 3000 mg/L Na2S2O5) en la cinética de oxidación de CN- con un flujo constante de aire de 276 NL/h; 400 RPM; pH 10.0 y 50 mg/L Cu2+. Se observa que un aumento en la concentración inicial de Na2S2O5 no influye significativamente en la remoción de CN-, ya que al trabajar con una concentración de 1000 mg/L Na2S2O5 se remueve 76 % de CN- en 480 min, mientras que con 2000 y 3000 mg/L Na2S2O5 se remueve el 84 % de CN-. Por otro lado la Fig. 2 indica que los 1000 mg/L de Na2S2O5 se consumen completamente en 120 min, los 2000 mg/L en 150 min y los 3000 mg/L en 240 min, lo que significa que a partir de los tiempos mencionados la oxidación del CN- se da solo en presencia de O2 como se muestra en la Ecuación 8. Esto demuestra que el exceso de Na2S2O5 no aumenta la velocidad de oxidación de CN- a CNO-. Según los estudios realizados por Breuer et al. (2011) esto se debe a que el exceso de - obtenido del Na2S2O5 se - oxida de acuerdo a la Ecuación 9, mientras que el solo reacciona con la cantidad estequiométrica requerida como se indica en la Ecuación 2. - → - (9) Esto sugiere que el Na2S2O5 se debería mantener a una - concentración constante a fin de que el este presente durante todo el proceso de oxidación. En la Fig. 3 se presenta la cinética de oxidación de CN - a las mismas condiciones de los ensayos de Fig. 2 pero manteniendo contante las concentraciones de 500, 1000 y 2000 mg/L Na2S2O5. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Estudio de la Detoxificación de Efluentes Cianurados por Oxidación con Dióxido de Azufre, Aire y Catalizadores de Cobre _________________________________________________________________________________________________________________________ 53 van añadiendo paulatinamente de acuerdo a las necesidades del proceso) logrando un 98 % de remoción de CN- en 210 min. Sin embargo, con base en la Fig. 2 al añadir al inicio del proceso 3000 mg/L Na2S2O5 se remueve aproximadamente el 84 % de CN- en 480 min. Esto demuestra que al ir añadiendo paulatinamente el Na2S2O5 de acuerdo a las necesidades del proceso de oxidación se minimiza la reacción directa del - con el O2 y por ende se evita el consumo innecesario de este gas, el cual limita la velocidad de oxidación del CNTabla 1. Resultados de la cinética de oxidación de CN- de soluciones sintéticas de 265 mg/L CN- a pH 10.0; 50 mg/L Cu2+; 276 NL/h y 400 RPM con diferentes dosificaciones constantes de Na2S2O5 Figura 2. Influencia de la dosificación inicial de Na2S2O5 en la oxidación de CN- y consumo de Na2S2O5 en función del tiempo en soluciones sintéticas de 265 mg/L CN- (pH 10,0; 50 mg/L Cu2+; 276 N L/h aire y 400 RPM) [CN-] (mg/L) [N2S2O5]constante (mg/L) Consumo promedio [N2S2O5] (mg/L) Oxidación CN- (%) Tiempo oxidación (min) 500 1000 2000 2 500 3000 5 500 98 98 98 350 210 220 3.1.3 Influencia de la dosificación de los catalizadores de Cu en la oxidación de CN- 300 Los estudios realizados por Breuer et al. (2011), indican que el Cu2+ obtenido a partir del CuSO4 actúa como un 250 [Na2S2O5]consta [Na2S2O5] = 500 mg/L nte= 500 mg/L 200 [Na2S2O5]consta [Na2S2O5] = 1000 mg/L nte= 1 000 mg/L 150 [Na2S2O5] = 2000 mg/L [Na2S2O5]consta nte= 2 000 mg/L 100 - catalizador en la oxidación del con O2, facilitando la - transferencia de electrones del al O2. En la Fig. 4 se presenta la influencia de la dosificación de Cu2+ (50, 40 y 20 mg/L) en la oxidación del CN-, al emplear una concentración constante de 1000 mg/L de Na2S2O5; 400 RPM y 276 NL/h aire. Además, señala que el CN- se oxida paralelamente con el - 50 0 0 100 200 Tiempo (min) 300 400 Figura 3. Influencia de la dosificación constante de Na2S2O5 en la oxidación de CN- en función del tiempo en soluciones sintéticas de 265 mg/L CN(pH 10,0; 50 mg/L Cu2+; 276 N L/h aire y 400 RPM) Se observa que con una concentración constante de 500 mg/L Na2S2O5 se logra oxidar el 98 % de en 350 min, mientras que con 1000 y 2000 mg/L Na2S2O5 se oxida la misma cantidad de CN- en 210 y 220 min, respectivamente. Esto demuestra que los mejores resultados en la remoción de CN- se obtienen al usar una concentración constante de 1000 mg/L Na2S2O5 y que el exceso de este agente reductor no contribuye a la remoción del CN-. En la Tabla 1 se muestran los resultados de la cinética de oxidación de CN- y se detalla el consumo promedio de Na2S2O5 de los ensayos presentados en la Fig. 3. El empleo de una concentración constante de 1000 mg/L Na2S2O5, equivale a usar un promedio de 3000 mg/L Na2S2O5 (que se de acuerdo a la Ecuación 2 y que al añadir Cu2+ a la solución cianurada se forma que actúa como catalizador de esta reacción. Por esta razón en la Fig. 4 se observa que a medida que aumenta la cantidad de Cu2+ empleado en el proceso de oxidación, disminuye el tiempo de remoción de CN-. En los últimos minutos del proceso se observó que la solución cianurada que inicialmente era transparente empezó a tornarse celeste como se observa en la Fig. 5, lo cual se debe presumiblemente a la precipitación del Cu en forma de hidróxido según la Ecuación 6. Esto sugiere que inicialmente se oxida el CN- presente en la solución, posteriormente el y finalmente precipita el Cu liberado del complejo cianurado de Cu. Adams (1994) señala que El Cu1+ y el Cu2+ catalizan la oxidación del cianuro con CAG en presencia de O2, y a su vez aumentan la capacidad de adsorción de este catalizador. Esto se atribuye al incremento de sitios activos en la superficie del CAG y a la formación de especies complejas de cianuro. Revista Politécnica - Septiembre 2015, Vol. 34, No. 2 54 Campos C.*; De la Torre E.* _______________________________________________________________________________________________________________________________ en los sitios activos del CAG, que al ser impregnado con Cu actúa como un catalizador redox en presencia de aire. [CN-] 300 (mg/L) 2+ [Cu ]= 20 mg/L [Cu2+]=20 mg/L 250 Por otro lado, al introducir por separado 50 g/L de CAG con 50 mg/L Cu2+ se remueve el 98 % de CN- en 195 min, con 50 mg/L de Cu2+ en 210 min, mientras que con 30 g/L de CAG impregnado con CuSO4 se alcanza este porcentaje de remoción en 85 min. Esto muestra que el mejor efecto catalítico en la oxidación del CN - se obtiene con CAG impregnado con CuSO4 debido al efecto catalizador del CAG y del y las propiedades de adsorbentes del CAG. [Cu2+]= 40 mg/L [Cu2+]=40 mg/L 200 2+ [Cu ]= 50 mg/L [Cu2+]=50 mg/L 150 100 50 [CN-] 300 (mg/L) 0 50 g g/LCA CAG imp. CuSO4 50 impregnado CuSO4 50 CA +virgen + Cu 502+ 50 g/L g/L CAG 50 mg/L mg/L Cu2+ g/L CAG virgen 5050g/L CA virgen 250 0 100 200 300 400 500 Tiempo (min) Figura 4. Influencia de la dosificación de Cu2+ en la oxidación de CN- en función del tiempo en soluciones sintéticas de 265 CN- (pH 10.0; 1000 mg/L Na2S2O5, 50 mg/L Cu2+; 400 RPM y 276 NL/h aire) 200 2+ mg/L Cu 5050mg/L Cu2+ 150 imp. CuSO4 3030gg/L CACAG impregnado CuSO4 100 50 0 0 (a) (b) 40 80 120 160 200 240 280 320 360 400 Tiempo (min) Figura 6. Influencia de la dosificación de CAG impregnado con CuSO4 (0,24 % Cu), CAG virgen (malla 4×8, No. Iodo 523 mg I2/g CAG) y Cu2+ en la oxidación de CN- en función del tiempo de soluciones sintéticas de 265 mg/L CN- (pH 10.0; 1000 mg/L Na2S2O5, 400 RPM y 276 NL/h aire) En la Fig. 7 se presentan las cinéticas de oxidación de CN- en una solución al emplear de 1 a 50 g/L de CAG impregnado con Cu0, todos los ensayos se realizaron a pH 10.0; 1000 mg/L Na2S2O5; 400 RPM y 276 NL/h aire. (c) (d) Figura 5. Oxidación de CN- y precipitación de Cu. (a) Inicio del proceso, (b) Final del proceso (precipitación de Cu), (c) Sedimentación del precipitado Cu y (d) Precipitado Cu En la Fig. 6 se presenta la cinética de oxidación de CN- al emplear 50 y 30 g/L de CAG impregnado con CuSO4 (0.24% Cu en peso). Además, se muestra la cinética de oxidación con 50 g/L de CAG virgen, 50 g/L de CAG virgen con 50 mg/L de Cu2+ (obtenido a partir de CuSO4) y 50 mg/L Cu2+. Todos los ensayos se llevaron a cabo a pH 10.0; 1000 mg/L Na2S2O5; 276 NL/h aire y 400 RPM. Se observa que al trabajar con 50 g/L de CAG virgen se logra oxidar el 98 % de CN- en 360 min mientras que al usar 50 g/L de CAG impregnado con CuSO4 se remueve esta misma cantidad de CN- en 40 min, lo que indica que el CAG activado impregnado con CuSO4 mejora hasta 9 veces el tiempo de oxidación de CN-. Según Deveci et al. (2006) este comportamiento se debe a la formación de cargas positivas Además, con el objetivo de estudiar el efecto catalizador del Cu0 se realizó un ensayo con 10 g/L de Cu0 granular (0.6×0.8 mm). Los ensayos con CAG impregnado con Cu0 de muestran que la velocidad de remoción de CN- es muy rápida desde el inicio del proceso hasta el final del mismo y que se incrementa en función de la cantidad de catalizador empleado. Al usar 50 g/L de catalizador se remueve el 98 % de CN- en 8 min, mientras que con 5 g/L este porcentaje de remoción se alcanza en 80 min. Por otra lado, se tiene que el empleo de 10 g/L Cu0 granular presenta la cinética más lenta en la remoción de CN- de la Fig. 7, con el 98 % de oxidación CNen 220 min que es un valor similar al obtenido con 1 g/L de CAG impregnado con Cu0. A fin de realizar un análisis comparativo de todos los catalizadores empleados en la oxidación de CN - y definir el que mejor se ajuste a las necesidades del proceso para posteriormente tratar los efluentes cianurados industriales del proceso de cianuración. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Estudio de la Detoxificación de Efluentes Cianurados por Oxidación con Dióxido de Azufre, Aire y Catalizadores de Cobre _________________________________________________________________________________________________________________________ 55 CAG virgen con 50 mg/L Cu2+ y con los 10 g/L Cu0 granular se llega a 0.89 mg/L Cu. [CN-] 300 (mg/L) 250 Tabla 2. Resultados de la cinética de primer orden con respecto al CN- de la oxidación de soluciones sintéticas de 265 mg/L CN- (pH 10,0; 1000 mg/L Na2S2O5; 400 RPM y 276 NL/h aire con diferentes catalizadores de Cu) 200 150 100 Parámetros 50 0 0 20 40 60 80 100 120 140 160 180 200 220 240 Tiempo (min) g/L CAG impregnado con Cu0 5050g/L CA impregnado 0 granular g/L Cu 1010g/L Cu0 30 g/L CAG impregando con 3020g/L CA impegnado g/L CAG impregnado conCu 0 2010g/L CA impregnado g/L CAG impregnado con Cu0 105 g/L g/LCAG CA impregando Cu 0 impregnadodo con g/LCA CAGimpregnado impregnado con Cu0 5 1g/L Cu0 Figura 7. Influencia de la dosificación de CAG impregnado Cu0 (20 % Cu0); CAG virgen (malla 4×8, No. Iodo 523 mg I2/g) y Cu0 granular (0.6×0.8 mm) en la oxidación de CN- en función del tiempo de soluciones sintéticas de 265 mg/L CN- (pH 10.0; 1000 mg/L Na2S2O5; 400 RPM y 276 NL/h aire) En la Tabla 2 se presentan un resumen de los tiempos de oxidación obtenidos con los diferentes catalizadores y la cantidad de Cu residual proveniente de los mismos antes y despúes de la precipitación de Cu. Todos los ensayos se realizaron a pH 10.0, una concentración constante de 1000 mg/L Na2S2O5, 276 NL/h aire y 400 RPM,que constituyen los mejores parámetros en el proceso. Se observa que los mejores resultados en la remoción de CN- se obtienen al emplear CAG impregnado con Cu0. Sin embargo, al usar concentraciones superiores a 5 g/L de este catalizador, se obtienen concentraciones de Cu residual superiores a 20 mg/L Cu luego de la precipitación, siendo este un valor que excede el permitido por TULAS que establece un límite permisible de 1.0 mg/L Cu. Esto se debe a que el contenido de Cu0 en el CAG impregnado con Cu0 es alto, por ende la cantidad depositada en la solución también lo es. Además, se observa que al usar 5 g/L de CAG impregnado con Cu0 y 30 g/L de CAG impregnado con CuSO4 se obtienen tiempos similares de remoción del 98 % (85 y 80 min, respectivamente). En cuanto al contenido de Cu residual se observa que con CAG impregnado con Cu0 quedan 1.22 mg/L de Cu luego de la precipitación y con 30 g/L de CAG impregnado con CuSO4 quedan 0.88 mg/L Cu, que se ajustan a la normativa ambiental vigente, por lo que se considera que son catalizadores aptos para el proceso. Por otra parte, el uso de 50 g/L de CAG virgen con 50 mg/L Cu2+ es equivalente a emplear 10 g/L Cu0 granular, con un porcentaje de remoción del 98 % CN- en 195 min. En cuanto al contenido de Cu residual después de la precipitación, se tienen 0.40 mg/L Cu al emplear 50 g/L de 50 mg/L Cu2+ 50 g/L CAG virgen + 50 mg/L Cu2+ 50 g/L CAG impregnado CuSO4 30 g/L CAG impregnado CuSO4 10 g/L Cu0 granular 50 g/L CAG impregnado Cu0 30 g/L CAG impregnado Cu0 20 g/L CAG impregnado Cu0 10 g/L CAG impregnado Cu0 5 g/L CAG impregnado Cu0 1 g/L CAG impregnado Cu0 Tiempo de oxidación del 98% de (min) 210 Antes precipitación --- Después Precipitación 1.17 195 --- 0.40 40 61.70 1.51 [Cu]residual (mg/L) 85 57.00 0.88 195 67.50 0.89 8 179.30 38.47 15 155.00 32.86 25 138.50 28.40 40 134.80 22.69 80 90.10 1.22 220 54.60 1.01 Estos catalizadores cumplen con los requerimientos del proceso de oxidación de efluentes cianurados, sin embargo el tiempo de remoción de CN-duplica al empleado por los 5 g/L de CAG impregnado Cu0 y 30 g/L de CAG impregnado CuSO4 (80 y 85 min), por lo que se descarta su empleo en el tratamiento de efluentes cianurados. Finalmente, se analizan los catalizadores de Cu que presentaron los menores tiempos en la remoción de CN-, siendo estos los 50 mg/L Cu2+ y el 1 g/L de CAG impregnado con Cu0 con tiempos de remoción del 98 % de CN- de 210 y 220 min, respectivamente. En cuanto al contenido de Cu residual de la solución cianurada, con 50 mg/L Cu2+ se llega a 1.17 mg/L Cu luego de la precipitación y con 1 g/L de CAG impregnado con Cu0 se solubilizan 54.60 mg/L Cu y luego de la precipitación se tiene 1.01 mg/L Cu. Esto demuestra que en presencia de CAG a pesar de tener mayor cantidad de Cu solubilizado, luego de la precipitación la cantidad de Cu residual es menor a cuando solo se emplea CuSO4 (50 mg/L Cu2+) en solución. Siendo esto un indicio que el CAG impregnado con Cu0 adsorbe en su superficie complejos cianurados de Cu incluso en mínimas cantidades. Estos catalizadores tampoco pueden ser considerados como candidatos en la remoción de CN- de los efluentes industriales, ya que su cinética de reacción es muy lenta. Luego de haber realizado un análisis de cada uno de los catalizadores de Cu se concluye que los mejores resultados tanto en el tiempo de oxidación de CN- como en la precipitación de Cu, se obtienen con 5 g/L de CAG impregnado Cu0 y 30 g/L de CAG impregnado con CuSO4 con 80 y 85 min de tiempo oxidación, respectivamente. Por Revista Politécnica - Septiembre 2015, Vol. 34, No. 2 56 Campos C.*; De la Torre E.* _______________________________________________________________________________________________________________________________ lo que para seleccionar el mejor catalizador se tomarán en cuenta tres factores. Primero, 5 min de diferencia en el proceso de oxidación, a nivel industrial representan costos energéticos, de mantenimiento y mano de obra. Segundo, el empleo de grandes cantidades de CAG genera problemas en la agitación ya que no se tiene un movimiento uniforme del catalizador y además el CAG tiende a romperse por el rozamiento generado entre sí y finalmente se debe considerar que el CAG virgen bordea un costo de 1.00 USD en el mercado, por lo que se debe emplear la menor cantidad de catalizador posible para reducir costos. Con todo lo expuesto, se concluye que el tratamiento de efluentes cianurados se realizará con 5 g/L de CAG impregnado con Cu0. 3.1.4 Detoxificación de efluentes industriales del proceso de cianuración En la Fig. 8 se presentan las cinéticas de oxidación de CN- de lo fl nt cian rado d la compañía min ra “ r na .A.” y “Paz Borja” bicada n l antón amilo Ponc Enríquez. Estos efluentes provienen del proceso de cianuración y su tratamiento tiene como objetivo determinar la eficacia del método INCO modificado que se ha desarrollado en este estudio. [CN-]300 (mg/L) Efluente Orenas Efluente Cianurado Orenas 250 Efluente Paz Borja Efluente Cianurado Paz Borja 200 Efluente Solución Sintético Sintética (265 mg/L NaCN 500 mg/L 150 100 50 0 0 20 40 60 80 100 Tiempo (min) Figura 8. Oxidación de CN- en función del tiempo de efluentes cianurados y una solución sintética de 265.4 mg/L CN- (pH 10.0; 5 g/L CAG impregnado Cu0; 1000 mg/L Na 2S2O 5; 400 RPM y 276 NL/h aire) El tratamiento de los efluentes cianurados industriales se realizó con base en los mejores resultados obtenidos en el tratamiento de detoxificación de soluciones sintéticas de NaCN (pH 10.0; 5 g/L CAG impregnado Cu0; 1000 mg/L Na2S2O5; 276 NL/h aire y 400 RPM). En la Fig. 8 se observa que el efluente cianurado Orenas con 159.2 mg/L CN- inicial requiere 45 min para remover el CN-, mientras que el efluente cianurado Paz Borja con 58.4 mg/L CN- inicial necesita 20 min, lo que indica que el tiempo de oxidación es directamente proporcional al contenido inicial de CNpresente en los efluentes. Los efluentes industriales tienen presencia de metales (Cu, Ni, Cd, Zn, Fe, Ag, Au, etc.) que forman complejos metálicos con el CN-. Por ende, el método volumétrico empleado para determinar el CN- resulta ser ineficiente en la evaluación de la detoxificación de los efluentes cianurados, ya que no mide los complejos de cianuro y solo detecta concentraciones mayores a 5 mg/L CN-. Razón por la cual, se midió el contenido de CN-, CNWAD y CNTOTAL por el método colorimétrico piridina-pirazolona con un límite de detección de 0.002 a 0.204 mg/L de CN-, cuyos resultados se presentan en la Tabla 3. Tabla 3. Concentración de las especies de cianuro en efluentes cianurados y una solución de 265 mg/L CN- en la oxidación de CN-(pH 10.0; 5 g/L CAG impregnado Cu0; 1000 mg/L Na2S2O5, 400 RPM y 276 NL/h aire) Muestra Efluente Cianurado Orenas Efluente Cianurado Paz Borja Solución Sintética NaCN Tiempo de Oxidación (min) CNTOTAL 0 432.50 315.00 159.20 45 5.26 4.62 2.47 90 0.06 0.02 <0.01 0 112.50 87.50 58.40 20 3.50 2.20 1.20 40 0.05 0.02 <0.01 0 265.40 265.40 265.40 80 0.28 0.26 0.12 CNWAD (mg/L) La Tabla 3 muestra que luego de los tiempos de oxidación determinados con el método volumétrico en el tratamiento de los efluentes cianurados, aún se tiene presencia de cianuro en concentraciones superiores a 0.1 mg/L CNTOTAL, por lo que se duplicó el tiempo de oxidación de los dos efluentes. Esto permitió llegar a la concentración de CNTOTAL establecida por el TULAS. Por otro lado, se tiene que el cianuro se remueve más rápido en forma de complejos cianurados que en forma de CN-, ya que según los datos de la Tabla 3, el efluente Orenas contenía inicialmente 432.50 mg/L CNTOTAL y con 90 min de tratamiento se redujo a 0.06 mg/L CNTOTAL. Mientras que al tratar el efluente sintético de NaCN con 265.4 mg/L CNTOTAL luego de 80 min se tiene 0.28 mg/L CNTOTAL. En el tratamiento del efluente Orenas, se observó que a partir de los 45 min (tiempo hasta el que se pudo medir el contenido de CN-), el efluente empezó a tornarse de un color pardo intenso, y finalmente hubo la presencia de un precipitado café oscuro que teóricamente corresponde a los metales librados del CNWAD. La Fig. 9 se presenta un esquema del proceso de oxidación de este efluente, donde se evidencian las observaciones descritas. Sin embargo, en el caso del efluente Paz Borja al final del proceso el agua se tornó de un color pardo claro pero no hubo presencia de precipitado. Con el objetivo de entender el comportamiento de los efluentes cianurados se analizó por absorción atómica el contenido de metales (Au, Ag, Cu, Cd, Zn, Ni y Fe) antes y después del proceso de oxidación, cuyos resultados se muestran en la Tabla 4. Además, se muestran los límites permisibles de descarga de metales según lo establecido en el TULAS. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 57 Estudio de la Detoxificación de Efluentes Cianurados por Oxidación con Dióxido de Azufre, Aire y Catalizadores de Cobre _________________________________________________________________________________________________________________________ Los datos de la Tabla 4 indican que antes del proceso de oxidación los contenidos de Cu (113.00 mg/L) y Fe (28.20 mg/L) del efluente Orenas estaban fuera de la norma ambiental (1.00 mg/L Cu y 10.0 mg/L Fe). Tabla 4. Concentración de metales en efluentes cianurados y una solución sintética de 265 mg/L CN- antes y después de la oxidación de CN- (pH 10,0; 5 g/L CAG impr. con Cu0; 1000 mg/L Na2S2O5; 400 RPM y 276 NL/h aire Efluente embargo, conforme se fue dando el proceso de oxidación este contenido disminuyó a 0.36 mg/L Cu y 0.11 mg/L Fe en 90 min. En cuanto a los contenidos de Cd, Zn y Ni se tiene que desde el inicio del proceso cumplen con la norma ambiental vigente, sin embargo su contenido también disminuyó a medida que se dio la oxidación. Por otro lado, se observa que el contenido de Ag es de 0.54 mg/L y luego de 90 min de oxidación se reduce a 0.06 mg/L, mientras que el contenido de Au no disminuye con el transcurso del tiempo. Cabe indicar, que el contenido de Ag y el Au no están normados en el TULAS pero su contenido es de gran importancia por su gran valor comercial. Orenas Paz Borja Solución Sintética NaCN Tiempo (min) Au Ag Cu Cd Zn Ni Fe (mg/L) 0 0.31 0.54 113.00 0.02 2.17 1.25 28.20 45 0.27 0.20 10.01 <0.01 <0.01 0.12 0.41 90 0.27 0.06 0.36 <0.01 <0.01 0.10 0.11 0 0.14 0.81 10.59 <0.01 0.27 0.90 5.20 20 0.14 0.24 20.39 <0.01 0.15 0.74 3.78 40 0.13 0.20 24.08 <0.01 0.07 0.48 3.49 0 -- -- 0.00 --- --- --- --- 80 -- -- 1.22 --- --- --- --- -- -- 1.00 0.02 5.00 2.00 10.0 *Límite de descarga TULAS Cabe indicar que el costo del tratamiento de efluentes cianurados por el método presentado en este estudio es de 6.70 USD/m3 de efluente detoxificado. Al comparar el pecio de este tratamiento con otros procesos de oxidación de cianuro como el peróxido de hidrógeno y ácido de caro que tienen un costo promedio de 4 USD/Kg cianuro total, se tendría un costo de 1.73 USD/m3 de efluente tratado [1]. (a) (b) (c) (d) Lo cual indica que el método planteado en este estudio es 3.87 veces más caro que otros métodos encontrados en el mercado. Sin embargo, se debe considerar que el método INCO modificado con CAG impregnado con Cu0 a diferencia del ácido de caro y peróxido de hidrógeno permite llegar a los límites permisible de descarga de cianuro de total y de metales (Cu, Fe, Zn, Cd y Ni) establecidos en el TULAS, por lo que no necesita de otros métodos de detoxificación complementarios. Figura 9. Oxidación del efluente cianurado Orenas (a) Inicio del proceso, (b) Final del proceso (precipitación de metales), (c) Sedimentación del precipitado (d) Precipitado de metales En cuanto al efluente Paz Borja se observa que al inicio del proceso todos los metales (Cd, Ni, Zn y Fe) a excepción del Cu (10.59 mg/L) cumplen con la normativa ambiental. Al igual que en el caso del efluente Orenas los contenidos de Cd, Zn, Ni, Fe y Ag disminuyen con el transcurso del proceso de oxidación, mientras que el Au no se remueve. Sin embargo, el contenido de Cu aumenta con el pasar del tiempo de 10.59 mg/L a 24.08 mg/L en 40 min. Esto puede atribuirse al bajo contenido de Fe presente en el efluente Paz Borja (3.49 mg/L Fe) en comparación al efluente Orenas (28.2 mg/L Fe), ya que de acuerdo a la información bibliográfica el método INCO oxida el CNWAD a CNO- y los metales liberados precipitan en forma de hidróxidos, mientras que los complejos cianurados de Fe se reducen a estado ferroso y precipitan como sales metálicas de ferrocianuro de Cu, Ni o Zn. Por esta razón algunos autores sugieren el empleo de cloruro férrico (FeCl3) como fuente de Fe, para precipitar todos los metales del efluente [6]. 4. CONCLUSIONES En el tratamiento de soluciones sintéticas de 500 mg/L NaCN (265 mg/L CN-) a pH 10.0; 50 mg/L Cu2+; 400 RPM; 276 NL/h aire y una concentración inicial de 3000 mg/L Na2S2O5 se obtuvo el 84 % de remoción de CN - en 480 min. Mientras que al usar una concentración constante de 1000 mg/L Na2S2O5 (equivalente a añadir al inicio del proceso 3000 mg/L Na2S2O5) se tuvo el 98 % de remoción de CN- en 210 min, debido a que al ir añadiendo paulatinamente el Na2S2O5 de acuerdo a las necesidades del proceso se minimiza la reacción directa del - con el O2. Los tiempos de oxidación del 98 % de de las soluciones sintéticas de 500 mg/L NaCN bajo las mejores condiciones de operación del proceso INCO (pH 10.0; 400 RPM; 276 NL/h aire y una concentración constante de 1000 mg/L Na2S2O5) fueron 210 min con 50 mg/L Cu2+ obtenido a partir de CuSO4, 40 min con 50 g/L de CAG impregnado con CuSO4, 8 min con 50 g/L de CAG impregnado con Cu0 y 195 min con 10 g/L Cu0 granular. Revista Politécnica - Septiembre 2015, Vol. 34, No. 2 58 Campos C.*; De la Torre E.* _______________________________________________________________________________________________________________________________ El método INCO permitió oxidar el CNWAD y precipitar el Cu presente en las soluciones sintéticas de 500 mg/L NaCN. Los mejores resultados en el tratamiento de detoxificación de estas soluciones se obtuvieron a pH 10,0; 1000 mg/L Na2S2O5 (concentración constante); 400 RPM; 276 NL/h aire; y 5 g/L CAG impregnado con Cu0, llegando a 0.28 mg/L CNTOTAL y 1.22 mg/L Cu en 80 min. Al emplear dosificaciones mayores a 5 g/L de CAG impregnado con Cu0, el contenido de Cu luego de la precipitación fue mayor a 20 mg/L, que es un valor superior al establecido por la normativa ambiental vigente (1 mg/L Cu). [10] W. Sacher and A. Acosta. “La minería a gran escala en Ecuador: Análisis y datos estadísticos sobre la minería industrial en el Ecuador”, 3rd ed, vol. 1. Transmission Systems for Communications, Western Electric Co., Winston-Salem, NC, 1985, 2012, pp. 44–60. La detoxificación de los efluentes cianurados industriales mediante el método INCO modificado (276 NL/h aire; pH 10.0; concentración constante de 1000 mg/L Na2S2O5 y 5 g/L CAG impregnado con Cu0) permitió remover el CNTOTAL del efluente Orenas de 432.50 mg/L a 0.06 mg/L CNTOTAL en 90 min y del efluente Paz Borja de 112.50 mg/L a 0.05 mg/L CNTOTAL en 40 min. Es decir, se alcanzó concentraciones inferiores a 0.1 mg/L de CNTOTAL que es el límite de descarga a un cuerpo de agua dulce según lo establecido en el TULAS. El proceso INCO modificado con CAG impregnado con Cu0 precipita los metales (Cu, Cd, Zn, Ag, Ni y Fe) paulatinamente conforme se va dando la oxidación de cianuro WAD, llegando a los límites establecidos por el TULAS. El costo del tratamiento de efluentes cianurados por oxidación con SO2, aire y CAG impregnado con Cu0 es de 6.70 USD/m3 de efluente detoxificado. REFERENCIAS [1] D. Pesántez., E. de la Torre., and A. Guevara. (2008). Influencia del ión cúprico y del cobre metálico en la oxidación del cianuro libre con aire y carbón activado. Revista Politécnica. 1 (1), pp. 1-7. Available: http://bibdigital.epn.edu.ec/handle/15000/4337 [2] [3] [4] [5] [6] [7] [8] [9] M. Adams (1994). Removal of cyanide from solution using activated carbon. Minerals Engineering, 7 (9), pp. 1165-1177. Available: doi: 10.1016/0892-6875(94)90004-3 M. Botz (2001). Overview of cyanide treatment methods. Mining Environment Management. Mining Journal Ltd., London, UK, pp. 28 P. Breuer, C. Jeffery. C and R. Meakin. (2011). Fundamental inv tigation of th /air, p roxid and caro’ acid cyanid destruction processes. Parker CRC for Integrated Hydrometallurgy Solutions, CSIRO Minerals Down Under National Research Flagship, CSIRO Process Science and Engineering, Australia, pp. 1-13. H. Deveci, E.Y. Yazici, I. Alp and T. Uslu. (2006). Removal of cyanide from aqueous solution by plain and metal-impregnated activat d carbon’, Int rnational Journal of mineral processing. 1 (79), pp. 198-208. Available: doi:10.1016/j.minpro.2006.03.002 N. Kuyucak and A. Akcil. (2013). Cyanide and removal options from effluents in gold mining and metallurgical processes. Minerals Engineering, 1 (50), pp. 19-20. Available: doi: 10.1016/j.mineng.2013.05.027 J. ar d n and L. o . “Th ch mi try of gold xtraction, oci ty for ining”, nd ed., vol.1, Metallurgy and Exploration Inc., 2006, pp. 50-80. T. Mudder, M. Botz and A. Smith. “ hemistry and treatment of cyanidation wastes”, 2nd ed, vol.1, Mining Journal Books Ltd., 2001, pp. 327-333. J. Oleson and H. Lin. (2004). Modeling of SO 2/air cyanide destruction proc ‘, E Ann al ting, D nv r, olorado, pp. 1-6. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Recuperación de Níquel, Vanadio y Molibdeno del Catalizador Agotado de la Unidad de Craqueo Catalítico Fluidizado (FCC) _________________________________________________________________________________________________________________________ Recuperación de Níquel, Vanadio y Molibdeno del Catalizador Agotado de la Unidad de Craqueo Catalítico Fluidizado (FCC) Barrera A.*; Endara D.*; De la Torre E.*; Manangón L.* *Escuela Politécnica Nacional, Facultad de Ingeniería Química, Departamento de Metalurgia Extractiva DEMEX, Quito, Ecuador e-mail: {ana.barrera; diana.endara; ernesto.delatorre; eliana.manangon}@epn.edu.ec Resumen: Se realizó el estudio de la lixiviación del catalizador agotado de la unidad de Craqueo Catalítico Fluidizado (FCC) de la Refinería Esmeraldas con el objetivo de recuperar níquel, vanadio y molibdeno. Se caracterizó física, química y mineralógicamente el catalizador agotado para confirmar la presencia de los metales de interés. Se realizó ensayos de lixiviación usando ácidos orgánicos: oxálico, cítrico y tartárico; inorgánicos: sulfúrico, clorhídrico, fluorhídrico y nítrico y bases: hidróxido de sodio y peróxido de hidrógeno. Se varió condiciones como: concentración del agente lixiviante: 50, 100, 150, 200 y 250 g/L, tamaño de partícula: catalizador 106 µm y pulverizado, calcinación del catalizador a 700, 800 y 950 °C, porcentaje de sólidos: 1, 10, 20, 30 % y tiempo del proceso 0, 0.5, 1, 3, 6, 12, 24 horas. Las mejores recuperaciones de níquel 70 %, vanadio 67 % y molibdeno 56 % fueron al trabajar con ácido sulfúrico a 250 g/L, tamaño de partícula de 106 µm, sin calcinación, 10 % de sólidos durante 3 horas. Se realizó ensayos de extracción por solventes y flotación iónica para separar los metales (Ni, V y Mo) de las soluciones ricas obtenidas en la lixiviación. El molibdeno se obtuvo mediante extracción por solventes usando tributil fosfato (TBP 0,5 mol/L) diluido en heptano, el metal contenido en la fase orgánica se separó usando ácido clorhídrico 0,1 mol/L. La flotación iónica se empleó para recuperar níquel y vanadio, metales presentes en la fase acuosa que se obtuvo del ensayo de extracción por solventes. Se usó como reactivo colector etilxantato de sodio al 5 % y como espumante aceite de pino al 100 %. Con estos métodos la recuperación de los metales fue más eficiente Ni 86 %, V 80 % y Mo 70 %. La torta que se obtuvo en los ensayos de lixiviación fue estabilizada mediante encapsulamiento en hormigón sustituyendo 10 % por cemento en la dosificación. Palabras clave: Catalizador agotado, recuperación, níquel, vanadio y molibdeno. Abstract: In this research, the recovery of nickel (Ni), vanadium (V) and molybdenum (Mo) from the spent catalyst originated at the Fluid Catalytic Cracking unit (FCC) of the Esmeraldas refinery was evaluated. The FCC catalyst was characterized mineralogically, physically and chemically to establish the content of Ni, V, and Mo. Leaching trials were conducted using organic acids (oxalic, citric, and tartaric), inorganic acids (sulfuric, hydrochloric, and hydrofluoric), and bases (sodium hydroxide and hydrogen peroxide). The process was studied by working at different conditions including: leaching agent concentration (50, 100, 150, 200 and 250 g/L), particle size (30 and 106 µm), calcination temperature (700, 800 and 950 °C), pulp density (1, 10, 20 and 30 %), and processing time (0.0, 0.5, 1.0, 3.0, 6.0, 12.0 and 24.0 h). The highest recovery percentages were 70 % Ni, 67 % V, and 56 % Mo, and the best working conditions were sulfuric acid 250 g/L as leaching agent, particle size 106 µm, no previous calcination, 10 % pulp density, and processing time of 3 hours. Solvent extraction and ion flotation trials were performed to recover the metals from the leach liquors. In the solvent extraction process, tributyl phosphate (0.5 mol/L) was used to recover Mo, which was stripped by mixing the organic phase with hydrochloric acid (0.1 mol/L). Ni and V were recovered from the aqueous phase by ion flotation. Sodium ethyl xanthate 5 % was used as a collector and pine oil 100 % as a frother. The extraction values for these methods were 86 % Ni, 80 % V, and 70 % Mo. The solid residues obtained from the leaching tests were stabilized to immobilize the toxic components by replacing 10 % of cement in the composition of concrete. Keywords: Spent catalyst, recovery, nickel, vanadium and molybdenum. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 59 60 Barrera A.*; Endara D.*; De la Torre E.*; Manangón L.* _______________________________________________________________________________________________________________________________ 1. INTRODUCCIÓN 1.1. Catalizadores agotados En muchas industrias químicas especialmente en las industrias de refinación de petróleo y petroquímica, grandes cantidades de catalizadores sólidos se utilizan de forma rutinaria. Los catalizadores sólidos contienen metales, óxidos metálicos o sulfuros y requieren su reemplazo después de un corto tiempo de funcionamiento. [3] En las unidades de craqueo catalítico de lecho fluidizado (FCC), donde se realiza el proceso de conversión más importante de la refinación de petróleo se generan anualmente alrededor de 400 000 toneladas de catalizadores agotados en el mundo. La eliminación inadecuada de estos residuos peligrosos puede causar problemas ambientales debido a su contenido de metales. Metales como el platino, molibdeno, níquel, vanadio, entre otros se pueden extraer a partir del catalizador agotado antes de su eliminación. [5] En el pasado los catalizadores agotados eran depositados en vertederos sin ningún tipo de tratamiento. Hoy en día, gracias a regulaciones ambientales algunos de estos residuos se han catalogado como tóxicos y peligrosos, exigiendo que se les dé tratamiento antes de su disposición final. Estos residuos representan una grave amenaza para el medio ambiente debido a la variedad de materiales utilizados en su producción, a su vez son también una fuente valiosa de diversos tipos de metales. [7] Actualmente, los catalizadores agotados se gestionan industrialmente a través de la regeneración para extender su vida útil, el depósito en vertederos para su disposición final y el reciclado de metales valiosos. Sin embargo, la regeneración de catalizadores agotados solo puede ser aplicada un número limitado de veces y en sistemas catalíticos específicos. [4] Los metales pesados pueden estar presentes como parte del catalizador original (por ejemplo, cobalto o níquel en catalizadores de hidroprocesamiento) o pueden acumularse sobre el catalizador durante el uso (por ejemplo, níquel o vanadio en catalizadores de FCC). En efecto, estos materiales de desecho, que contienen altas concentraciones de metales, pueden ser considerados como "minerales artificiales" y pueden servir como materias primas secundarias con la consiguiente reducción en la demanda de recursos minerales primarios. [5] Todos estos metales se pueden recuperar, de este modo, el reciclaje de catalizadores agotados traería beneficios tangibles para la ecología y la economía. Además, esto ayudaría a disminuir la cantidad de residuos tóxicos depositados en vertederos, con lo que se reduciría el consumo de energía y la contaminación ambiental que se produciría en comparación con las tecnologías de obtención de metales a partir de materias primas (menas). [13] La recuperación de metales de los catalizadores agotados se puede lograr a través de dos métodos principalmente: hidrometalurgia y pirometalurgia. La hidrometalurgia disuelve los metales presentes en el catalizador por lixiviación usando ácidos o bases, de esta manera se obtienen compuestos metálicos comerciales. La pirometalurgia utiliza tratamientos térmicos tal como calcinación o fundición para separar los metales de los catalizadores agotados, sin embargo, esto conlleva altos costos de operación y poca rentabilidad. [2] 2. METODOLOGÍA EXPERIMENTAL 2.1. Caracterización del catalizador agotado Las propiedades físicas del catalizador agotado se determinaron mediante ensayos de densidad real y aparente. El análisis granulométrico se realizó en tamices Tyler compuestos por mallas estándar ajustadas a un vibrotamiz ATM ARROW siguiendo la norma ASTM C136-05. El área superficial, tamaño y volumen de poro se determinaron utilizando el equipo de isotermas de adsorción BET (Quantachrome NovaWin), se desgasificó durante 3 horas las muestras con nitrógeno seco como gas de medición, se trabajó con un rango de P/Po de 0.05 a 0.3. Para determinar la composición química del catalizador, se realizó un análisis semi cuantitativo en el microscopio electrónico de barrido MEB-EDX (Tescan) con analizador de rayos X Quantax (Bruker), además se realizó una disgregación ácida en microondas, analizando el contenido mediante la técnica de absorción atómica en un equipo AAnalyst 300 marca Perkin Elmer. Para la caracterización toxicológica (TCLP) se siguió el procedimiento EPA 1311. La composición mineralógica se determinó usando un difractómetro de rayos X modelo D8 Advance (Bruker). [10] 2.2. Lixiviación Se preparó soluciones de 100 mL de volumen con cada agente lixiviante a las diferentes concentraciones, se colocó 1 gramo de catalizador agotado para tener una pulpa con 1 % de sólidos. Las pulpas formadas se agitaron durante 24 horas a 750 rpm. Se evaluó la concentración de los agentes lixiviantes: ácido oxálico, cítrico, tartárico, sulfúrico, nítrico, clorhídrico, fluorhídrico, hidróxido de sodio y peróxido de hidrógeno (50, 100, 150, 200 y 250 g/L), tamaño de partícula (material entero y pulverizado), temperatura de calcinación del catalizador agotado (700, 800 y 950 °C), porcentaje de sólidos (1, 10, 20, 30 %) y tiempo de lixiviación (0.0, 0.5, 1.0, 3.0, 6.0, 12 y 24 h). Al finalizar cada ensayo se filtró la pulpa, obteniéndose una solución fuerte y una torta. La torta fue lavada y filtrada nuevamente, usando 50 mL de agua destilada, obteniéndose una solución débil. Las soluciones se analizaron mediante absorción atómica. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Recuperación de Níquel, Vanadio y Molibdeno del Catalizador Agotado de la Unidad de Craqueo Catalítico Fluidizado (FCC) _________________________________________________________________________________________________________________________ 2.3. Extracción por solventes El molibdeno se obtuvo al realizar ensayos de extracción por solventes usando el extractante tributil fosfato (TBP) a diferentes concentraciones: 0.1, 0.5, 1.0 y 3.0 (mol/L), con una relación fase orgánica-fase acuosa (A/O) de 1, durante 30 minutos. El metal se separó de la fase orgánica añadiendo ácido clorhídrico diluido (0.1 mol/L). [6] 2.4. Flotación iónica La flotación iónica se empleó para recuperar níquel y vanadio presentes en la fase acuosa proveniente de la extracción por solventes. Para ello se usó una celda Denver de 1 litro de capacidad, con agitación de 0 a 2800 RPM y con inyección de aire. Como reactivo colector se usó etilxantato de sodio (EXNa) a 150 y 300 g/m3 y como espumante aceite de pino a 30 g/m3, el proceso duró 40 minutos. 2.5. Cristalización En los ensayos de extracción por solventes y flotación iónica se obtuvieron soluciones cargadas de los metales de interés: níquel, vanadio y molibdeno. En la fase orgánica formada en la extracción por solventes se logró recuperar molibdeno. El concentrado de flotación contenía níquel y el relave vanadio. Estas tres soluciones fueron cristalizadas obteniéndose productos secos cuyo contenido se analizó por difracción de rayos X. Es importante mencionar que estos análisis son semi cuantitativos debido a que los sólidos obtenidos presentan una naturaleza amorfa. 2.6. Tratamiento de la torta lixiviada La torta que se obtuvo en los ensayos de lixiviación se estabilizó mediante encapsulamiento en hormigón para inmovilizar los componentes tóxicos. Se prepararon probetas de hormigón con porcentajes de sustitución de cemento por torta lixiviada de 10 y 20 %. Para la evaluación del hormigón se realizaron ensayos de resistencia a la compresión a los 7 y 28 días. El hormigón que se desea obtener es aquel que no requiera de un esfuerzo de compresión mayor a 210 kg/cm2. 3. RESULTADOS 3.1. Caracterización del catalizador agotado El catalizador agotado presentó las siguientes propiedades físicas mostradas en la Tabla 1. El 80 % del catalizador agotado presentó un tamaño de partícula menor a 106 µm, la densidad aparente del catalizador agotado considera los espacios entre las partículas 61 Tabla 1. Propiedades físicas del catalizador agotado Propiedad física Valor Granulometría d80 (µm) 106 Densidad real (g/cm3) 1.01 Densidad aparente (g/cm3) 0.95 Área superficial (m2/g) 187 Tamaño de poro (Å) 7.90 Volumen de poro (cm3/g) 0.08 siendo por tanto menor que la densidad de la propia partícula, es decir, que la densidad real. [11] El tamaño de poro es de 7.9 Å por lo que se trata de microporos, el volumen de poro es de 0.08 cm3/g esto se determinó usando la metodología de la teoría de la curva –t. La composición química del catalizador agotado se presenta en la Tabla 2. Los valores más altos que corresponden a aluminio y silicio se deben a que la matriz del catalizador es una masa de sílice SiO2 y alúmina Al2O3. Los valores de níquel, vanadio y molibdeno son representativos y resultan atractivos para ser recuperados. La caracterización mineralógica del catalizador agotado identificó los siguientes componentes cristalinos: faujasita alrededor del 80 %, caolinita, bohemita y muscovita alrededor del 10 %, los metales de interés, están presentes en forma de sulfuros: millerita, patronita y molibdenita en cantidades pequeñas por debajo del 2 %. La caracterización toxicológica reportó que las concentraciones de mercurio y plomo en 2.7 y 1.6 veces respectivamente, exceden a los límites permisibles, como estos metales se consideran peligrosos para el medio ambiente y la salud de los seres humanos, se considera al catalizador agotado como un residuo peligroso. Tabla 2. Composición química del catalizador agotado Elemento Contenido (%) Aluminio 30.4 Silicio 29.7 Vanadio 2.2 Molibdeno 1.6 Titanio 1.4 Níquel 1.2 3.2. Influencia del agente lixiviante y su concentración En los ensayos realizados usando ácido oxálico, cítrico, tartárico, hidróxido de sodio y peróxido de hidrógeno a 50, 100 y 150 g/L, los porcentajes de recuperación de níquel, vanadio y molibdeno no alcanzaron valores mayores al 50 %. Se podría seguir trabajando con soluciones más concentradas de los ácidos orgánicos pero esto no es posible debido a que dichas soluciones a altas concentraciones tienden a saturarse y una vez concluida la lixiviación al obtener las soluciones fuertes estas se cristalizan complicando su lectura en el equipo de absorción atómica. En la Fig. 1 se presentan los resultados de las recuperaciones de los mestales de interés usando ácido sulfúrico (H2SO4), Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 62 Barrera A.*; Endara D.*; De la Torre E.*; Manangón L.* _______________________________________________________________________________________________________________________________ nítrico (HNO3), clorhídrico (HCl) y fluorhídrico (HF) a 150 g/L. El ácido fluorhídrico es el que posee la mejor afinidad con todos los metales de interés obteniendo: níquel 88.2 %, vanadio 87.2 % y molibdeno 85.1 %. % Recuperación del metal 100 En estudios realizados por Mishra, Chaudhury, Kim y Ahn (2010) [9] se recomienda realizar ensayos de lixiviación añadiendo peróxido de hidrógeno a diferentes concentraciones: 10, 20 y 30 g/L a soluciones de ácido sulfúrico para aumentar la eficiencia en las extracciones de níquel, vanadio y molibdeno. Como previamente ya se demostró que los mejores resultados usando ácido sulfúrico fueron a una concentración de 150 g/L, se prepararon nuevas soluciones añadiendo peróxido de hidrógeno, obteniéndose los resultados indicados en la Fig. 2. También se realizaron ensayos de lixiviación con el catalizador agotado usando como agente lixiviante una base (hidróxido de sodio). En la Fig. 3 se muestran los resultados obtenidos. Se puede apreciar que en los ensayos realizados se obtuvieron recuperaciones favorables de níquel y vanadio, con valores de alrededor del 70 % cuando se trabajó a una concentración de 150 g/L. El níquel metálico no es soluble en hidróxido de sodio [12] y por lo tanto se deberían haber alcanzado recuperaciones bajas 60 Níquel 40 Vanadio Molibdeno 20 0 Ac. Sulfúrico y Ac. Sulfúrico y Ac. Sulfúrico y H2O2 10 g/L H2O2 20 g/L H2O2 30 g/L Figura 2. Recuperación de níquel, vanadio y molibdeno en los ensayos de lixiviación del catalizador agotado con ácido sulfúrico a 150 g/L y peróxido de hidrógeno (10, 20 y 30 g/L) y 1 % de sólidos . 100 80 % Recuperación del metal Las recuperaciones de los metales de interés son muy parecidas, en todos los casos se tiene un ligero aumento en los valores de tan solo 1 o 2 %. Sin embargo son valores bajos y no se justifica el uso de este reactivo oxidante. 80 60 Níquel Vanadio 40 Molibdeno 20 100 0 90 Sosa 50 g/L Sosa 100 g/L Sosa 150 g/L % Recuperación del metal 80 Figura 3. Recuperación de níquel, vanadio y molibdeno en los ensayos de lixiviación del catalizador agotado con hidróxido de sodio a 50, 100 y 150 g/L y 1 % de sólidos 70 60 Níquel 50 Vanadio 40 Molibdeno 30 20 10 0 H2SO4 HNO3 HCl HF Figura 1. Recuperación de níquel, vanadio y molibdeno en los ensayos de lixiviación usando ácido sulfúrico, nítrico, clorhídrico y fluorhídrico a 150 g/L y 1 % de sólidos Los resultados mostrados en la Fig. 3 presentan recuperaciones altas de este metal, esto se logra debido a que el níquel se encuentra en el gasóleo como un complejo metalporfirínico, es decir está asociado a las porfirinas que son un grupo de moléculas orgánicas muy amplio y muy estudiado, por lo tanto al ponerle en contacto con una base se logrará un pH alcalino que favorecerá su disolución. [8] Comparando todos los ensayos de lixiviación realizados con ácidos orgánicos, inorgánicos y bases, la mejor recuperación se obtuvo al trabajar con ácido fluorhídrico a una concentración de 150 g/L. Sin embargo se eligió como mejor agente lixiviante al ácido sulfúrico, que presentó los mejores resultados de Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Recuperación de Níquel, Vanadio y Molibdeno del Catalizador Agotado de la Unidad de Craqueo Catalítico Fluidizado (FCC) _________________________________________________________________________________________________________________________ recuperación después del ácido fluorhídrico, ya que su manipulación a nivel de laboratorio e industrial es más fácil que cuando se trabaja con los demás ácidos, además su costo es económicamente aceptable y presenta una selectividad alta con los metales de interés. Para aumentar la eficiencia en la recuperación de los metales se trabajó con soluciones de ácido sulfúrico a concentraciones de 200 y 250 g/L. Los resultados se presentan en la Fig. 4. Las máximas recuperaciones de níquel, vanadio, molibdeno son: 90 %, 92 %, y 86 % respectivamente, esto se logró al trabajar con ácido sulfúrico a 250 g/L y 1 % de sólidos. 100 catalizador se procedió a disgregarlo ácidamente para que mediante un análisis de absorción atómica se puedan determinar los cambios en su composición. El catalizador agotado calcinado a 800 °C es el que presenta los mejores resultados puesto que las concentraciones de níquel y molibdeno aumentan, sin embargo lo mismo no sucede con el vanadio esto se debe a que en el catalizador se encuentra como un sulfuro y a partir de 650 °C se transformó en óxido y empezó a volatizarse. Con el catalizador calcinado a 800 °C se realizó ensayos de lixiviación usando ácido sulfúrico a 200 g/L y 1 % de sólidos, obteniendo las siguientes recuperaciones: níquel 13 %, vanadio 55 % y molibdeno 51 %. La recuperación de níquel es baja debido a que la cantidad de níquel metálico presente en el mismo se transformó a óxido de níquel, el cual no es soluble en medio ácido. Con los resultados obtenidos en los dos ensayos se concluye que no es necesario darle un tratamiento térmico previo al catalizador agotado para su posterior lixiviación. 80 % Recuperación del metal 63 60 Niquel Vanadio 40 Molibdeno 20 0 Acido sulfúrico 200 g/L Acido sulfúrico 250 g/L 3.5. Influencia del porcentaje de sólidos Los resultados de los ensayos de lixiviación variando el porcentaje de sólidos, se presentan en la Fig. 5. El factor más importante y el que limita la lixiviación de los metales de interés del catalizador agotado es la relación que existe entre la concentración del agente lixiviante y el porcentaje de sólidos. A medida que aumenta el porcentaje de sólidos las recuperaciones de los metales disminuyen, ya que se tendría menor cantidad de ácido sulfúrico disponible para lixiviar una mayor cantidad de catalizador agotado. Figura 4. Recuperación de níquel, vanadio y molibdeno en los ensayos de lixiviación del catalizador agotado con ácido sulfúrico a 200 y 250 g/L y 1 % de sólidos 100 Se analizó la influencia del tamaño de partícula usando el catalizador agotado a 106 y 30 µm, las dos muestras se lixiviaron en soluciones de ácido sulfúrico a 200 y 250 g/L. Los porcentajes de recuperación de los metales de interés en los ensayos realizados son muy parecidos, cuando se trabaja con el catalizador a 30 µm se logra un pequeño aumento en la recuperación, mismo que no justifica los costos que se tendrían al realizar una pulverización tanto a nivel de laboratorio como a nivel industrial. % Recuperación del metal 90 3.3. Influencia del tamaño de partícula 80 70 60 50 Níquel 40 Vanadio 30 Molibdeno 20 10 0 1 3.4. Influencia del catalizador agotado 10 20 30 Porcentaje de sólidos Se calcinó el catalizador agotado a 3 temperaturas: 700, 800 y 950 °C con el fin de remover las impurezas presentes en el mismo y mejorar los porcentajes de recuperación de los metales en los ensayos de lixiviación. Una vez calcinado el Figura 5. Recuperación de níquel, vanadio y molibdeno en los ensayos de lixiviación del catalizador agotado con ácido sulfúrico a 250 g/L, a diferente porcentaje de sólidos Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 64 Barrera A.*; Endara D.*; De la Torre E.*; Manangón L.* _______________________________________________________________________________________________________________________________ 3.6. Determinación del tiempo óptimo de lixiviación En la Fig. 6 se presenta la cinética de lixiviación de níquel, vanadio y molibdeno trabajando con ácido sulfúrico a 250 g/L, 10 % de sólidos, durante 24 horas. Las mejores recuperaciones se logran a las tres horas de iniciado el ensayo de lixiviación y son: 70 % de níquel, 68 % de vanadio y 56 % de molibdeno. A partir de las 3 horas las recuperaciones de los metales de interés tienden a mantenerse en un valor constante, por lo que no será necesario seguir lixiviando el material debido a que se tendría un consumo innecesario de energía. a diferentes concentraciones: 0.1; 0.5; 1 y 3 mol/L, con una relación fase acuosa-fase orgánica de 1, el proceso se llevó a cabo en dos etapas: extracción y reextracción. Los resultados de la etapa de extracción se muestran en la Figura 7. 100 % Recuperación de molibdeno (extracción) Al trabajar con un porcentaje de sólidos del 1 % las recuperaciones de los metales alcanzan valores superiores al 80 %, sin embargo cuando se trabaja con 10 % de sólidos las recuperaciones de los metales disminuyen pero la concentración de los mismos aumenta porque se ingresa mayor cantidad de catalizador agotado en el proceso. 80 60 40 20 0 0 3.7. Resultados de los ensayos de extracción por solventes y flotación iónica empleados para separar níquel, vanadio y molibdeno Se realizaron ensayos de extracción por solventes y flotación iónica con el fin de purificar, concentrar y separar níquel, vanadio y molibdeno de las soluciones enriquecidas obtenidas en los ensayos de lixiviación del catalizador agotado. 100 % Recuperación del metal 90 80 70 60 50 Níquel 40 Vanadio 30 Molibdeno 20 0,5 1 1,5 2 2,5 3 Concentración de tributil fosfato (mol/L) Figura 7. Recuperación de molibdeno en los ensayos de extracción por solventes de las soluciones lixiviadas usando TBP a diferente concentración: etapa de extracción En la Fig. 7 se muestra la influencia de la concentración del agente extractante TBP sobre la extracción del molibdeno. A la concentración de 0.1 mol/L de TBP se logra una recuperación de 40 % de molibdeno, a 0.5 mol/L de TBP se obtiene 70 % de recuperación, a partir de esta concentración se tiene un aumento constante en la recuperación de molibdeno, logrando el valor más alto 75 % a una concentración de 3 mol/L de TBP. En trabajos anteriores se usaron como extractantes TBP, Cyanex 921, y Alamine 308 [9]. Se demostró que Cyanex 291 extraía la mayor cantidad de molibdeno de las soluciones lixiviadas alcanzando valores mayores al 90 %. Sin embargo al realizar la segunda etapa de la extracción por solventes, no se logró separar el extractante Cyanex 921 de la fase orgánica que contenía al ión metálico, por lo que se sugirió usar TBP que también alcanzó recuperaciones altas y su separación es muy fácil. 10 0 0 3 6 9 12 15 18 21 24 Tiempo (h) Figura 6. Recuperación de níquel, vanadio y molibdeno en los ensayos de lixiviación del catalizador agotado con ácido sulfúrico a 250 g/L, 10 % de sólidos, variando el tiempo 3.7.1. Extracción por solventes La extracción por solventes se realizó con el fin de separar el molibdeno, se usó como extractante orgánico Tributil fosfato La razón por la cual la separación de TBP es eficiente se debe al hecho de que contiene grupos atractores de electrones que proporcionan menor basicidad hacia las especies de metal, mientras que Cyanex 291 contiene grupos donadores de electrones. [6] Se empleó ácido clorhídrico a diferentes concentraciones: 0.1; 0.5; 1.0 y 1.5 mol/L para separar el TBP de la solución orgánica, obteniendo el molibdeno en solución para su posterior cristalización. Los resultados se presentan en la Figura 8, se alcanzó una recuperación de molibdeno del 90 % a una concentración de 0.1 mol/L, a concentraciones más altas de ácido clorhídrico se tiene recuperaciones constantes. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Recuperación de Níquel, Vanadio y Molibdeno del Catalizador Agotado de la Unidad de Craqueo Catalítico Fluidizado (FCC) _________________________________________________________________________________________________________________________ 100 % Recuperación de Níquel Acumulado 100 80 % Recuperación molibdeno (re-extracción) 65 60 40 20 80 60 40 20 0 0 0 0,5 1 1,5 0 15 Figura 8. Recuperación de molibdeno en los ensayos de extracción por solventes de las soluciones lixiviadas usando TBP a diferente concentración: etapa de reextracción 3.7.2 Flotación iónica En los ensayos de flotación iónica se usó como reactivo colector etil xantato de sodio (EXNa 5 %) a dos concentraciones: 150 y 300 g/m3. Como reactivo espumante se utilizó aceite de pino 100 % a una sola concentración: 30 g/m3. Al trabajar con EXNa 150 g/m3 se obtiene una recuperación acumulada de níquel del 75 % en un volumen acumulado del 60 %, mientras que usando EXNa 300 g/m3 se logró una recuperación de níquel más eficiente del 86 % en un volumen acumulado del 70 %, por lo que el relave final tiene un volumen de tan solo 30 %. Los resultados del ensayo de flotación iónica se muestran en la Fig. 9. El lixiviado usado para los ensayos de flotación contenía níquel y vanadio en solución, al lograr concentrar el níquel, en el relave quedó disuelto el vanadio. El reactivo fundamental dentro del proceso de flotación es el etilxantato de sodio ya que permite que se forme una película hidrofóbica sobre la partícula del metal de interés. La afinidad que tiene el colector (EXNa) por el níquel es la razón por la que se obtiene altas recuperaciones, los iones de níquel presentes en la solución del lixiviado precipitaron en forma de complejos al añadir este colector. 30 45 60 75 90 % Volumen acumulado Concentración ácido clorhídrico (mol/L) Figura 9. Recuperación de níquel en los ensayos de flotación iónica usando EXNa 300 g/m3, aceite de pino 30 g/ton, pH=3.5 acondicionamiento se trabajó a 900 RPM y en la etapa de flotación a 1300 RPM, con esta velocidad de agitación se formaron burbujas con un tamaño óptimo que mejoró el área de contacto entre los iones de níquel y la fase gaseosa logrando colectar de forma eficiente el metal de interés. [1] 3.4. Cristalización En las soluciones cristalizadas de los procesos de extracción por solventes y flotación iónica se logró identificar la forma mineralógica en la que están presentes los metales de interés. En el concentrado de flotación cristalizado, se identificó que alrededor del 95 % se encuentra como sulfato de níquel hexahidratado (NiSO4 6H2O), en el relave de flotación cristalizado se encontró que más del 90 % está como pentóxido de vanadio (V2O5) y en la solución orgánica cristalizada se identificó que alrededor del 70 % está como trióxido de molibdeno (MoO3). Es importante mencionar que estos análisis son semi cuantitativos debido a que los sólidos obtenidos presentan una naturaleza amorfa. 3.5 Tratamiento de la torta lixiviada La torta obtenida en la lixiviación se logró encapsular en hormigón sustituyendo en la dosificación 10 % de este sólido residual por cemento, a los 28 días se alcanzó una resistencia a la compresión de 194.78 kg/cm2. 4. CONCLUSIONES El aceite de pino creó una espuma que fue capaz de mantener las burbujas cargadas del metal de interés (níquel) hasta que fueron removidas de la celda de flotación. La composición química elemental del catalizador agotado determinó que los valores de níquel, vanadio y molibdeno son representativos y resultan atractivos para ser recuperados. Un factor fundamental dentro del proceso de flotación fue la formación de burbujas proporcionada por la inyección de aire y la agitación dentro de la celda. Por ello en la etapa del El área superficial, tamaño y volumen de poro presentaron valores menores a los que tiene un catalizador fresco debido a Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 66 Barrera A.*; Endara D.*; De la Torre E.*; Manangón L.* _______________________________________________________________________________________________________________________________ la formación de coque y el depósito de contaminantes en el catalizador durante su uso. metales El análisis toxicológico del catalizador agotado demostró que es un residuo peligroso porque las concentraciones de mercurio y plomo exceden en 2.7 y 1.6 veces respectivamente, a los límites permisibles establecidos por los estándares U.S EPA 40CFR 261.24. Las recuperaciones más altas de níquel, vanadio y molibdeno determinadas en los ensayos de lixiviación son: 70 %, 67 % y 56 % respectivamente, esto se logró trabajando con ácido sulfúrico como agente lixiviante a una concentración de 250 g/L, con el catalizador agotado sin realizar tratamiento térmico previo, con un tamaño de partícula de 106 µm, al 10 % de sólidos y durante 3 horas. REFERENCIAS [1] [2] [3] [4] [5] El factor más importante y el que limita la lixiviación eficiente de níquel, vanadio y molibdeno es la relación que existe entre la concentración del agente lixiviante y el porcentaje de sólidos. A medida que aumenta el porcentaje de sólidos las recuperaciones de los metales disminuyen, ya que se tiene menor cantidad de ácido sulfúrico disponible para lixiviar una mayor cantidad de catalizador agotado. La recuperación más eficiente de molibdeno mediante la técnica de extracción por solventes es de 70 %, usando el extractante tributil fosfato (TBP) a 0,5 mol/L diluido en heptano, con una relación fase acuosa y fase orgánica de 1. [6] [7] [8] [9] La flotación iónica logró recuperar 86 % de níquel y 80 % de vanadio, usando como reactivo colector etil xantato de sodio 300 g/m3 y como espumante aceite de pino 30 g/m3, a un pH de 3.5 durante 40 minutos. En el concentrado de flotación cristalizado, se identificó que alrededor del 95 % se encuentra como sulfato de níquel hexahidratado (NiSO4 6H2O), en el relave de flotación cristalizado se encontró que más del 90 % está como pentóxido de vanadio (V2O5) y en la solución orgánica cristalizada se identificó que alrededor del 70 % está como trióxido de molibdeno (MoO3). La torta lixiviada fue estabilizada mediante encapsulamiento en hormigón sustituyendo 10 % de este sólido residual por cemento en la dosificación para inmovilizar los componentes tóxicos. [10] [11] [12] [13] R. Alcalá, and Y. Cabaleiro, Obtención por flotación iónica del Ni y Co en licores obtenidos de la lixiviación ácida de las colas de Nicaro. Presented at: V Simposio Minería y Metalurgia: MINIMETAL. [Online]. Available: http://www.redciencia.cu/geobiblio/paper/2013_Alcala_Farina_MIN5P10.pdf. F. Alonso, S. Ramírez, J. Ancheyta, and M. Mavil, (2008). Alternativas para la recuperación de metales a partir de catalizadores gastados del hidrotratamiento de hidrocarburos pesados: un caso de estudio. Revista Internacional Contaminación Ambiental. [Online]. 24(2), pp. 55–69. Available: http://www.redalyc.org/articulo.oa?id=37024202. F. Amiri, I. Asghari, S. Mousavi and S. Tavassoli, (2013). Bioleaching of spent refinery catalysts: A review. Journal of Industrial and Engineering Chemistry. [Online]. 19(4), pp. 1069–1081. Available: http://www.sciencedirect.com/science/article/pii/S1226086X12004145 F. Amiri, S. Mousavi, S. Sheibani and S. Yaghmaei, (2011). Recovery of metals from spent refinery hydrocracking catalyst using adapted Aspergillus niger. Hydrometallurgy. [Online]. pp. 65–71. Available: http://www.sciencedirect.com/science/article/pii/S0304386X11001204 K. Aung and Y. Ting, (2005). Bioleaching of spent fluid catalytic cracking catalyst using Aspergillus niger. Journal of Biotechnology. [Online]. 116(2), pp. 159–170. Available: http://www.sciencedirect.com/science/article/pii/S0168165604005206. R. Banda, T. Nguyen, S. Sohn and M. Lee, (2013). Recovery of valuable metals and regeneration of acid from the leaching solution of spent HDS catalysts by solvent extraction. Hydrometallurgy. [Online] pp. 161–167. Available: http://www.sciencedirect.com/science/article/pii/S0304386X13000091 A. Fornalczyk, (2012). Industrial catalyst as a source of valuable metals. Journal of Achievements in Materials and Manufacturing Engineering. [Online]. 55(2), pp. 864–868. Available: http://www.journalamme.org/papers_vol55_2/58299.pdf. N. Márquez, F. Ysambertt and C. De la Cruz, Three analytical methods to isolate and characterize vanadium and nickel porphyrins from heavy crude oil. Available: http://www.sciencedirect.com/science/article/pii/S0003267099003049. D. Mishra, G. Chaudhury, D. Kim and J. Ahn, Recovery of metal values from spent petroleum catalyst using leaching-solvent extraction technique. Available: http://www.sciencedirect.com/science/article/pii/S0304386X09002941. J. Ricaurte, E. de la Torre, A. Guevara. (2014, Enero). Estudio Comparativo de la Recuperación de Zn de Polvos de Acería por Lixiviación con H2SO4 y HCl, electrodeposición electrolítica y bielectrolítica. Revista Politécnica. [Online]. 33(2), pp. 1-2. Available: http://www.revistapolitecnica.epn.edu.ec/ojs2/index.php/revista_polite cnica2/article/view/156/pdf. R. Sadeghbeige, Fluid Catalytic Cracking Handbook, ButterworthHeinemann publications, 2000, vol. 2, pp. 93–95. A. Vogel, Química Analítica Cualitativa, 5ta. ed., Buenos Aires: Kapelusz, 1974, pp. 12-14. L. Zeng and C. Cheng, A literature review of the recovery of molybdenum and vanadium from spent hydrodesulphurisation catalysts. Part I: Metallurgical processes. Available: http://www.researchgate.net/publication/257130795_A_literature_revie w_of_the_recovery_of_molybdenum_and_vanadium_from_spent_hydr odesulphurisation_catalysts_Part_I_Metallurgical_processes. RECONOCIMIENTO Al Departamento de Metalurgia Extractiva DEMEX de la Facultad de Ingeniería Química y Agroindustria de la Escuela Politécnica Nacional del Ecuador. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Extracción y Caracterización de la Fracción No Metálica de las Tarjetas de Circuitos Impresos de Computadoras Desechadas _________________________________________________________________________________________________________________________ Extracción y Caracterización de la Fracción No Metálica de las Tarjetas de Circuitos Impresos de Computadoras Desechadas Loyo C.*; Arroyo C.*; Aldás M.*; Montero R. ** * Escuela Politécnica Nacional, Centro de Investigaciones Aplicadas a Polímeros, Facultad de Ingeniería Química y Agroindustria, Quito, Ecuador e-mail: [email protected]; [email protected]; [email protected] ** Empresa Pública Yachay, Dirección de Investigación Científica y Transferencia de Tecnología e-mail: [email protected] Resumen: Los residuos electrónicos han sido en los último años una de las principales fuentes de contaminación a nivel mundial, debido principalmente a la gran producción y comercialización de este tipo de equipos. Uno de los componentes fundamentales y más difíciles de reciclar debido a la complejidad de su mezcla son las tarjetas de circuitos impresos (PCBs). Este proyecto tiene como objetivo extraer y caracterizar la fracción no metálica (FNM) de las PCBs con el propósito de determinar una forma práctica de reciclaje de dichos residuos. Primeramente se retiraron los puertos de salida, baterías y capacitores de las tarjetas, después fueron cizalladas y molidas en un molino de martillos para ser enviadas a la separación gravimétrica, finalmente se tomaron dos tamaños de partícula uno entre 0.15 y 0.075 mm y el otro menor a 0.075 mm, con el fin de determinar la influencia del tamaño de partícula en su composición. Esta fracción fue caracterizada mediante espectroscopía de infrarrojo (FTIR), calorimetría diferencial de barrido (DSC), termogravimetría (TGA) y análisis para la determinación de lixiviados (TCLP). Las PCBs están compuestas principalmente por resina epóxica y fibra de vidrio. Además, se determinó que los residuos de mayor tamaño contiene 67.22 % de fibra de vidrio mientras que la de menor tamaño contiene 78.78 %. Los metales que lixiviaron en mayor concentración fueron el zinc, cobre y plomo y en menores cantidades el cobalto, plata y níquel. Una manera económica y práctica de reciclar la FNM de PCBs sería mediante la reutilización física, es decir, como carga en materiales compuestos debido a la gran cantidad de fibra de vidrio contenida principalmente en el material de menor tamaño. No es recomendable realizar una combustión de los residuos ya que se obtiene una gran cantidad de cenizas y se necesita una alta temperatura para su descomposición. Así mismo, no es recomendable su disposición en desechos sanitarios debido a la lixiviación de los metales pesados. Palabras clave: Tarjetas de circuitos impresos, fracción no metálica, caracterización, espectroscopía de infrarrojo, calorimetría diferencial de barrido, termogravimetría, reciclaje físico, reciclaje. Abstract: Electronic waste has been one of the main sources of pollution worldwide in the last years, due to largescale of production and commercialization of this kind of equipment. One of the most important components and more difficult to recycle due to the complexity of mixture are the printed circuit boards. The aim of this project is to extract and characterize the non-metallic fraction (NMF) of the PCBs in order to determine a practical way to recycle this waste. First, ports, batteries, and capacitors were removed. Then, they were sheared and ground in a hammer mill in order to do the gravimetric separation. Finally, two particle sizes, one between 0.15 and 0.075 mm and another less than 0.075 mm, were taken to determine the influence of particle size composition. This fraction was characterized by infrared spectroscopy (FTIR), differential scanning calorimetry (DSC), thermogravimetry (TGA) and analysis for determining leachate. The PCBs are mainly composed of epoxy resin and fiberglass. In addition, it was determined that bigger waste fraction contains 67.22 % fiberglass while smaller contains 78.78 %. The metals were leached in higher concentration were zinc, copper and lead; and cobalt, silver and nickel in minor amounts. Physical recycling is an economical and practical way to recycle the PCBs as a filler in composites because of the amount of fiberglass contained in the NMF. It is not recommended to perform a combustion of waste due to the big quantity of ash obtained and high temperature needed for decomposition. It is also not recommended for disposal in landfill due to the leaching of heavy metals. Keywords: Printed circuit boards, non-metallic fraction, characterization, infrared spectroscopy, differential scanning calorimetry, thermogravimetry, physical recycling, recycling. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 67 68 Loyo C.*; Arroyo C.*; Aldás M.*; Montero R. ** _______________________________________________________________________________________________________________________________ 1. INTRODUCCIÓN En los últimos años ha existido un gran incremento en la producción de aparatos eléctricos y electrónicos; los cuales, una vez terminada su vida útil pasan a formar parte de los desechos eléctricos y electrónicos. En el año de 1990 se generaron alrededor de 20 millones de toneladas de desechos mientras que el año 2015 se estima que existirán 75 millones de toneladas. Si esta gran cantidad de desechos no son tratados adecuadamente pueden producir grandes impactos ambientales [6]. El factor más importante para determinar el tipo de reciclaje que se debe llevar a cabo para los desechos eléctricos y electrónicos, es su composición. Sin embargo, el progreso en cuanto a producción de aparatos eléctricos y electrónicos ha llevado a las industrias a utilizar una gran variedad de materiales para su fabricación, provocando de esta manera que su separación y posterior reciclaje se dificulte [10]. Las tarjetas de circuitos impresos (PCB por sus siglas en inglés) son la plataforma sobre la cual los componentes microelectrónicos se montan. Las PCBs proporcionan las interconexiones eléctricas entre sus componentes. Las PCBs se encuentran en prácticamente todos los aparatos eléctricos y electrónicos [4]. Además, se encuentran en todos los sistemas de armamento y equipos aeroespaciales, siendo así una parte esencial del funcionamiento de los equipos. Las placas de circuitos impresos son la base de la industria electrónica, siendo su producción crucial para la fabricación y venta de más de un billón de equipos electrónicos al año [9]. Las placas de circuitos impresos son una mezcla compleja de fibra de vidrio, resina polimérica y múltiples tipos de metales. Esta mezcla compleja y sus características hacen de su reciclaje uno de los más difíciles de realizar. Sin embargo, el reciclaje de estas placas no solo es importante ambientalmente, sino también es importante económicamente ya que está compuesta de varios metales valiosos. Entre los más importantes se encuentran el oro y la plata, los cuales están en un rango entre 244 a 857 g/t para el oro y entre 520 a 2901 g/t para la plata [2, 3, 5]. ser eliminadas, además de ser peligrosas para la salud y el medioambiente [6, 11]. Por estos motivos, el presente trabajo pretende extraer y caracterizar la fracción no metálica de las tarjetas de circuitos impresos de computadoras desechadas, para poder determinar el potencial de reciclaje que tienen estos desechos. 2. METODOLOGÍA 2.1 Obtención de la fracción no metálica de las tarjetas de circuitos impresos de computadoras desechadas Inicialmente fueron seleccionadas las tarjetas de circuitos impresos de los residuos electrónicos recolectados por empresas dedicadas a esta actividad. Se seleccionaron tarjetas de circuitos impresos de computadoras desechadas. Posteriormente se retiraron los puertos de salida USB, audio, videos, serie, paralelo y VGA de las tarjetas y además se extrajeron las baterías y capacitores. Mediante la utilización de una cizalla se cortaron las tarjetas en 8 partes de aproximadamente 6 cm de lado. Estos pedazos de tarjetas obtenidos fueron molidos a través de un molino de martillos, el cual está conformado por una tolva de alimentación, una cámara de trituración, un juego de cuchillas metálicas y una malla metálica. Una vez realizada la molienda se separó el producto obtenido en dos partes de acuerdo a su tamaño mediante un tamiz. El material con tamaño menor a 1.18 mm fue llevado a una mesa de separación gravimétrica con el fin de separar la fracción no metálica de la fracción metálica de las tarjetas de circuitos impresos. El material con tamaño mayor a 1.18 mm fue reprocesado en el molino de martillos. Para mejorar la separación gravimétrica se adicionó agua, por lo cual se tuvo que realizar un posterior secado tanto de la fracción metálica como de la fracción no metálica. Del proceso de separación se obtuvieron tres fracciones: la fracción metálica, la fracción no metálica y la fracción de mixtos, esta última fue reprocesada en la mesa de separación gravimétrica con el fin de obtener una mayor cantidad de fracción no metálica. El proceso de recuperación de la fracción metálica de las PCBs a nivel mundial ya está muy avanzado. Sin embargo, la recuperación y reciclaje de la fracción no metálica no ha sido aún implementada, pese a que entre el 70 % y 80 % de las PCBs corresponde a la parte no metálica. Esta fracción está compuesta por fibra de vidrio (65 %), resina epóxica (32 %), impurezas (cobre <3 %, soldadura <0.1 %) y retardantes a la llama bromados. Esta falta de exploración en el reciclaje de la fracción no metálica se debe a que éste proceso no constituye un beneficio altamente económico como lo es el reciclaje de la fracción metálica [4, 5]. Finalmente la fracción no metálica fue tamizada a través de las mallas número 100 y 200. Se obtuvieron dos tamaños de partícula: el primero comprendido entre 0.15 y 0.075 mm y el segundo con tamaño menor a 0.075 mm. Estos dos tamaños de partícula fueron caracterizados posteriormente por separado. La disposición y recuperación de los residuos de aparatos eléctricos y electrónicos tienen altas implicaciones ambientales, en especial la fracción no metálica de dichos residuos, debido a que esta contiene substancias peligrosas como cadmio, plomo o retardantes a la llama bromados. Estas sustancias demandan altos costos de producción para Primero se llevó a cabo la eliminación de la humedad tanto del material con tamaño de partícula 0.15-0.075 mm como del material con tamaño <0.075 mm, para lo cual se lo colocó en la estufa a 100 °C durante dos horas. Debido a que la muestra es de tamaño de partícula muy fina, se llevó a cabo 2.2 Caracterización de la fracción no metálica de tarjetas de circuitos impresos 2.2.1 Análisis de Espectroscopia de Infrarrojo (FTIR) Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Extracción y Caracterización de la Fracción No Metálica de las Tarjetas de Circuitos Impresos de Computadoras Desechadas _________________________________________________________________________________________________________________________ la formación de una pastilla de la mezcla del material de análisis con bromuro de potasio, en una relación 1:100. Las pastillas formadas de la muestras de la fracción no metálica de las tarjetas de circuitos impresos fueron analizadas por transmitancia desde los 4 000 cm-1 hasta los 400 cm-1 con el espectrofotómetro de infrarrojo. Para obtener una buena resolución de los espectros se llevó a cabo una normalización, suavizado de los picos y corrección de la línea base, mediante el software del equipo. Se realizaron un total de tres ensayos de espectroscopía de infrarrojo. 2.2.2 Análisis de Calorimetría Diferencial de Barrido (DSC) Con el propósito de determinar las propiedades térmicas de la fracción no metálica de las tarjetas de circuitos impresos se llevó a cabo el análisis de Calorimetría Diferencial de Barrido. Los ensayos se realizaron en atmósfera de nitrógeno con un flujo de 20 mL/min con 5 barridos: primer calentamiento (desde temperatura ambiente hasta 250 °C a una tasa de calentamiento de 10 °C/min), paso isotermal (a 250 °C durante 10 minutos), primer enfriamiento (desde 250 °C hasta temperatura ambiente), segundo calentamiento (desde temperatura ambiente hasta 250 °C a una tasa de calentamiento de 10 °C/min), y segundo enfriamiento (desde 250 °C hasta temperatura ambiente). Se realizaron un total de tres ensayos de calorimetría diferencial de barrido. 2.2.3 Análisis de Termogravimetría (TGA) Con el propósito de determinar la temperatura de descomposición y la composición en cuanto a materia orgánica e inorgánica se realizó el análisis de termogravimetría. Para realizar este análisis se tomó aproximadamente 10 mg de muestra y se lo colocó en el porta muestra del equipo de termogravimetría. El procedimiento que se llevó a cabo fue con base en la norma ASTM E2105 [1]. La prueba se llevó a cabo con una tasa de calentamiento de 10 °C/min hasta alcanzar los 900 °C y un flujo de nitrógeno de 50 mL/min. Se realizaron un total de tres ensayos de termogravimetría. 2.2.4 Análisis de determinación de toxicidad de lixiviados (TCLP) Para realizar el análisis de toxicidad de lixiviados se tomó una muestra representativa de ambos tamaños de partícula, después se procedió a colocar 10 g de muestra con 200 mL de agua destilada, posteriormente se reguló el pH de la solución en 4 y se dejó en agitación continua durante 21 horas. Este ensayo se realizó con referencia al método EPA 1311. Una vez terminada la agitación se realizó a caracterización de los metales lixiviados mediante el equipo de Espectrofotometría de Absorción Atómica. 69 3. RESULTADOS Y DISCUSIÓN 3.1 Obtención de la fracción no metálica de las tarjetas de circuitos impresos de computadoras desechadas Los puertos de salida, capacitores y batería corresponden a 11.27 % del peso total de las tarjetas de circuitos impresos seleccionadas. Estos componentes fueron removidos debido a que los puertos de salida están compuestos en su totalidad por metales, los cuales ocasionarían daños considerables en los equipos de molienda; mientras que los capacitores y baterías contienen sustancias altamente tóxicas que necesitan un tratamiento diferente. En la Tabla 1 se presentan los resultados de las dos fracciones de tamaño obtenidas después de la molienda en el molino de martillos. Pese a que el molino de martillos no es el más adecuado para moler este tipo de residuos, se obtuvo casi la mitad del material con tamaño menor a 1.18 mm, el cual fue llevado a la separación gravimétrica. Solo el material que tenía un tamaño menor a 1.18 mm pudo ser llevado a la separación gravimétrica debido principalmente a que en el material con mayor tamaño no se separaba de una manera eficiente su parte metálica de su parte no metálica. Además, se apreciaron aglomerados metálicos que tenían un tamaño mayor a 1.18 mm, estos aglomerados se formaron dentro del molino. Como muestra la Tabla 1, el material llevado a la separación gravimétrica correspondía al 43.42 %, mientras que el restante 58.58 % fue reprocesado en el molino de martillos para incrementar la extracción de la fracción no metálica de las tarjetas de circuitos impresos. En la Tabla 2 se observan los resultados obtenidos de fracción metálica y fracción no metálica obtenida después de realizar el proceso de separación gravimétrica Tabla 1. Resultados de las fracciones obtenidas después de la molienda en el molino de martillos Peso de Peso de Peso de tarjetas sin material con material con capacitores. puertos tamaño tamaño de salida y baterías mayor 1.18 menor 1.18 (g) mm (g) mm (g) 15 303.63 8 659.42 6 644.22 100.00 % 56.58 % 43.42 % Porcentajes Tabla 2. Resultados obtenidos después de la separación gravimétrica Material total procesado en la separación gravimétrica (g) Fracción metálica (g) Fracción no metálica (g) 6 17.09 2 092.84 4 324.25 100.00 % 32.61 % 67.39 % Porcentaje Según un estudio realizado por [4], entre el 70 % y 80 % del peso total de las tarjetas de circuitos impresos corresponde a la fracción no metálica. En la Tabla 2 se puede observar que se obtuvo un 67.39 % de fracción no metálica. Esta variación con respecto al estudio citado se debe principalmente a pérdidas de material no metálico durante la separación Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 70 Loyo C.*; Arroyo C.*; Aldás M.*; Montero R. ** _______________________________________________________________________________________________________________________________ gravimétrica. Como se puede observar en la Fig. 1, en la fracción metálica obtenida existe aún una cantidad de material no metálico que no pudo ser separado. Asimismo, se perdió una parte del material no metálico que no se sedimentó en el agua utilizada para mejorar la separación gravimétrica. En la Tabla 3 se muestran los porcentajes de las fracciones de tamaño 0.15-0.075 mm y <0.075 mm obtenidas mediante tamizado de la fracción no metálica. Se puede observar que la mayor cantidad de material corresponde al de tamaño mayor a 0.15 mm. Este material puede ser molido mediante un molino de discos ya que el contenido de metales es mínimo. Además, un molino de discos permite obtener un tamaño deseado mediante el ajuste de la separación de sus discos. También se obtuvo mayor cantidad de material con tamaño de partícula <0.075 mm respecto al material con tamaño de partícula entre 0.15-0.075 mm. Esto es conveniente si se desea realizar un reciclaje físico ya que, como se discutirá posteriormente, este material contiene una mayor cantidad de fibra de vidrio que permite reforzar materiales compuestos. Figura 1. Fracción metálica obtenida después de la separación gravimétrica Tabla 3. Resultados de los pesos obtenidos en el tamizado con malla número 100 y 200 Peso de Material Material con Material con fracción con tamaño tamaño tamaño entre no mayor a menor a 0.15 mm y metálica 0.15 mm 0.075 mm 0.075 mm (g) (g) (g) (g) 4 324.25 2 221.67 960.19 1 142.39 Porcentaje 100.00 % 51.38 % 22.20 % 26.42 % Porcentaje 100.00 % 51.38 % Figura 2.Espectro infrarrojo de la fracción no metálica de las tarjetas de circuitos impresos con tamaño de partícula de tamaño 0.15-0.075 mm 48.62 % 3.2 Caracterización de la fracción no metálica de tarjetas de circuitos impresos 3.2.1 Análisis de Espectroscopía de Infrarrojo (FTIR) En la Fig. 2 se muestran un espectro infrarrojo obtenido por transmitancia, en un rango de 4 000 hasta los 400 cm-1, de la fracción no metálica de las tarjetas de circuitos impresos. El espectro infrarrojo obtenido no se logró comparar con un espectro estándar disponible en la biblioteca del software del equipo utilizado. Esto sucede porque existe una gran variedad de componentes en las tarjetas de circuitos impresos. Sin embargo, se lograron apreciar los principales componentes de las tarjetas los cuales se señalan en la Fig. 2: resina epóxica y fibra de vidrio (representada por los grupos silicato). 3.2.2 Análisis de Calorimetría Diferencial de Barrido (DSC) En la Fig. 3 se muestra el termograma realizado desde temperatura ambiente hasta 250 °C, para la fracción no metálica de las tarjetas de circuitos impresos de computadoras con el fin de determinar la temperatura de transición vítrea. Se puede observar que la temperatura de transición vítrea de la fracción no metálica es aproximadamente 133.4 °C, la cual no difiere significativamente con la temperatura de transición vítrea indicada en bibliografía que corresponde a 140 °C [8]. Figura 3. Termograma de la fracción no metálica de las tarjetas de circuitos impresos Esta diferencia se debe principalmente a que la temperatura indicada en bibliografía se realizó de compuestos que contenía solamente fibra de vidrio y resina epóxica, mientras que el material analizado en el proyecto puede contener trazas de metales como cobre, plomo, níquel y compuestos bromados. 3.2.3 Análisis de Termogravimetría (TGA) En las Fig. 4 y Fig. 5 se presentan los termogramas obtenidos de la fracción no metálica de tamaño 0.15-0.075 mm y <0.075 mm.Los diagramas termogravimétricos nos indican la estabilidad térmica que tiene la muestra, la cual es muy importante tanto para el reciclaje físico como para el reciclaje químico [3, 7]. Para el caso de los residuos utilizados en este Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Extracción y Caracterización de la Fracción No Metálica de las Tarjetas de Circuitos Impresos de Computadoras Desechadas _________________________________________________________________________________________________________________________ proyecto podemos observar que tienen una alta estabilidad térmica ya que los materiales gruesos y finos empiezan a descomponerse a los 291 °C y 269 °C respectivamente. Es decir no hay ningún peligro de descomposición en los procesos de reciclaje físico que se realizan comúnmente, en los cuales se trabaja generalmente a temperatura ambiente o máximo a 100 °C. Además, en las Fig. 4 y Fig. 5 se puede observar que se alcanzó la mayor degradación para gruesos y finos a los 346 °C y 351 °C respectivamente. Figura 4. Diagrama termogravimétrico obtenido de la fracción no metálica de tarjetas de circuitos impresos con tamaño de partícula de tamaño 0.150.075 mm Finalmente se puede observar que la descomposición de la mayor parte de la resina epóxica se obtiene a una temperatura para gruesos y finos de 464 °C y 443 °C respectivamente. En la Tabla 4 se presentan los resultados del análisis termogravimétrico para ambos tamaños de partícula analizados en cuanto a porcentaje en peso de material orgánico e inorgánico para las 3 repeticiones realizadas. Los componentes mayoritarios de la fracción no metálica de las tarjetas de circuitos impresos son la resina epóxica y la fibra de vidrio, por lo que se puede observar en la Tabla 4 que existe una mayor cantidad de material inorgánico. Además, en el material con menor tamaño de partícula se observa que existe una mayor cantidad de material inorgánico (que corresponde en su mayoría a la fibra de vidrio). Esto se debe principalmente a que la fibra se encuentra liberada en este tamaño de partícula. Es importante determinar el contenido de material inorgánico, ya que en caso de realizarse un reciclaje físico de estos residuos como refuerzo, conviene tener la mayor cantidad de esta fibra para mejor ciertas propiedades mecánicas del material reforzado, como las de tracción, flexión o abrasión. 71 3.2.4 Análisis de determinación de toxicidad de lixiviados (TCLP) En la Tabla 5 se presentan los resultados obtenidos del análisis de toxicidad de lixiviados para cada tamaño de partícula. Tabla 4. Resultados del análisis termogravimetría para ambos tamaños de partícula Material orgánico Material inorgánico (%) (%) 0.15-0.075 <0.075 0.15-0.075 <0.075 mm mm mm mm M1 40.61 20.55 59.39 79.45 M2 26.92 20.54 73.08 79.46 M3 30.81 22.56 69.19 77.44 Promedio 32.78 21.22 67.22 78.78 Tabla 5. Resultados del análisis de toxicidad de lixiviados Tamaño de partícula 0.075-0.15 mm <0.075 mm Elemento Concentración (mg/L) Concentración (mg/L) 2.34 3.79 Zinc 20.43 38.22 Cobre 43.01 27.99 Plomo 0.01 0.01 Cobalto 0.02 0.03 Plata 0.07 0.23 Níquel <0.01 <0.01 Cromo <0.01 <0.01 Cadmio <0.1 <0.1 Bario <0.1 <0.1 Arsénico <0.1 <0.1 Mercurio En la Tabla 5 se puede observar que para ambos tamaños de partícula las concentraciones más altas de metales obtenidas en los lixiviados pertenecen al zinc, cobre y plomo. Sin embargo, el único valor que excede los valores máximos permitidos para ensayos de lixiviación según la norma técnica de suelo de la ordenanza metropolitana es la concentración de plomo, cuyo valor máximo permisible es de 0.2 mg/L. Se observó además que, en general, para el material con partículas de tamaño <0.075 mm existe una mayor concentración de metales. Esto se debe a que a menor tamaño de partícula los metales se liberan de la matriz polimérica, por lo que aumenta su facilidad de lixiviación. 4. CONCLUSIONES Mediante separación gravimétrica se determinó que las tarjetas de circuitos impresos utilizadas contenían un 32.61 % de fracción metálica y 67.39 % de fracción no metálica. Figura 5. Diagrama termogravimétrico obtenido de la fracción no metálica de tarjetas de circuitos impresos con tamaño de partícula < 0.075 mm Mediante FTIR se logró determinar que la fracción no metálica de las tarjetas de circuitos impresos están compuestas principalmente por resina epóxica y fibra de vidrio. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 72 Loyo C.*; Arroyo C.*; Aldás M.*; Montero R. ** _______________________________________________________________________________________________________________________________ Mediante TGA se determinó que la fracción no metálica de tarjetas de circuitos impresos de tamaño 0.15-0.075 mm tiene 67.22 % de material inorgánico (fibra de vidrio) y 32.78 % de material orgánico (resina epóxica); y para el material de tamaño <0.075 mm se obtuvo un 78.78 % de material inorgánico y 21.22 % de material orgánico. Mediante el TCLP se determinó que la fracción no metálica de las tarjetas de circuitos impresos superan los límites permisibles de lixiviación de plomo por lo tanto no es recomendable su disposición en rellenos sanitarios. La fracción no metálica de las tarjetas de circuitos impresos puede ser utilizada para un reciclaje físico, es decir incorporarlos como carga dentro de una matriz para formular un material compuesto. No es recomendable realizar un reciclaje químico de la fracción no metálica de las tarjetas de circuitos impresos, ya que las temperaturas de descomposición son muy altas. REFERENCIAS [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] ASTM E2105. (2010). Standard Practice for General Techniques of Thermogravimetric Analysis (TGA) Coupled With Infrared Analysis (TGA/IR). EstadosUnidos. E. De la Torre, A. Guevara y S. Espinoza. (2009). Los teléfonos celulares una nueva mina de metales preciosos, factible de valorizar mediante tostación y lixiviación con cianuro. Revista Politécnica. 30(1). 21-28 I. De Marco, B. M. Caballero, M. J. Chomón, M. F. Laresgoiti, A. Torres, G. Fernández y S. Arnaiz, (2008). Pyrolysis of electrical and electronic wastes. Journal of Analytical and Applied Pyrolysis, 82(2), 179-183. J. Guo, J. Li, Q. Rao y Z. Zu. (2008). Phenolic Molding Compound Filled with Nonmetals of Waste PCBs. Environmental Science &Technology, 42(2), 624-628. J. Guo, J. Guo y Z Xu. (2009). Recycling of non-metallic fractions from waste printed circuit boards: A review. Journal of Hazardous Materials, 168(2-3), 567-590. V. Goodship y A. Stevels. (2012). Waste electrical and electronic equipment (WEEE) handbook. (1ra Ed). Conwall, Reino Unido: TJ International Ltd. N. H. Hoe, A. Salmiaton y H. Hizam. (2014). Catalytic Pyrolysis and a Pyrolysis Kinetic Study of Shredded Printed Circuit Board for Fuel Recovery. Bulletin of Chemical Reaction Engineering & Catalysis, 9(3), 224-240. E. Kaisersberger, S. Knappe, H. Mohler y S. Rahner. (2004). TA for Polymer Engineering: DSC TG DMA TMA. (1ra Ed).Wurzburg, Alemania: Netzsch-Geratebau. J. La Dou (2006). Printed circuit board industry. International Journal of Hygiene and Environmental Health, 209(3), 211-219. M. Oguchi, H. Sakanamura y A. Terazono. (2013). Toxic metals in WEEE: Characterization and substance flow analysis in waste treatment processes. Science of the Total Environmental, 463(8), 1124-1132. P. A. Wager, M. Schluep, E. Muller y R. Gloor. (2011). RoHS regulated Substances in Mixed Plastics from Waste Electrical and Electronic Equipment. Environmental Science & Technology, 46(2), 628-635. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Modelo de Deconvolución para la Cuantificación de los Componentes del Caucho Vulcanizado Presente en los Neumáticos Fuera de Uso _________________________________________________________________________________________________________________________ 73 Modelo de Deconvolución para la Cuantificación de los Componentes del Caucho Vulcanizado Presente en los Neumáticos Fuera de Uso Meza I.*; De la Torre E.* *Escuela Politécnica Nacional, Facultad de Ingeniería Química y Agroindustria, Quito, Ecuador e-mail: [email protected] Resumen: Se efectuaron ensayos termogravimétricos a distintas velocidades de calentamiento y con diferentes tamaños de partícula con el objeto determinar los parámetros cinéticos y la composición del caucho de llantas. Se determinó que existen tres zonas de degradación entre 146-549 °C en las cuales se descomponen los distintos componentes de forma independiente e irreversible. Se encontró que el componente más resistente a la degradación fue el caucho sintético, seguido del caucho natural y como componente más volátil los plastificantes y aditivos, manifestando energías de activación de 135.4, 123.9 y 47.7 KJ mol-1 respectivamente. El modelo de deconvolución empleado, determinó que el caucho sintético se encuentra en mayor proporción con un 60.8 %, seguido por el caucho natural con un 32.8 % y por último los plastificantes y aditivos comprenden un 6.1 %. Palabras clave: Neumáticos fuera de uso (NFU’s), deconvolución, pirólisis, caucho natural, caucho sintético. Abstract: Thermogravimetric tests were conducted under different heating rates and particle sizes in order to determine the kinetic parameters and the rubber tire composition. Three stages of degradation between 146-549 °C were found, in which components decompose in an independently and irreversible way. Synthetic rubber is the toughest component to be degraded, followed by natural rubber; plasticizer and additives were found as the most volatile components. The mentioned materials have the following activation energies: 135.4, 123.9 and 47.7kJmol-1, respectively. The employed deconvolution model determined that tires are mainly composed of synthetic rubber in a greater proportion with 60.8%, followed by natural rubber with 32.8 % and finally plasticizers and additives with 6.1 %. Keywords: End Life tires (ELTs), deconvolution, pyrolysis, natural rubber (NR), synthetic rubber. 1. INTRODUCCIÓN Se estima que alrededor de 250 millones de coches y camiones se desechan cada año en la Unión Europea, lo que representa alrededor de 2.6 millones de toneladas de neumáticos. Cantidades comparables son generadas en Europa del Este, América del Norte, América Latina, Asia y el Medio Oriente. Por lo tanto, se calcula que en todo el mundo, más de mil millones de neumáticos de desecho se generan anualmente [6, 14]. La acumulación de neumáticos usados en un campo abierto resulta en un desperdicio de un valioso recurso energético, además de un potencial riesgo para la salud, puesto que son un medio propicio para la proliferación de plagas que transmiten enfermedades tropicales tales como dengue, paludismo, etc.; así como, un peligro latente por la posibilidad de que se produzca un incendio de grandes proporciones [1, 4]. Varias soluciones para reducir la disposición final en vertederos se han desarrollado. Prácticas de reciclaje de neumáticos, tales como el recauchutado, molienda y el uso de neumáticos como aditivo para las pistas deportivas o carreteras de asfalto son algunos de los tratamientos que se realizan actualmente. Sin embargo, todos ellos tienen desventajas significativas o limitaciones. El poder calorífico de los neumáticos oscila entre 2837 MJ/kg, valor comparable al que presentan ciertos carbones de alto rango, por lo que procesos térmicos, como la combustión, pirólisis y gasificación han sido considerados como métodos alternos para la recuperación de energía y materias primas a partir de llantas de desecho [1]. Varios estudios han demostrado de igual manera que el residuo solido producto de la pirólisis de los NFU’s, puede tener aplicaciones, tales como precursor de carbón activado. Debido a su importante contenido de carbono [7] Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 74 Meza I.*; de la Torre E.* _______________________________________________________________________________________________________________________________ 2. MODELIZACIÓN MATÉMATICA DEL PROCESO DE DESCOMPOSICIÓN TÉRMICA Aunque varios estudios fundamentales sobre la pirólisis de llantas de desecho se han realizado en los últimos años, los mecanismos implicados en el proceso de pirólisis aún no se entienden completamente. Modelar la cinética del proceso de degradación térmica de los neumáticos puede proporcionar una visión de los mecanismos responsables de la pirólisis de neumáticos y predecir posibles dificultades en un reactor de pirólisis [8]. El hecho de que la pirólisis de es un fenómeno multietapa y multicomponente está ampliamente aceptado [14]. El modelado de la pirólisis de los neumáticos depende casi exclusivamente de los datos de TGA (análisis termogravimétrico) y DTG, donde se usan las ecuaciones del modelo para la deconvolución de los picos en los termogramas [3]. Un gran número de modelos cinéticos se han desarrollado para el estudio de mecanismo de pirólisis y parámetros cinéticos [10]. n n dWa dWi k iWi dt i 1 dt a i 1 Donde Wa = Wt - W∞ corresponde a la porción degradable del peso de llanta a cualquier tiempo; Wt al peso del caucho a cualquier tiempo; W∞ corresponde al peso del carbón residual; Wi es el peso del componente degradable i a cualquier tiempo; ki es la constante cinética correspondiente al componente i, cabe recalcar que Wa y dWa/dt, pueden ser directamente obtenidos de las curvas TGA y DTG respectivamente. Como se sabe el caucho está compuesto de varios componentes, y cada uno de ellos exhibe sus propios parámetros cinéticos característicos. Además, existen zonas de temperatura propias en las que reflejan la descomposición única de cada componente. La descripción cinética de las curvas TGA y DTG para n componentes se muestra a continuación: La mayoría de modelos de pirólisis asumen una velocidad de reacción limitada por la reacción química y no consideran las limitaciones de transferencia de masa y de calor en un reactor. Por lo tanto, la velocidad de reacción se describe mediante la siguiente ecuación: (1) r kC n Donde: r k C n Velocidad de reacción Constante de reacción Concentración másica Orden de la reacción Referencia [16], afirma que para hacer uso de este modelo matemático, se debe tomar en cuenta que: i. Los componentes mayoritarios como plastificantes, caucho natural y cauchos sintéticos, se descomponen térmicamente de forma independiente, sin interacciones entre ellos. ii. Cada reacción de descomposición sigue un solo mecanismo y es irreversible. iii. Solo los datos de la curva DTG son necesarios para estimar la composición inicial del neumático, así como la velocidad de volatilización de cada componente, además de sus parámetros cinéticos 2.1 Modelo cinético para la deconvolución de los componentes de un NFU’s El modelo que se explica a continuación es una transcripción de un modelo previamente desarrollado [16]. La ecuación general que expresa la velocidad de descomposición o degradación térmica de los NFU’s es descrita como: (2) n n dWa dWi E Ai exp i Wi dT RT i 1 dT i 1 (3) Siendo β igual la velocidad de calentamiento (dT/dt). Se obtiene (4) dividiendo (3) para –Wa, y luego aplicando logaritmo natural a ambas partes: dWa n E W ln Ai exp i i ln RT Wa Wn dT i 1 (4) Se puede asumir que el caucho de los neumáticos está compuesto por dos compuestos mayoritarios, los cuales se descomponen con reacciones independientes y además poseen diferentes velocidades de degradación con respecto a la temperatura. El compuesto 1 ha sido considerado como más difícil de descomponer, sugiriendo su degradación a la zona de altas temperaturas. Se considera que en la zona de altas temperaturas solo se descompone el menos volátil (componente 1), debido a que el componente 2 para esta etapa ya está totalmente degradado. La región de temperaturas medias es responsable de la descomposición de ambos componentes, mientras que en la zona de bajas temperaturas se considera que solo el componente 2 se degrada. Para el caso de 2 componentes, (4) puede ser escrita como: E1 W1 A1 exp dWa RT Wa ln ln Wa dT A exp E 2 W2 2 RT Wa (5) Para la zona de altas temperatura el contenido del componente 2 es despreciable por ende W 2 ≈ 0 y Wa = W1 .Por lo cual (5) se reescribe: Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Modelo de Deconvolución para la Cuantificación de los Componentes del Caucho Vulcanizado Presente en los Neumáticos Fuera de Uso _________________________________________________________________________________________________________________________ dW1 E 1 ln A1 1 ln R T W1 dT (6) Por otra parte la ecuación correspondiente a la zona de bajas temperaturas, la descomposición del componente 1 se considera despreciable comparándola con respecto al componente 2. Por ende se considera la tasa de descomposición despreciable dW1/dT ≈ 0. dW2 E 1 (7) ln A2 2 ln W dT R T 2 Tomando el lado izquierdo de (5) y graficando versus el reciproco de la temperatura, dos líneas rectas pueden ser graficadas haciendo uso de (6) y (7), para las zonas de altas y bajas temperaturas respectivamente. La pendiente de la curva de alta temperatura permite obtener la energía de activación, mientras que la intersección con el Eje “y” arroja el valor del factor de frecuencia para el componente 1. Sin embargo, para poder obtener los parámetros cinéticos del segundo componente se requiere del cálculo de W 2 y dW2/dT ya que su obtención grafica no es posible a través de las curvas TGA y DTG. La ecuación cinética del componente 1 puede ser descrito como: dW1 E A1 exp 1 W1 dT RT (8) Integrando (8) se tiene: W1 dW1 A1 T E exp 1 dT dT 0 RT W1.0 (9) La ecuación (9) se resuelve aproximando el lado derecho de a: E1 A RT exp dT 1 0 E1 RT A1 T 2 (10) 2 RT E1 2 RT 1 exp 4 E1 E1 RT Así, W1 es expresado como: W1 W1.0 A1 RT 2 E 1 2 RT 2 RT exp 1 4 E E 1 1 E1 exp RT W1.0 A1 RTh 2 E1 2 RTh 2 RTh Wa exp 1 4 E E 1 1 E1 exp RT h 75 (12) De la misma forma se puede proceder para obtener la velocidad de descomposición del componente 2, restado dW1/dT de la curva de DTG dWa/dT y considerar que W2 se calcula a través de la sustracción de W 1 de Wa. Por tanto el peso inicial del segundo componente se enuncia como: W2.0 A2 RTm 2 E 2 RTm 2 RTm Wa W1 exp 1 4 E2 E2 exp E 2 RT m 2 (13) Donde Tm corresponde a la temperatura en la zona de temperaturas medias; W2 puede ser expresado de la siguiente manera: 2 A RT 2 2 RT RT 2 1 4 E2 E 2 E 2 (14) W2 W2.0 exp E2 exp RT 3. METODOLOGIA EXPERIMENTAL En el presente trabajo se desarrollaron ensayos de termogravimetría para estudiar la influencia de la velocidad de calentamiento y el tamaño de partícula en el proceso de pirólisis. Así como, el desarrollo de un modelo matemático de deconvolución que permita cuantificar los componentes principales del caucho vulcanizado presente en los neumáticos de desecho. 3.1 Caracterización de la materia prima y definición del método de reducción de tamaño de los NFU’s (11) Sabiendo que W1 es igual a Wa a las altas temperaturas W1.0 se obtenido de: En la caracterización se analizaron las siguientes propiedades: densidad, contenidos de humedad, volátiles, cenizas y carbón fijo. El análisis elemental de los componentes químicos de los NFU’s se realizó mediante microscopía electrónica de barrido (MEB-EDX Tescan con analizador de rayos X Quantax (Bruker)). El método de reducción tamaño consistió en separar las partes del neumático con contenido metálico, de los flancos zona que posee una malla textil. El caucho presente en la sección lateral del neumático es aquel que se utilizó dentro de Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 76 Meza I.*; de la Torre E.* _______________________________________________________________________________________________________________________________ este estudio. Durante esta fase se intentó dividir la llanta en fracción de menor tamaño con el uso de varios instrumentos (tijeras, cuchillas, etc.). Sin embargo, la amoladora fue la herramienta que mostró mejores resultados en el fraccionamiento de la llanta, posteriormente se siguió reduciendo de tamaño el material de forma manual hasta obtener un material de tamaño homogéneo de entre 2-3 cm. 3.2 Evaluación del comportamiento de los NFU’s en el proceso de pirólisis y deconvolución de sus componentes. Para el estudio cinético de la pirólisis y la deconvolución de los componentes presentes en las llantas de desecho, se empleó el método de termo-gravimetría haciendo uso de una termobalanza (Shimadzu TGA-50). Todos los ensayos se realizaron bajo una atmósfera inerte de nitrógeno con un flujo de gas de 50 mL/min y un peso aproximado de muestra de 18 mg. Así mismo, para determinar la influencia del tamaño de partícula y la velocidad de calentamiento dentro del proceso de pirólisis; se trabajó con dos distintos tamaños de partícula (polvo < 1mm y trozos entre 2-5 mm) a tres diferentes velocidades de calentamiento (5, 10 y 20 K/min). 4. RESULTADOS Y DISCUSIÓN 4.1 Caracterización de la materia prima La Tabla 1 reporta los elementos químicos existentes en la muestra de caucho analizada. Este tipo de ensayo es semicuantitativo y realiza una lectura superficial dentro de toda la muestra. Los resultados reflejaron un alto contenido de carbono del 92.2 %, porcentaje esperado, debido a que el caucho es una mezcla de polímeros, donde el carbono es elemento preponderante. Además es un valor comparable con los resultados obtenidos por varios autores, que proponen un contenido de carbono que oscila entre un 80 a 90 % del total [9, 11, 16, 17]. A pesar de que el análisis reportó valores similares a los bibliográficos, el contenido de hidrógeno no fue determinado dentro de la muestra, debido que el método empleado no permite cuantificar este tipo de elementos. Asimismo, cabe recalcar que el porcentaje no reportado dentro de este estudio podría corresponder al hidrógeno reportado en otros estudios. En la Tabla 2 se observa que es un material con un alto contenido de material volátil, el cual posee un 33 % de material sólido del total del peso del caucho. Por otra parte su contenido de humedad es bastante bajo lo que demuestra que es un material con baja permeabilidad. El bajo contenido de cenizas presente en el residuo sólido permitió considerar a los neumáticos como una materia prima viable para la producción de carbón de alta pureza. La densidad aparente fue calculada para un tamaño de partícula entre 2-3 cm., mientras que la densidad real se evaluó con caucho en polvo. Tabla 1. Análisis químico de trozo de caucho de NFU’s mediante MEBEDX Elemento Contenido en peso (%) Carbono 92.2 Azufre 1.5 Los resultados obtenidos se muestran en la Tabla 3. Tabla 2. Contenidos de humedad, volátiles, cenizas y carbón fijo Contenido (%) Humedad 0.9 Volátiles 66.4 Cenizas 1.9 Carbón fijo 31.6 Tabla 3. Densidad real y densidad aparente de los NFU´s Densidad real (g/mL) 1.098±0.003 Densidad aparente (2-3 cm) (g/mL) 0.423±0.045 La desviación estándar reportada en los resultados de densidad real y densidad aparente, demuestra que los ensayos son reproducible y que el error entre las medidas realizadas no es representativo, lo que permite definir al caucho de llanta como un material voluminoso y de baja densidad. 4.2 Influencia de la velocidad de calentamiento y tamaño de partícula dentro los ensayos de pirólisis. En la Fig. 1 se exhiben tres zonas de degradación entre 146-549 °C dentro de las cuales se considera que descomponen, los distintos componentes de forma independiente e irreversible. Estos resultados son análogos a los reportados por [16], que establece temperaturas iniciales de degradación de 120 a 152 °C y temperaturas finales de proceso entre 455 y 520 °C. Estos rangos de temperatura se obtuvieron al trabajar con velocidades de calentamiento entre 1 y 40 K/min. En la Fig. 2 se puede observar que la curva DTG del caucho en trozos se degrada a mayor temperatura, desplazándose hacia la derecha. Además los tres picos presentes en la curva DTG se presentan a mayores temperaturas, lo que constituye mayores tiempos de residencia a causa de la resistencia a la transferencia de calor al interior de la partícula. Referencia [2], asevera que el incremento en el tamaño de grano genera mayores gradiente de temperatura y por ende mayores tiempos de residencia para que el material volátil pueda ser gasificado. Por otra parte, las curva de la pérdida de peso del caucho en trozos en la zona de medias y altas temperatura muestra un ligero desplazamiento hacia la derecha del gráfico con Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 77 Modelo de Deconvolución para la Cuantificación de los Componentes del Caucho Vulcanizado Presente en los Neumáticos Fuera de Uso _________________________________________________________________________________________________________________________ Figura 1. Análisis TGA y DTG de trozos de caucho (dp=2-5 mm) con β=10 K/min Figura 2. Análisis comparativo de curvas TGA y DTG del polvo de caucho y trozos de caucho. respecto a la curva del caucho en polvo, lo que se traduce como un incremento en las temperaturas de degradación. Referencia [16], atribuye este fenómeno al detrimento en la velocidad de volatilización de los plastificantes. La influencia de la velocidad de calentamiento se puede observar en la Fig. 3, donde se obtiene una conversión total del material volátil a los 20 min de residencia para una velocidad de calentamiento de β=20 K/min, mientras que se con una β= 5 K/min se logra obtener la misma conversión recién a los 90 min trascurridos. De tal forma se puede aseverar que el incremento en la tasa de calentamiento acelera el proceso de degradación del caucho provocando una marcada disminución en los tiempos de residencia para la generación del residuo sólido carbonoso. 4.3 Determinación de los parámetros cinéticos de los principales componentes del caucho vulcanizado de NFU’s. La Fig. 4 ilustra gráficamente el procedimiento seguido para determinar los parámetros cinéticos de los componentes. Los valores de la pendiente de cada una de las linealizaciones corresponde a las energías de activación de cada componente Figura 3. Comparación de la conversión del NFU’s en trozos (dp=2-5mm) para distintas velocidades de calentamiento en ensayo de TGA con flujo de N2=50 mL/min Figura 4. Expresión gráfica de la estimación de los parámetros cinéticos de la pirólisis de NFU’s trozos para β=20 K/min (-Ea/R), mientras que los valores de los respectivos factores de frecuencia se obtienen de la intersección de las curvas con el eje de las abscisas. Los parámetros cinéticos obtenidos para el caucho en trozos proveniente de la banda lateral de los NFU’s se detalla en la Tabla 4. Como se puede observar el componente 1 posee una mayor energía de activación con respecto a los otros dos componentes. Es aquel que se degrada en la zona de altas temperaturas requiriendo de mayor energía para su degradación. Mientras que el tercer componente presenta el valor más bajo respecto a la energía de activación requiriendo de bajas temperatura de degradación correspondiente al componente más volátil. Tabla 4. Parámetros cinéticos de los componentes para la pirolisis del caucho para diferentes velocidades de calentamiento Componente 1 Componente 2 Componente 3 Velocidad de calentamiento Ea Ax10-9 Ea Ax10-9 Ea Ax10-4 5 (K/min) 135.7 1.17 117.4 0.46 45.6 0.31 10 (K/min) 137.7 2.11 124.2 2.79 48.6 1.1 20 (K/min) 132.9 1.17 130.0 11.20 48.9 2.3 Promedio 135.4 1.48 123.9 4.81 47.7 1.3 Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 78 Meza I.*; de la Torre E.* _______________________________________________________________________________________________________________________________ Ea: Energía de activación (kJ/mol) A: Coeficiente pre-exponencial (min-1) con valor de Ea reportados en la bibliografía para cauchos sintéticos como el BR (194 a 215 KJ/mol) y SBR (120 a 145 KJ/mol), por lo cual se podría suponer que el material correspondiente al tercer componente es el SBR [16]. Que plantea un valor promedio de 63.6 % respecto del caucho de la banda lateral y un 69.5 % para el caucho proveniente de la banda de rodadura. Tabla 5. Contenido inicial en peso de cada componente en el caucho evaluado para distintas velocidades de calentamiento Figura 5. Deconvolución de los diferentes componentes que conforman el caucho de los NFU’s (trozos dp=2-5 mm) para β=20 K/min Los estudios de termobalanza determinaron que el caucho de llantas está conformado por tres componentes mayoritarios que corresponden a los plastificantes y aditivos, al caucho natural y al caucho sintético. Además se determinó que el caucho sintético es el compuesto menos volátil, debido a que se degradó en la zona de altas temperaturas reportando una energía de activación de 135.4 KJ mol-1, seguido del caucho natural que se volatiliza en la zona media de temperaturas con una energía de activación de 123.8 KJ mol-1. Finalmente los plastificantes y aditivos que son los más volátiles mostrando la menor energía de activación de 47.7 KJ mol-1. En la Fig. 5 se puede observar el método de deconvolución empleado para la determinar la composición de los componentes mayoritarios del caucho de NFU’s. Se muestra en color rojo la curva correspondiente al componente más resistente a la degradación, que corresponde al caucho sintético. Por otra parte la curva de color amarillo hace referencia al comportamiento de una mezcla de caucho natural y sintético dentro de la zona de temperaturas medias. Finalmente, en color verde se define la curva referente a los aceites y plastificantes, los cuales presentan su degradación en la zona de bajas temperaturas. 4.4 Determinación de los componentes principales a través del modelo de deconvolución . A partir de esta modelización se realizó la cuantificación másica de los componentes mayoritarios presentes en el caucho de las llantas de desecho. En la Tabla 5 se encuentran tabulados los datos referentes con el peso inicial calculado para cada uno de los tres componentes de los NFU’s mediante la deconvolución. Se pudo determinar que el caucho sintético es el que se encuentra presente en mayor proporción con un contenido promedio de 60.8 % con respecto del peso total del caucho vulcanizado. Al observar la energía de activación reportada para el tercer componente de 135.4 kJ/mol, y al compararla Velocidad de calentamiento Componente 1 (%) Componente 2 (%) Componente 3 (%) 5 (K/min) 58.1 34.1 7.6 10 (K/min) 60.6 32.9 6.2 20 (K/min) 63.7 31.5 4.4 Promedio 60.8 32.8 6.1 La degradación que sufre el segundo componente se ha atribuido al comportamiento como una mezcla de NR y SBR. Sin embargo se estimó que el componente que se degrada mayoritariamente en esta zona es el caucho natural y que el aporte del caucho sintético es menor. Por ende el peso inicial calculado para el segundo componente se lo atribuye al NR obteniendo un valor promedio de 32.8 %. Al contrastar con los resultados obtenidos, se observa que el contenido de caucho natural presente en el caucho de la banda lateral es de apenas un 22.5 % y de 21.3 % para la banda de rodadura [16]. Estos resultados reflejan una vez más la complejidad y limitaciones presentes en la zona de temperaturas medias. Al comparar los resultados referentes al peso inicial del tercer componente, respecto a lo expuesto [16], se pudo establecer que el contenido de plastificantes, aditivos y aceites presentes en la banda de lateral, en ambos casos es inferior al 10 %. Planteando una estrecha concordancia entre ambos resultados. Referencia [16] considera que el componente 3, está conformado por los aceites de procesamiento de cada elastómero los cuales exhiben similares parámetros cinéticos. 4.5 Comparación de la curva DTG experimental respecto de la curva teórica obtenida a través del modelo matemático A través del desarrollo del modelo matemático para la determinación de los parámetros cinéticos y la deconvolución de los componentes mayoritarios de la llantas, se reconstruyó la curva de la derivada de la pérdida de peso (DTG), dicha curva teórica se comparó con la curva obtenido experimentalmente. Lo que permitió validar el ajuste del modelo con respecto a la experimentación. Se pudo observar que para bajas velocidades de reacción tanto la curva calculada como la experimental se ajustan en la mayoría de su trayectoria como se muestra en la Fig.6. Sin embargo, respecto del segundo y tercer pico los datos experimentales muestran cierta dispersión con relación a la curva calculada, lo cual se vuelve más evidente conforme se incrementa la velocidad de calentamiento. Dicho fenómeno puede ser atribuido a la influencia de la velocidad de calentamiento que genera una degradación del material más acelerada, produciendo un incremento en las magnitudes de los picos de Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Modelo de Deconvolución para la Cuantificación de los Componentes del Caucho Vulcanizado Presente en los Neumáticos Fuera de Uso _________________________________________________________________________________________________________________________ las curvas DTG. No obstante, se puede apreciar claramente que ambas curvas guardan una misma tendencia. [2] [3] [4] [5] [6] Figura 6. Comparación entre la curva DTG experimental y calculada para la pirólisis de NFU´s en trozos con β=20 K/min [7] 5. CONCLUSIONES [8] Se determinó que el caucho de NFU’s contiene 31.6 % de carbón fijo, 66.4 % de material volátil, 1.9 % de cenizas y 0.9 % de humedad. Demostrando que es un material con baja permeabilidad y que posee un gran contenido de material volátil. Se demostró que trabajar con tamaños de partícula superiores produciría mayores tiempos de residencia. Mientras que el incremento en la tasa de calentamiento acelera el proceso de degradación del caucho provocando una marcada disminución en los tiempos de residencia. Los resultados de los ensayos en TGA y DTG para tres distintas velocidades de calentamiento, exhibieron tres etapas de degradación para la pirólisis de caucho. Esto demostró que la primera etapa se presenta entre 146 y 329 °C con un pico máximo a 258 °C, la segunda etapa ocurre entre 318 y 439 °C con un pico máximo a 393 °C y la tercera etapa se encuentra entre 428 y 549 °C con un pico máximo a 458 °C. Los estudios de termobalanza determinaron que el caucho vulcanizado de los NFU’s está conformado por tres componentes mayoritarios. Además, se mostró que el componente más resistente a la degradación fue el caucho sintético, seguido del caucho natural y como componente más volátil los plastificantes y aditivos, manifestando energías de activación de 135.4; 123.9 y 47.7 KJ mol-1 respectivamente. [9] [10] [11] [12] [13] [14] [15] [16] [17] thermobalance. Chemical Engineering Journal, 126(6), 79-85, 2007. Available: doi:10.1016/j.cej.2006.08.031 A. Gómez, W. Klose, y S. Rincón. Pirolisis de Biomasa, Cuesco de palma de aceite, Kessel Univesity Press, ISBN 978-3-89958-457-8, 2008. Available: http://www.uni-kassel.de/upress/online/frei/978-389958-457-8.volltext.frei.pdf.(Junio, 2013) A. Quek, y R. Balasubramanian. Mathematical modeling of rubber tire pyrolysis. Journal of Analytical and Applied Pyrolysis, 95(1), 1-13, 2012. Available: http://dx.doi.org/10.1016/j.jaap.2012.01.012 B. Suárez, M. C. Ling-Ling, y S. Pérez. Proceso de 12.000 t/año de NFU’s en Gran Canaria. INGENIERIA QUIMICA-MADRID, (409), 162-175, 2004. Available: http://www.inese.es/html/files/pdf/amb /iq/409/11-ARTICULOEN.pdf, (Junio, 2013) D. W. Brazier y G. H. Nickel. Thermoanalytical methods in vulcanizate analysis II. Derivative thermogravimetric analysis. Rubber Chemistry and Technology, 48(4), 661-677, 1975. Available: doi: http://dx.doi.org/10.5254/1.3539668 E. Aylón, R. Murillo, A. Fernández-Colino, A. Aranda, T. García, M. S. Callén, y A. M. Mastral. Emissions from the combustion of gasphase products at tyre pyrolysis. Journal of analytical and applied pyrolysis, 79(1), 210-214, 2013. Available: doi:10.1016/j.jaap.2006.10.009 E. de la Torre, y A. Guevara. Estudio del proceso para la producción de carbón activado a partir de cuescos de palma africana. Revista Politécnica. 27(2), 27-37, 2007. E. L. Mui, D. C. Ko, y G. McKay, (2004). Production of active carbons from waste tyres––a review. Carbon, 42(14), 2789-2805. doi:10.1016/j.carbon.2004.06.023 F. Colina, I. Caballero y J. Costa. Diseño básico de hornos rotatorios para el tratamiento de minerales. INGENIERIA QUIMICA-MADRID, 34(392), 107-111, 2002. H. Teng, M. A. Serio, M.A. Wojtowicz, R. Bassilakis, y P. R. Solomon. Reprocessing of used tires into activated carbon and other products. Industrial & engineering chemistry research, 34(9), 31023111, 1995. Available: doi: 10.1021/ie00048a023 J. Yang, S. Kaliaguine, y C. Roy. Improved quantitative determination of elastomers in tire rubber by kinetic simulation of DTG curves. Rubber chemistry and technology, 66(2), 213-229, 1993. doi; http://dx.doi.org/10.5254/1.3538307 K. Unapumnuk, T. C. Keener, M. Lu, y S. J. Khang. Pyrolysis behavior of tire-derived fuels at different temperatures and heating rates. Journal of the Air & Waste Management Association, 56(5), 618-627, 2006. Available: doi:10.1080/10473289.2006.10464481 P. Ariyadejwanich, W. Tanthapanichakoon, K. Nakagawa, S. R. Mukai, y H. Tamon. Preparation and characterization of mesoporous activated carbon from waste tires. Carbon, 41(1), 157-164, 2003. Avalaible: doi: http://dx.doi.org/10.1016/S0008-6223(02)00267-1 P. Williams. Waste treatment and disposal. (2da. ed.). West Sussex, England: John Wiley & Sons Ltd, 2005. Rosa C. Miranda, Ciro C. Segovia, y César A. Sosa. Pirólisis de Llantas Usadas: Estudio Cinético e Influencia de Variables de Operación. Información tecnológica, 17(2), 7-14, 2006. Available: doi: http://dx.doi.org/10.4067/S0718-07642006000200003 S. Kim, J. K. Park, y H. D. Chun. Pyrolysis kinetics of scrap tire rubbers. I: Using DTG and TGA. Journal of environmental engineering, 121(7), 507-514, 1995. Available: doi: http://dx.doi.org/10.1061/(ASCE)0733-9372(1995)121:7(507) Z. Koreňová, J. Haydary, J. Annus, J. Markoš, y L. U. Jelemenský. Pore structure of pyrolyzed scrap tires. Chemical Papers, 62(1), 86-91, 2008. Available: doi: 10.2478/s11696-007-0083-7 A partir del modelo matemático se estableció que el caucho sintético es el que se presenta en mayor proporción con un porcentaje del 60.8 %, seguido por el caucho natural con un valor del 32.8 % y por último los plastificantes y aditivos que conforman un 6.1 %. REFERENCIAS [1] 79 A. Aranda, R. Murillo, García, M. Callén, y A. Mastral. Steam activation of tyrepyrolytic carbon black: Kinetic study in a Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 80 Reciclaje de Poliestireno Expandido por el Método de Disolución Precipitación _______________________________________________________________________________________________________________________________ Reciclaje de Poliestireno Expandido por el Método de Disolución Precipitación Saltos P.*; Chango I.*; Aldás M.*; Quiroz F.* * Escuela Politécnica Nacional, Centro de Investigaciones Aplicadas a Polímeros, Facultad de Ingeniería Química y Agroindustria, Quito, Ecuador [email protected]; {jose.chango; miguel.aldas; francisco.quiroz}@epn.edu.ec Resumen: En el presente proyecto se estudió el proceso de reciclaje de poliestireno expandido (EPS) por el método de disolución – precipitación, como alternativa para minimizar el impacto ambiental posconsumo. Se desarrolló el reciclaje de empaques de EPS de electrodomésticos variando las condiciones del método con el fin de recuperar poliestireno. Los residuos de EPS recolectados y fragmentados, se disolvieron en tetrahidrofurano (THF) y se precipitaron con etilenglicol (EG), bajo agitación mecánica continua. El polímero precipitado se separó por filtración y posterior secado, molienda y lavado. El producto así tratado se lo caracterizó estructuralmente por espectroscopía infrarroja, térmicamente por calorimetría diferencial de barrido y termogravimetría. Además, se determinó el índice de fluidez para estimar su capacidad de procesamiento. De los ensayos realizados se obtuvieron polímeros estructuralmente semejantes al poliestireno, con un grado de contaminación menor al 1.6 % de agente precipitante, presentan un índice de fluidez entre 15.56 y 23.60 g/10 min por lo que pueden ser reprocesados por inyección y extrusión, la temperatura de transición vítrea y el peso molecular son similares a los residuos de EPS. El ensayo con mejores resultados fue la disolución 30 % EPS y precipitación con una relación volumétrica 1/3 THF/EG, ofreciendo un polímero sin contaminación por el etilenglicol remanente. Palabras clave: poliestireno expandido; reciclaje; disolución – precipitación; poliestireno reciclado. Abstract: In this project the recycling process of expanded polystyrene (EPS) was studied by the method of dissolution - precipitation, as an alternative to minimize the environmental impact post-consumer. Thus, recycling of EPS packaging was developed varying the conditions of the method in order to recover polystyrene reprocessing. The EPS waste collected and fragmented were dissolved in tetrahydrofuran (THF) and precipitated with ethylene glycol (EG) under continuous mechanical stirring. The precipitate polymer was recovered with filtration, through drying, crushing and washing. The recovered polymers are structural characterized by the infrared spectroscopy, thermal by differential scanning calorimetry and thermogravimetry. Melt flow index was measured in order to estimate its processability. The tests performed indicate that the polymers obtained are structural identical to polystyrene with a lower degree of contamination to 1.6 % of precipitant, they have a melt index between 15.56 and 23.60 g/10 min so can be reprocessed by injection and extrusion, the glass transition temperature and molecular weight are similar to the waste EPS. The best performing test was 30 % EPS solution and precipitation with a volume ratio 1/3 THF/EG, offer a polymer free ethylene contamination. Keywords: expanded polystyrene; recycling; dissolution – precipitation; recycled polystyrene 1. INTRODUCCIÓN El aumento en el consumo de los plásticos implica la búsqueda de un tratamiento de reciclaje de sus desechos, que permita convertirlos de una amenaza ambiental a un recurso para la industria, solucionando este problema [8, 11]. Uno de los materiales ampliamente usado como empaque es el poliestireno expandido (EPS). Se trata de un material ligero y bajo costo. De modo que, el consumo global de EPS ya excede los 3 millones de toneladas con un incremento del 6 % al año [1, 14]. El peso ligero del EPS es una ventaja en el ámbito del empaque; pero este aspecto resulta ser una complicación en el proceso de reciclaje debido a la dificultad de transporte que implica este desecho voluminoso. Además, se debe considerar la necesidad de disponer materiales que puedan ser desechados con el menor impacto ambiental y no comprometan la salud humana [7]. Un camión de residuos con una capacidad de 70 m3 sólo podría transportar entre 700 - 1700 kg de EPS [13], lo que implica un elevado costo de transporte hacia los lugares de recuperación [4]. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 81 Saltos P.*; Chango I.*; Aldás M.*; Quiroz F.* ______________________________________________________________________________________________________________________________ Sin embargo, el EPS tiene gran potencial para ser reciclado debido a que existe en grandes cantidades y la separación de otros residuos es simple por su baja densidad y fácil identificación. Adicionalmente, la industria ha desarrollado equipos para la compactación y densificación del EPS, de modo que la ejecución de diferentes técnicas de reciclaje sea viable con el fin de solucionar el problema ambiental que representa. Es recomendable que su reprocesamiento se realice cerca de los sitios de recolección [2, 4]. En el proceso de reciclaje los desechos de EPS pierden su característica de espuma, una vez compactados se pueden fabricar juguetes, artículos de oficina, madera plástica, recipientes y hasta EPS nuevamente. Si los desechos son fraccionados pueden ser usados en la construcción, procesos de floculación como sustrato y la obtención de composites de fibras naturales, carbonatos y nanopartículas con buenas propiedades mecánicas [9, 11]. El EPS postconsumo puede ser reciclado mecánicamente, que consiste en la reducción de tamaño de sus desechos por métodos físicos de pulverización, compactación o densificación. Una vez que el EPS ha perdido su volumen puede ser implementado como materia prima para el moldeo de productos cotidianos hechos con poliestireno o producción de materiales de construcción. En la industria es considerado una técnica factible a gran escala, prácticamente limpia y de bajo costo [2, 13]. El reciclaje químico permite la depolimerización del monómero o una degradación del poliestireno en otros materiales secundarios, se realiza en un rango de temperatura ente 300 – 450 °C en presencia de una atmósfera de nitrógeno, clorofluorocarbono, propano o similares, con catalizadores óxidos ácidos o básicos [6, 9]. Existe además el reciclaje energético, visto como una alternativa al reciclaje mecánico, para aprovechar el alto poder calórico del EPS y así convertirlo en energía mediante la combustión de los desechos. Este proceso no es el más aconsejable por la generación de emisiones gaseosas y al requerimiento de reactores cónicos de lecho fluidizado [4, 8, 9]. Se han desarrollado otras técnicas como el reciclaje térmico y el método de disolución. Con esta última técnica se reduce el tamaño del EPS en más de 100 veces, al eliminar todo el aire que contiene su estructura sin degradación de las cadenas poliméricas, por lo que tiene una gran potencialidad [5]. 2. MATERIALES Y MÉTODOS Por medio de espectroscopía infrarroja por HATR (Horizontal Attenuated Total Reflection) se determinó la estructura y composición de los desechos de EPS. El peso molecular se estimó por viscosimetría bajo el procedimiento de la norma ASTM D2857 – 95 (2007) para soluciones de EPS en metil etil cetona. La temperatura de transición vítrea (Tg) se evaluó por medio de calorimetría diferencial de barrido (DSC) en atmósfera de nitrógeno y a 20 °C/min. Por análisis termogravimétrico (TGA) se determinó la descomposición térmica del EPS recolectado con una velocidad de calentamiento de 20 ºC/min hasta 800 °C en atmósfera de nitrógeno. 2.2 Desarrollo del proceso de reciclaje de EPS por disoluciónprecipitación Se utilizó como base la metodología descrita en la patente inventada por M. Notari y F. Rivetti [10], con la diferencia que se la realizó a temperatura ambiente y presión atmosférica. Las condiciones para el proceso fueron establecidas por medio de pruebas preliminares, que determinaron operaciones adicionales para mejorar el proceso. 2.2.1 Método de disolución – precipitación El EPS homogeneizado se disolvió en tetrahidrofurano (THF), variando el contenido de polímero en las proporciones de 10, 20, 30 y 40 % bajo agitación mecánica constante. En cada porcentaje de mezcla se recuperó el poliestireno disuelto por precipitación empleando como agente precipitante al etilenglicol (EG). El EG se adicionó lentamente y bajo agitación en las siguientes relaciones volumétricas: 1/1, 1/2 y 1/3 THF/EG. Al término de la adición se separó por filtración la solución THF – EG y el precipitado. Se secó durante 1 hora a 100 °C y se redujo de tamaño para deshacer los poros de la estructura del precipitado. Se lavó con isopropanol durante 1 hora y con agitación magnética para cada muestra con el fin de que el alcohol arrastre el EG remanente. Nuevamente, se secó a 100 °C durante 11 horas para evaporar residuos de la solución alcohol - EG contenidos en el polímero molido. 2.2.2 Caracterización del polímero recuperado Con la finalidad de comparar las características estructurales, nivel de contaminación y propiedades térmicas del poliestireno recuperado se lo caracterizó con las mismas técnicas descritas para el material de partida (EPS). 2.1 Caracterización del EPS recolectado Se recolectaron los desechos de EPS procedentes de empaques de electrodomésticos y fueron reducidos de tamaño en un molino de cuchillas. Una vez homogeneizado el material fragmentado, se lo caracteriza con el fin de establecer parámetros comparativos entre el material que va a ser reciclado y el producto de su reciclaje. Se establecieron ciertas diferencias en la aplicación de la viscosimetría, se determinó el índice de fluidez (MFI) para aproximar el peso molecular de los polímeros recolectados tomando como referencia el peso molecular del EPS recolectado. El MFI permitió validar las propiedades que tiene el polímero como materia prima para reprocesamiento. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 82 Reciclaje de Poliestireno Expandido por el Método de Disolución Precipitación _______________________________________________________________________________________________________________________________ Por medio de TGA no solo se obtuvo la temperatura de degradación de los productos recuperados, sino también la pérdida en peso que tiene la muestra en función del avance del calentamiento. 2.2.3 Recuperación del solvente, agente precipitante y reactivo de lavado La recuperación de los reactivos empleados en el proceso es relevante, no solo por la connotación ambiental, sino también por la reducción en el consumo de reactivos. Por lo tanto, se aprovecha la diferencia significativa entre los puntos de ebullición de los componentes de la solución THF – EG y de la solución alcohol – EG, para su separación por destilación fraccionada a 54 °C y 72 °C, respectivamente. Figura 1.Espectro del EPS recolectado 3.1.3 Termogravimetría (TGA) Al finalizar las operaciones se midió la cantidad de destilado y residuo recuperados en cada destilación, lo cual permitió determinar su rendimiento. Se evaluó la pureza química de los productos de la destilación mediante espectroscopía infrarroja por Transformadas de Fouriere (FTIR) y el índice de refracción. Por FTIR se realizó el análisis estructural del destilado y el residuo de cada destilación y a partir del índice de refracción se estimó la concentración de THF, EG e isopropanol en los reactivos recuperados. El termograma en la Fig. 2 indica que la descomposición del EPS recolectado inicia a 276,44 °C hasta que se registra la máxima pérdida de peso a los 430,02 °C, punto de la degradación térmica donde solamente queda materia inorgánica. El EPS recolectado presenta 0,1 % de materia inorgánica y de acuerdo con el termograma no existe una descomposición térmica adicional en el poliestireno. 3. RESULTADOS Y DISCUSIÓN 3.1 Caracterización de los residuos de poliestireno expandido recolectados 3.1.1 Espectroscopía infrarroja por Transformadas de Fourier (FTIR) En el espectro de la Fig. 1 se señalan los grupos funcionales presentes en la estructura de los residuos de EPS. Se observan las zonas de absorción entre 3100 – 3000 cm-1 y 1600 – 1450 cm-1 típicas del anillo bencénico y la región propia del poliestireno entre 760 - 700 cm-1 correspondiente al anillo monosustiuido con sus respectivos sobretonos en la región de 2000 a 1650 cm-1. Además, se evidencia absorción en las bandas a 2850 y 2919 cm-1 referente a los estiramientos de las moléculas CH2. La zona entre 1400 y 400 cm-1 corresponde a la huella dactilar de la molécula, patrón de frecuencias útil en la comparación con el espectro bibliográfico, que nos indica que los desechos de EPS recolectados presentan las bandas características de la macromolécula de poliestireno. Figura 2.Termograma correspondiente al análisis termogravimétrico del EPS recolectado 3.1.4 Calorimetría diferencial de barrido (DSC) Mediante calorimetría diferencial de barrido (DSC) se determinó que la temperatura de transición vítrea (Tg) del EPS recolectado es de 108 °C. Este valor está dentro del rango bibliográfico [10] que es de 107 °C ± 2 °C; cualquier aumento sobre los 100 °C que es el valor más conocido de Tg del poliestireno, puede ser consecuencia del agente expansor u otros aditivos que se usan en la fabricación del EPS. 3.2 Caracterización del polímero recuperado 3.2.1 Espectroscopía infrarroja por Transformadas de Fourier (FTIR) 3.1.2 Viscosimetría Se determinó que el peso molecular viscosimétrico del EPS recolectado es igual a Mw=107 510 g·mol-1, valor que se obtiene de la ecuación de Mark – Houwink. Se encontró que el incremento de la viscosidad de la solución metil etil cetona – EPS, por molécula de poliestireno es de 0,5879 mL/g [3]. En la Fig. 3 se presentan los espectros de los polímeros recuperados por los ensayos disolución - precipitación: 10 % EPS - 1/1 THF/EG, 10 % EPS – 1/2 THF/EG y 10 % EPS – 1/3 THF/EG. Esta figura es un ejemplo del análisis por FTIR que se hizo a cada producto. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 83 Saltos P.*; Chango I.*; Aldás M.*; Quiroz F.* ______________________________________________________________________________________________________________________________ La banda correspondiente al estiramiento entre los enlaces CH y el anillo aromático se localiza en alrededor de 3026 cm-1 para los diferentes espectros. Se evidencian los estiramientos de la molécula de CH2 cerca de 2918 y 2842 cm-1. La presencia del anillo bencénico se constata por los modos vibratorios existentes en 1601, 1492, y 1451 cm-1. Con respecto a las bandas representativas del polímero se ubican en 747 y 690 cm-1 para los productos recuperados. En los espectros de los polímeros obtenidos se aprecia la descompensación atmosférica provocada tanto por la absorción de CO2 cercana a 2330 cm-1 con una evolución positiva y por la absorción de agua a 3919 y 3462 cm-1. En general, todos los polímeros recuperados corresponden al poliestireno, debido a la presencia de las bandas de absorción características de la macromolécula. La comparación de cada grupo de espectros con el estándar bibliográfico del poliestireno avala este resultado. Por lo tanto, el método de reciclaje sugerido permite recuperar poliestireno a partir de EPS, sin registro de otro componente por análisis de FTIR. En consecuencia, también existe similitud entre los espectros de los polímeros recuperados y el espectro del EPS recolectado, debido a que ambos corresponden al poliestireno. Date: 08/04/2015 100.0 90 80 844.01 70 1744.13 3461.89 2329.96 60 906.16 1363.07 3919.06 1600.72 %T 50 2841.75 1026.90 3026.11 40 1491.83 2917.97 1450.94 30 20 747.37 10 690.44 3.2 4000.0 3600 3200 2800 2400 2000 1800 cm-1 1600 1400 1200 1000 800 630.0 AP0067.s p - 08/04/2015 - AP0067 POLYSTYRENE DC-OTI0341-2014 precipitado10%1.1 rep.006 - 30/09/2014 DC-OTI0341-2014 Precipitado10%1.2rep.006 - 21/10/2014 DC-OTI0341-2014 Precipitado10%1.3rep.006 - 21/10/2014 Figura 3. Espectros de los polímeros recuperados por los ensayos de disolución 10 % EPS y precipitación al 1/1, 1/2 y 1/3 THF/EG 3.2.2 Termogravimetría (TGA) remanente y las temperaturas de degradación térmica de los polímeros reciclados. En la Fig. 4 se presenta el termograma obtenido del producto más contaminado por precipitante remanente junto al termograma del poliestireno libre de EG. En la investigación, los polímeros que contenían EG remanente en su estructura presentaron un termograma similar al gráfico a) de la Fig. 4, donde se visualizan dos desviaciones con respecto a la línea base, la primera se identificó como la evaporación del EG remanente debido a su proximidad con la temperatura de ebullición (185 °C) del componente; y la segunda desviación representa la descomposición del poliestireno reciclado. Aquellos poliestirenos que se encontraron libres de EG remanente, presentan un termograma similar al gráfico b) de la Fig. 4, donde sólo se muestra la descomposición del poliestireno. Existen dos productos que entran en esta categoría, obtenidos por la disolución del 20 y 30 % de EPS en THF y la precipitación con una relación volumétrica de 1/3 THF/EG. Sin embargo, entre estos dos, se considera que las condiciones más adecuadas serán las que permitan reciclar mayor cantidad de desechos y convertirlos en poliestireno no contaminado, en este caso 30 % EPS – 1/3 THF/EG. Acerca de la degradación térmica de los diferentes poliestirenos se da en un rango de (232 – 261) °C hasta (475 – 483) °C. La cantidad de EG remanente en la estructura de cada polímero se relaciona con el inicio de la degradación del mismo, de modo que en cada grupo de disolución existe una tendencia descendente de la temperatura de descomposición cuando menor es el contenido de contaminación del poliestireno. Con respecto al punto final de la degradación térmica de los polímeros recuperados, no se aprecia cambios relevantes entre cada ensayo. En general, si se compara las temperaturas de inicio y fin de la degradación térmica de los poliestirenos reciclados con temperaturas de inicio (276,44 °C) y fin (430,02 °C) de la degradación del EPS recolectado, se deduce que existe un adelanto en el inicio y un retardo en el punto final de la degradación de los productos recuperados. Por tanto, la presencia de precipitante remanente afecta la descomposición térmica de los polímeros. Para conocer los datos obtenidos en cada análisis termogravimétrico se dispone de la Tabla 1, donde se incluye el porcentaje en peso identificado como la evaporación del EG Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 84 Reciclaje de Poliestireno Expandido por el Método de Disolución Precipitación _______________________________________________________________________________________________________________________________ La cantidad de EG presente en la estructura del polímero recuperado tiende a disminuir cuanto mayor es la cantidad de agente precipitante utilizado para precipitar el poliestireno en la disolución; es decir, que usar una porción mayor de EG favorece la recuperación del poliestireno disuelto. Tabla 1. Variación de las temperaturas de degradación de los poliestirenos recuperados en cada ensayo Ensayo EPS (%) Experimentalmente, sin importar la proporción de EPS utilizada en la disolución, el fenómeno se repite en cada ensayo del método disolución – precipitación; los polímeros recuperados con la relación 1/1 THF/EG, se presentan como un fluido viscoso lo que dificulta su separación de la solución THF – EG y por tanto tienden a estar más contaminados; además, la solución filtrada luce blanquecina, lo que se presume como poliestireno sin precipitar. Al contrario, los polímeros recuperados por las relaciones 1/2 y 1/3 THF/EG son fáciles de separar, lucen como una sola masa compacta y elástica, la solución THF – EG luce transparente, por tanto es posible que el poliestireno ha sido precipitado totalmente. 10 20 30 40 a) Contenido de etilenglicol (%) Inicio de la descomposición térmica (°C) Temperatura de máxima pérdida de peso (°C) 1/1 1.55 261.40 476.41 1/2 1.32 254.79 480.18 1/3 1.07 254.74 476.17 1/1 0.72 254.27 480.66 1/2 0.25 251.72 475.18 1/3 0.00 231.94 479.22 1/1 0.25 240.43 480.35 1/2 0.23 237.27 480.69 1/3 0.00 236.74 477.65 1/1 0.71 237.94 483.10 1/2 0.38 234.70 480.03 Relación THF/EG b) Figura 4.Termogramas correspondientes al análisis por termogravimetría de los poliestirenos recuperados por: a) disolución 10 % EPS y precipitación 1/1 THF/EG, b) disolución 30 % EPS y precipitación 1/3 THF/EG Es por ello que los poliestirenos reciclados con 1/2 y 1/3 THF/EG, tienen menor contenido de remanente en su estructura. De lo que se puede inferir que la cantidad necesaria para precipitar el poliestireno en disolución es tres veces el volumen del solvente usado, de modo que se obtenga una masa compacta fácil de separar y aparte una solución THF – EG sin residuos de polímero, lo cual permitirá una recuperación más limpia de solvente y precipitante. 3.2.3 Viscosimetría Los resultados de la medición del MFI para cada poliestireno reciclado se tabulan en la Tabla 2. Los datos se muestran atípicos dentro de cada grupo de ensayos; por lo que en general se infiere que los polímeros obtenidos en las diferentes pruebas de reciclaje tienen un MFI entre 15.56 y 23.60 (g/10 min), según las condiciones del proceso. El MFI se incrementa al usar una cantidad mayor de EG para precipitar al polímero, por lo que los poliestirenos recuperados con una relación 1/3 THF/EG tienen un MFI mayor que aquellos productos precipitados con un relación 1/1 THF/EG. Tabla 2. Variación del índice de fluidez de los poliestirenos recuperados en cada ensayo Ensayo Índice de fluidez EPS Relación (g/10 min) (%) THF/EG 1/1 18.5901 ± 0.22 10 1/2 16.9551 ± 0.13 1/3 19.1386 ± 1.24 1/1 18.7404 ± 0.96 20 1/2 18.1961 ± 1.19 1/3 18.9342 ± 1.20 1/1 15.5615 ± 1.22 30 1/2 18.6357 ± 2.63 1/3 23.6044 ± 1.36 1/1 26.1183 ± 0.75 40 1/2 19.2147 ± 1.62 Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 85 Saltos P.*; Chango I.*; Aldás M.*; Quiroz F.* _______________________________________________________________________________________________________________________________ De acuerdo con la Fig. 5, los valores de MFI entre los poliestirenos reciclados no son iguales, pero se puede apreciar que están aproximadamente entre 16 y 21 g/10 min. Esta desviación es consecuencia de que en la precipitación las cadenas que precipitan primero son las de mayor tamaño molecular y conforme se adiciona EG continúan precipitando el resto de cadenas con tamaños moleculares más pequeños. De modo que, no en todos los ensayos se habrá conseguido la misma distribución molecular de acuerdo a la cantidad de agente precipitante agregado. Los polímeros con una amplia distribución de tamaños tienden a fluir más [12, 15]. En consecuencia, si el contenido de glicol incide en la Tg de los poliestirenos recuperados, entonces la relación de THF/EG utilizada en la precipitación también. Si se usa una cantidad mayor de agente precipitante, la Tg del polímero es similar para los grupos de disolución 20, 30 y 40 % EPS ensayados. No se considera el grupo del 40 % de EPS para la Fig. 5, porque es la condición menos eficiente, debido a que la concentración del polímero en la disolución impide una mezcla apropiada con el agente precipitante. La pequeña diferencia entre las Tg de los poliestirenos, no solo se atribuye a la cantidad de EG en su estructura sino también a que no todos los productos tienen el mismo peso molecular. Sin embargo, al no existir cambios relevantes en la Tg se presume que se ha alcanzado un cierto tamaño molecular de 107 510 g·mol-1 y por lo tanto los polímeros recuperados tienen un peso molecular cercano a éste. La comparación del MFI de los poliestirenos recuperados con el MFI de los poliestirenos comerciales indica que el poliestireno obtenido del reciclaje de EPS por este método puede ser reprocesado por inyección o extrusión para la elaboración de distintos productos como producción de EPS o poliestireno extruido, paneles aislantes y artículos desechables. Los valores de la Tg de los poliestirenos se encuentran en el rango propuesto por investigaciones anteriores [10], que es de 107 °C ± 2 °C. De la misma forma no exceden la Tg del EPS recolectado a partir del cual fueron recuperados en más de 0,7 °C. Tabla 3. Temperatura de transición vítrea de los poliestirenos recuperados del reciclaje de EPS Ensayo EPS (%) 10 20 30 Figura 5. MFI de los poliestirenos reciclados en función de la cantidad de agente precipitante (1/1, 1/2 y 1/3 THF/EG) La relación directa de la viscosidad con el peso molecular e inversa con el MFI implica que, si los poliestirenos recuperados tienen índices de fluidez altos presentarán una viscosidad de flujo baja y en consecuencia un peso molecular bajo y viceversa. Por lo tanto, el peso molecular de los productos del reciclaje estará alrededor de 107 510 g·mol-1, que es el peso molecular determinado para el EPS recolectado [1]. 3.2.4 Calorimetría diferencial de barrido (DSC) Los resultados de los análisis calorimétricos se pueden apreciar en la Tabla 3. Por medio de los datos tabulados se infiere que, los productos recuperados por el reciclaje de EPS tienen una Tg entre 108,3 a 108,7 °C. Con excepción de los polímeros con mayor contenido de etilengicol en su estructura que se obtuvieron por las condiciones disolución 10 % EPS y precipitación 1/1 y 1/2 THF/EG, que tienen una Tg menor e igual a 107,8 °C. 40 Contenido de etilenglicol (%) Temperatura de transición vítrea (°C) 1/1 1.55 107.8 1/2 1.32 107.8 1/3 1.07 108.4 1/1 0.72 108.3 1/2 0.25 108.6 1/3 0.00 108.6 1/1 0.25 108.4 1/2 0.23 108.6 1/3 0.00 108.7 1/1 0.71 108.3 1/2 0.38 108.4 Relación THF/EG 3.3 Caracterización de los productos de destilación 3.3.1 Espectroscopía infrarroja por Transformadas de Fourier (FTIR) a. Infrarrojo del destilado de la solución filtrada del proceso de disolución – precipitación En la Fig. 6 se puede observar el espectro del destilado, de acuerdo con el análisis por espectroscopía infrarroja el destilado de la solución THF – EG es THF, debido a que presenta una estrecha similitud con el espectro bibliográfico del compuesto. Puede contener algunas trazas de EG pero su presencia no es notoria en el espectro. La banda correspondiente a 3490 cm-1 es una evidencia de presencia de grupos OH-; aunque la molécula no presente en su estructura el hidroxilo aparece en el espectro tanto del destilado como en el bibliográfico debido a que el THF es un compuesto altamente higroscópico. Las bandas a 2974 y 2862 cm-1 pertenecen al estiramiento asimétrico y simétrico de los Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 86 Reciclaje de Poliestireno Expandido por el Método de Disolución Precipitación _______________________________________________________________________________________________________________________________ enlaces C-H, la flexión que sufre el grupo (CH2)4 en el plano se observa en 1460 cm-1. El estiramiento del anillo aparece en 1646 y 1365 cm-1. Con respecto al estiramiento del enlace C-O-C característico del THF como compuesto heterocíclico, se observa en 1182 y 1063 cm-1 con intensidad más fuerte. Finalmente, en 906 y 661 cm-1 se encuentran las bandas originadas por la flexión que sufren los enlaces C-H. b. Infrarrojo del residuo de la solución filtrada del proceso de disolución – precipitación En la Fig. 7 se observa el espectro del residuo de la destilación de la solución THF – EG, el cual registra bandas características del EG en concordancia con los modos vibratorios de sus moléculas constitutivas. Con relación al espectro bibliográfico, el residuo corresponde al EG, dada su semejanza estructural dada por las bandas características de los grupos CH2 en 1456 cm-1 con un tijereteo y torsión en 1257 y 1205 cm -1; aleteo a 1315 cm-1 y balanceo en las bandas de 881 y 860 cm-1. puro, tal como se esperaba. El residuo de isopropanol encontrado demuestra que no se han separado completamente los componentes de la solución filtrada en la destilación, de modo que el residuo recuperado equivale aún a la solución alcohol isopropílico – EG. En la Fig. 9, se observa que el espectro del residuo presenta por un lado las bandas concernientes a los alcoholes secundarios como la flexión del O-H en 1408 y 679 cm-1 y los estiramientos del C-C-O en las bandas de 1341, 1306, 1161, y 1129 cm-1. La presencia de los grupos metilo aparece en 2969 y 1463 cm-1 como estiramiento asimétrico, en 1379 cm-1 como la vibración simétrica en forma de “umbrella” y en 951 cm-1 como el estiramiento entre los enlaces CH3-C-CH3. Por otro lado, existen bandas propias del EG en el espectro del residuo, así como la torsión de la molécula de CH2 en 1204 cm1-, el estiramiento del C-C en 1086 cm-1, en 1039 cm1 el estiramiento del C-O y el balanceo del CH2 en las bandas de 884 y 862 cm-1. Con respecto a los modos vibratorios de los hidroxilos en la molécula del glicol aparecen en 3306 y 1408 cm-1, de igual forma como lo hacen en el espectro del isopropanol. Se visualiza a 3296 cm-1 el estiramiento y a 1415 cm-1 se da lugar una banda ancha de la flexión de los grupos hidroxilo de la molécula de EG. Los enlaces C-H del grupo CH2 sufren un estiramiento a 2936 y 2876 cm-1 y en 1043 cm-1 los enlaces C-O se estiran. c. Infrarrojo del destilado de la solución filtrada del proceso de lavado El espectro del destilado de la solución alcohol – EG (Fig. 8) muestra las bandas constitutivas del propanol y en comparación con el espectro bibliográfico se confirma que el producto recuperado en la destilación de la solución filtrada del lavado es isopropanol. No se evidencia contaminación del producto. En dicho espectro se aprecia el estiramiento del grupo hidroxilo en 3327 cm-1. La absorción propia de los alcoholes secundarios se diferencia en 1408 cm-1 por la flexión en el plano del O-H y en 679 cm-1 por la flexión fuera del plano del O-H. Se suman también los modos vibratorios del C-C-O en las bandas de 1341 y 1307 cm-1, estiramiento asimétrico en 1160 y 1128 cm-1, y en la longitud de 817 cm-1 el estiramiento simétrico. Figura 6. Espectro del destilado de la solución filtrada del proceso disolución – precipitación Se observa, además, las tensiones asimétricas y simétricas de las moléculas de CH2 que se encuentran en 2932 y 2882 cm-1, respectivamente. Los grupos metilo sufren un estiramiento asimétrico con una intensidad muy fuerte en 2970 y 1467cm-1, vibración simétrica tipo “umbrella” a 1379 cm-1 y un estiramiento en 951 cm-1 entre los enlaces CH3-C-CH3. d. Infrarrojo del residuo de la solución filtrada del proceso lavado La espectroscopía infrarroja del residuo recuperado de la solución alcohol – EG (Fig. 9) no corresponde al etilenglicol Figura 7. Espectro del residuo de la solución filtrada del proceso disolución – precipitación Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 87 Saltos P.*; Chango I.*; Aldás M.*; Quiroz F.* _______________________________________________________________________________________________________________________________ n: índice de refracción c: concentración de la solución THF – EG (%v/v) De acuerdo con dicha expresión y según los valores del índice de refracción en la Tabla 4, el destilado contiene 97.80 % de THF y 2.20 % de EG que ha sido arrastrado en la fracción. De la misma manera, el residuo es 95,60 % EG y retiene 4.40 % de THF. Adicionalmente, se encontró que el rendimiento de la destilación con respecto a la cantidad de solvente recuperado con base al THF que entra a la destilación, es alrededor del 75 %, por lo tanto existen pérdidas debido a la volatilidad del compuesto. Con respecto a la cantidad de agente precipitante el rendimiento es cercano al 99 % con base en la cantidad de EG que tiene la solución THF – EG destilada. Figura 8. Espectro del destilado de la solución filtrada del proceso de lavado Date: 08/10/2014 100.0 90 80 70 b. Pureza de los productos de la destilación de la solución alcohol – EG El contenido de isopropanol y EG en el destilado y en el residuo, productos de la destilación de la solución filtrada del proceso de lavado se deduce de la relación lineal entre el índice de refracción y la concentración de isopropanol en soluciones de EG, dada por (2): 𝑛 = −0.049 𝑐 + 1.4272 Donde n: índice de refracción c: concentración de la solución alcohol – EG (%v/v) 60 %T 50 40 30 (2) 20 10 3.1 4000.0 3000 2000 1500 1000 500 cm-1 DC-OTI0341-2014 res iduo_EG2.rev.s p - 08/10/2014 F59310.sp - 08/10/2014 - F59310 ISOPROPYL ALCOHOL OR0492.s p - 08/10/2014 - OR0492 OR0492.DX 1,2-ETHANEDIOL Figura 9. Espectro del residuo de la solución filtrada del proceso de lavado comparado con sus componentes La banda en 1463 cm-1 que aparece en el espectro del residuo puede atribuirse a la deformación asimétrica del grupo metilo presente en el isopropanol o también al tijereteo del CH 2 presente en el EG; el hecho de que sea una banda ancha superpone al CH3 y al CH2. Este último grupo, se aprecia en el rango de 2935 y 2877 cm-1 correspondiente a las tensiones asimétricas y simétricas. 3.3.2 Índice de refracción a. Pureza de los productos de la destilación La composición de los productos de la destilación se determinó por medio de la relación lineal entre el índice de refracción y la concentración de THF en soluciones de EG, representada por (1): 𝑛 = −0,0182 𝑐 + 1,4263 Donde (1) Sobre la base de esta relación lineal y de los valores del índice de refracción de la Tabla 5 se tiene que el destilado es alcohol isopropílico puro y el residuo de la solución filtrada contiene aún un 28.60 % de isopropanol por destilar y 71.40 % es EG. De esta manera, se corrobora la información estructural proporcionada por el análisis de espectroscopía infrarroja. 295.0 Sumada a la pureza, se plantea el rendimiento de la destilación con respecto a la cantidad del reactivo de lavado recuperado en base al isopropanol que entra a la destilación, que es alrededor del 92 %. Con respecto al EG recuperado no se establece un rendimiento debido a la contaminación que tiene el residuo de la destilación con alcohol. Tabla 4. Índice de refracción de los productos obtenidos por destilación de la solución THF – EG y compuestos puros Índice de refracción Destilado 1.4085 Residuo 1.4255 Tetrahidrofurano 1.4070 Etilenglicol 1.4260 Tabla 5. Índice de refracción de los productos obtenidos por destilación de la solución alcohol – EG y compuestos puros Índice de refracción Destilado 1.3775 Residuo 1.4135 Isopropanol 1.3770 Etilenglicol 1.4260 Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 88 Reciclaje de Poliestireno Expandido por el Método de Disolución Precipitación _______________________________________________________________________________________________________________________________ 4. CONCLUSIONES El proceso de reciclaje de poliestireno expandido (EPS) por el método de disolución – precipitación expuesto en esta investigación tuvo resultados positivos, se consiguió polímeros estructuralmente iguales al poliestireno. Es decir, que la técnica usada permitió obtener un producto para reprocesamiento a partir de desechos de EPS. La caracterización de los desechos de EPS recolectados demostró que el material usado para el reciclaje tiene un peso molecular aproximadamente de 107 510 g·mol-1, una temperatura de transición vítrea de 108 °C y se descomponen térmicamente a 276.44 °C hasta alcanzar los 430.02 °C. [5] M. García, I. Gracia, G. Duque, A. De Lucas y F. Rodríguez, «Study of the solubility and stability of polystyrene wastes in a dissolution recycling process,» Journal Waste Management, 29(6),, vol. 29, nº 6, pp. 1814-1818, 2009. [6] A. Kan y R. Demirboğa, «A new technique of processing for wasteexpanded polystyrene foams as aggregates,» Journal of Materials Processing Technology, vol. 209, nº 6, p. 2994–3000, 2009. [7] F. Layedra, S. Galeas, V. Guerrero. (2015). Estudio de la Biodegradación de un Material Compuesto Obtenido con Ácido Poliláctico (PLA) Reforzado con Fibra Corta de Abacá (Musatextilis). Revista Politécnica. [Online]. 35(3), pp. 126-136. Available: http://www.revistapolitecnica.epn.edu.ec/revista_archivos/ revista_volumen_35/TOMO_3.pdf [8] D. López, P. Rhenals, M. Tangarife, K. Vega, L. Rendón, Y. Vélez y M. Ramírez, «Tratamiento de residuos de Poliestireno expandido utilizando solventes,» 2013. [En línea]. Available: http://kosmos.upb.edu.co/web/ uploads/articulos/(A)_Ingeniar_2013_Tratami. El desarrollo del proceso de reciclaje a diferentes condiciones se llevó a cabalidad, el método sugerido permite procesar hasta un 30 % de desechos de EPS, concentraciones mayores en la disolución no son recomendables. La relación volumétrica 1/3 THF/EG fue la más adecuada para conseguir un poliestireno reciclado sin contenido de etilenglicol remanente. [9] T. Maharana, Y. Negi y B. Mohanty, «Recycling of Polystyrene,» Journal Polymer – Plastics Technology and Engineering, vol. 46, nº 7, pp. 729-736, 2007. Los poliestirenos recuperados por el método de disolución – precipitación presentaron un porcentaje bajo de contaminación, el contenido de agente precipitante remanente no superó el 1.6 % y también se obtuvieron poliestirenos no contaminados, como es de interés. [12] J. Rieger, «The glass transition temperature of polystyrene,» Journal of Thermal Analysis, vol. 46, nº 3-4, pp. 965-972, 1996. Los poliestirenos reciclados tienen un peso molecular similar al tamaño molecular de los desechos de EPS recolectados, un índice de fluidez que varía entre 15.56 y 23.60 g/10 min por lo que pueden ser reprocesados por inyección o extrusión para la elaboración de distintos productos. [10] M. Notari y F. Rivetti, «Patente WO 2005023922 A1,» Diciembre 2005. [En línea]. Available: http://www.google.st/patents/W O2005023922A1?cl=. [11] M. Poletto, J. Dettenborn, M. Zeni y J. Zaterra, «Characterization of composites based on expanded polystyrene wastes and wood flour,» Journal Waste Management, vol. 31, nº 4, p. 779–784, 2010. [13] M. Samper, M. Rico, S. Ferrandiz y L. J., «Reducción y Caracterización del Residuo de Poliestireno Expandido,» de En I Simposio Iberoamericano de Ingeniería de Residuos, Alcoy, España, 2008. [14] A. B. T. E. Sekharan R., «Utilization of waste expanded polystyrene: Blends with silica - filled natural rubber,» Journal Materials and Design , vol. 40, pp. 221-228, 2012. [15] T. Instruments, « Understanding Rheology of Thermoplastic Polymers,» 2013. [En línea]. Available: http://www.tainstruments.com/ pdf/literature/AAN013_V_1_U_Thermoplast.pdf . El análisis por DSC determinó que la temperatura de transición vítrea de los poliestirenos reciclados fue entre 108,3 a 108,7 °C, a excepción de los polímeros con mayor contenido de contaminación donde el remanente actúa como plastificante. Todos se encuentran dentro del rango bibliográfico. El proceso de reciclaje se complementó con la recuperación del solvente, agente precipitante y reactivo de lavado; por medio de destilación se consiguió tetrahidrofurano con pureza del 97,80 %; etilenglicol al 95,60 % e isopropanol puro. Se recuperó aproximadamente el 75 % de solvente, 92 % del reactivo de lavado y alrededor del 99 % de agente precipitante. REFERENCIAS [1] D. Achilias, A. Giannoulis y G. Papagergiou, «Recycling of polymers from plastic packaging materials using the dissolution - reprecipitation technique,» Polymer Bulletin, vol. 63, nº 3, pp. 449-445, 2009. [2] S. Acierno, C. Carotenuto y M. Pecce, «Compressive and Thermal Properties of Recycled EPS Foams,» Polymer - Plastics Technology and Engineering, vol. 49, nº 1, pp. 13-19, 2009. [3] ASTM. D2857–95, Standard Practice for Dilute Solution Viscosity of Polymers, 2007. [4] A. Barnetson, Expanded Polystyrene: Development, Processing, Applications and Key Issues, S. Rapra, Ed., Shrewsburry: Handbook of Polymer Foams, 2004, pp. 37-54. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Desarrollo de un Material Compuesto de Matriz de Poliuretano Rígido Reforzado con Fibra de Raquis De Palma Africana __________________________________________________________________________________________________________________________ 89 Desarrollo de un Material Compuesto de Matriz de Poliuretano Rígido Reforzado con Fibra de Raquis De Palma Africana Proaño A.*; Bonilla O.**; Aldás M.* *Escuela Politécnica Nacional, Centro de Investigaciones Aplicadas a Polímeros, Facultad de Ingeniería Química y Agroindustria, Quito, Ecuador e-mail: {andres.proanof; miguel.aldas}@epn.edu.ec ** Escuela Politécnica Nacional, Centro Textil Politécnico, Facultad de Ingeniería Química y Agroindustria, Quito, Ecuador e-mail: [email protected] Resumen: En el presente artículo se expone el desarrollo de un material compuesto de matriz de poliuretano rígido con fibra de raquis de palma africana. Como materia prima del poliuretano se utilizó: difenilmetano diisocianato y poliol polioxipropilénico, fabricados por la empresa Huntsman. La fibra de raquis de palma africana fue recolectada de una planta extractora de aceite. La fibra fue molida, tamizada y secada. Para mejorar la afinidad matriz-fibra, las fibras fueron sometidas a un proceso de acetilación con anhídrido acético. Se elaboraron materiales compuestos con concentraciones de carga en peso de fibra de: 0 %, 5 %, 10 % y 15 %. A los materiales compuestos desarrollados se realizaron ensayos mecánicos de flexión y tracción. Los materiales compuestos con 15 % en peso de fibra mostraron propiedades mecánicas iguales o superiores a las del material de poliuretano. Respecto al material compuesto formulado con fibra acetilada, éste presentó resultados ligeramente mayores en los ensayos mecánicos en comparación con el material formulado con fibra sin tratamiento químico. Para los materiales compuestos con 15 % en peso de fibra, se determinó el costo de producción y se comparó con el costo de producción del material de poliuretano. Palabras clave: Material compuesto, matriz polimérica, poliuretano rígido, fibra natural, raquis de palma africana Abstract: This article explains the development of a composite made from rigid polyurethane and oil empty fruit bunch fibre with the objective of reducing production cost. As rigid polyurethane raw material, it was used: diphenylmethane diisocyanate and polyoxypropylene polyol, manufactured by Huntsman. The oil palm empty fruit bunch fiber was collected from an oil extract plant. The fiber was milled, sieved and dried. In order to improve the compatibility between the matrix and the fiber, the fibers were subjected to a process of acetylation using acetic anhydride. Composites were prepared with different fiber ratios: 0-wt%, 5-wt%, 10-wt% and 15-wt%. The mechanical properties of the composites were tested, by tensile and flexural tests. The composites with 15-wt% of fiber showed equal or superior mechanical properties than the rigid polyurethane material. The composite formulated with acetylated fiber showed slightly higher results in the mechanical tests than the material formulated with untreated fiber. The cost of production of the composites with 15-wt% of fiber were determined, this cost was compared with the cost of production of the polyurethane material. Keywords: Composite, polymer matrix, rigid polyurethane, natural fiber, oil palm empty fruit bunch fiber. 1. INTRODUCCIÓN Desde las pasadas décadas, las fibras naturales han atraído el interés de investigadores e industrias debido a sus ventajas específicas en comparación con las fibras sintéticas (tales como: resistencia al impacto, tensión, desgaste y fatiga). La creciente preocupación mundial sobre el medio ambiente está alentando el uso de materiales de fuente renovable que no dañan la naturaleza y provienen de una fuente alterna de buen potencial económico. Actualmente las fibras naturales están siendo utilizadas para el refuerzo de matrices termoplásticas y termoestables [6]. Hoy en día, uno de los problemas ambientales que existe es el poco uso que se les da a los residuos agroindustriales lignocelulósicos como el tamo de arroz, residuos de cosechas de cereales, bagazo de caña de azúcar, aserrín, raquis de palma africana, subproductos del desfibrado de plantas como la cabuya, abacá, entre otros. Todos estos residuos en su mayoría son incinerados y devueltos al campo como fertilizante. Estos materiales pueden ser una fuente alternativa de materia prima para crear bienes con valor agregado [8]. La fibra de raquis de palma africana es un residuo lignocelulósico que se obtiene como desecho de la extracción de aceite de palma africana. La fibra de raquis de Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 90 Proaño A.*; Bonilla O.**; Aldás M.* _______________________________________________________________________________________________________________________________ palma africana se la utiliza como abono y alimento para los animales, generación de energía, en la fabricación de pulpa y papel, como relleno en tableros de madera y como refuerzo de hormigón armado [9]. Para la elaboración del material compuesto se utilizó las fibras que pasaron la malla #60. Por último, las fibras fueron secadas en una estufa durante 4 horas a 105 °C. 2.2 Tratamiento químico a las fibras Por otro lado, los polímeros reforzados con fibra natural han venido emergiendo en los últimos años como una opción amigable con el medio ambiente y económica en comparación con fibras sintéticas como la aramida, fibra de vidrio o carbono. Las fibras lignocelulósicas poseen factores que favorecen su uso, como: propiedades aceptables de esfuerzo, bajo costo, baja densidad, no abrasivos, mayor recuperación de energía, y biodegradables. Además, poseen buena estabilidad térmica y proporcionan excelente aislamiento para el calor y el ruido [2]. Los poliuretanos son ahora utilizados en varias aplicaciones tecnológicas debido al bajo costo de procesamiento, la facilidad con la que se puede obtener material en grandes dimensiones y la variedad de reactivos que conducen a productos con diferentes propiedades físicas, químicas y mecánicas. Dependiendo de la materia prima y de la composición, el poliuretano se puede utilizar para la fabricación de una amplia gama de productos tales como adhesivos, recubrimientos, elastómeros y espumas rígidas y flexibles. La espuma rígida de poliuretano es usada en el campo del transporte, tecnología de refrigeración y electrodomésticos, industria de la construcción, industria automotriz, empaque, refuerzo de alfombras y artículos deportivos [7]. El uso del poliuretano en aplicaciones que requieren elevadas propiedades mecánicas es muy limitado, es por esto que materiales compuestos de poliuretano se desarrollan con el fin de obtener un material compuesto con mayores propiedades [12]. De esta manera en este trabajo, se busca conseguir el reemplazo de materiales importados con materiales locales naturales, lo cual es una vía para incrementar la competitividad de los productos ecuatorianos. Con el objetivo de aprovechar los recursos naturales disponibles en el Ecuador, y en busca de alternativas que sean económicamente viables para el refuerzo de matrices poliméricas, se desarrollaron materiales compuestos con matrices de poliuretano reforzado con fibras de raquis de palma africana. 2. MATERIALES Y MÉTODO 2.1 Preparación de la fibra Se empleó fibra de raquis de palma africana que fue recolectada de una planta extractora de aceite de la provincia de Los Ríos. La fibra de raquis de palma africana se obtiene como resultado del proceso de extracción del aceite de palma, la fibra fue obtenida en forma de racimos húmedos. Las impurezas de las fibras fueron limpiadas manualmente. Primero, las fibras fueron molidas en un molino de cuchillas. Luego, las fibras fueron tamizadas durante 30 minutos, utilizando un juego de tamices de mallas #20, #30, #50 y #60. Se realizó un tratamiento químico de acetilación a las fibras, con dos objetivos: el primero es aumentar la naturaleza hidrofóbica de la fibra y el segundo es hacer más lisa la superficie de la fibra mediante la eliminación de la cera, con la finalidad de mejorar la interacción entre la matriz polimérica y las fibras [1]. Se utilizó anhídrido acético con 97 % de pureza, como agente acetilante. Las fibras fueron colocadas en un reactor de vidrio de 1 000 ml de capacidad y se añadió anhídrido acético hasta que las fibras estén totalmente cubiertas. El reactor fue colocado en un baño de aceite a 110 °C con un reflujo condensador durante 120 minutos. Terminado el proceso de acetilación, el reactor fue retirado del baño de aceite y las fibras fueron extraídas del reactor. Las fibras fueron lavadas con agua destilada y luego secadas a 105 °C durante 8 horas 2.3 Preparación del material compuesto Los constituyentes del material compuesto fueron: matriz de poliuretano rígido y raquis de palma africana. Se desarrollaron dos tipos de materiales compuestos: el primero fue un material de matriz de poliuretano rígido reforzado con fibra de raquis de palma africana y el segundo fue un material de matriz de poliuretano rígido reforzado con fibra de raquis de palma africana químicamente tratada. Como materia prima del poliuretano rígido se utilizó: difenilmetano diisocianato y poliol polioxipropilénico, materiales fabricados por la empresa Huntsman. Se utilizaron las condiciones de trabajo dadas por la empresa fabricante, como: relación de mezclado, tiempo de desmolde y temperatura del molde. La preparación del material compuesto, se realizó mediante moldeo manual, donde se añade manualmente las materias primas en un molde y así el producto toma su respectiva forma. El molde debe tener un sellado óptimo para asegurar que no se formen rebabas. Antes de añadir los componentes, se aplicó desmoldante por todas las paredes internas del molde, en este proyecto se utilizó parafina líquida. Primero se pesaron el poliol e isocianato, en distintos recipientes. La fibra natural, con el respectivo porcentaje en peso, fue añadida en el recipiente con poliol y se batió la mezcla. Luego, se añadió el isocianato y con el uso de un agitador se mezcló para que la fibra se pueda distribuir por todo el contenedor. La mezcla fue vertida en el molde y se procedió a cerrarlo. Pasados 15 minutos, se abrió el molde y se sacó el material compuesto. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Desarrollo de un Material Compuesto de Matriz de Poliuretano Rígido Reforzado con Fibra de Raquis De Palma Africana __________________________________________________________________________________________________________________________ 2.4 Caracterización del material compuesto Para los ensayos mecánicos se utilizó la máquina de ensayos universales para propiedades mecánicas marca Instron modelo 1011. El ensayo de flexión se realizó según la norma ASTM D7264: Pruebas estándar para propiedades de flexión para materiales compuestos con matriz polimérica. Se utilizaron probetas rectangulares de: 200 mm x 12 mm x 10 mm [5]. El ensayo de tracción se realizó según la norma ASTM D3039: Pruebas estándar para propiedades de tracción para materiales compuestos con matriz polimérica. Se utilizaron probetas rectangulares de: 100 mm x 10 mm x 12 mm. [4]. Posteriormente a la determinación de las propiedades mecánicas, se seleccionó un material compuesto cuyo porcentaje en peso de fibra presente mayores o iguales resultados que el material de poliuretano en los parámetros de resistencia a la flexión y resistencia a la tracción. Al material seleccionado se realizó el análisis de densidad según la norma ASTM D1622: Densidad de plásticos rígidos [3]. Por último se realizó el análisis de costos de producción del material compuesto. 2.5 Análisis de costos de producción del material compuesto Se realizó un análisis de costos de producción del material compuesto con 15 % en peso de fibra. Se realizó dicho análisis al material compuesto con fibra sin tratamiento químico. Dado que para el tratamiento químico se utilizó anhídrido acético, el cual es un reactivo regulado por el CONSEP y tiene un costo en el mercado de $56 por litro, no se realizó el análisis de costos para el material compuesto con fibra químicamente tratada, ya que el presente proyecto busca disminuir los costos de producción. Tabla 1. Variables y niveles para el desarrollo del material compuesto Material Variable Niveles Compuesto 5% Porcentaje en peso Material 10 % de la fibra compuesto de 15 % poliuretano con Sin tratamiento químico Tratamiento a la fibra de raquis fibra Con tratamiento químico El análisis de costos de producción tiene como finalidad establecer el precio de producción de un lote de 1 000 kg de material compuesto. Para el análisis de costos se consideró: el precio de preparación de las fibras y, el precio del poliol e isocianato. Sumados estos dos costos se determinó el costo real para producir un lote de 1 000 kg de material compuesto de matriz de poliuretano rígido reforzado con fibras 3. DISCUSIÓN DE RESULTADOS Al momento de producir el material compuesto se tomó en cuenta que el material no tenga fallas superficiales como burbujas o huecos, que el material tenga la forma del molde utilizado y que no pierda su forma con el pasar del tiempo (estabilidad dimensional). Además se cuidó de no realizar la agitación a grandes velocidades y de no aplicar en exceso el desmoldante. 3.1 Caracterización del material compuesto 3.1.1 Ensayo de flexión En la Fig. 1 se muestran los resultados obtenidos para el parámetro de la deformación en el punto de fluencia para el material compuesto con fibra de raquis de palma africana. Se puede apreciar que no existe una tendencia lineal, con 5 % en peso la deformación aumenta para ambos casos, con 10 % en peso la deformación disminuye en relación con el porcentaje de carga anterior. Y para el material con 15 % en peso se obtuvo la mayor deformación cuando se formuló con fibra tratada, mientras cuando se formuló con fibra sin tratar la deformación es ligeramente menor al del material de puro. Para este parámetro el material compuesto formulado con fibra químicamente tratada presenta mayores resultados que el material compuesto con fibra sin tratar. En cada una de las concentraciones en peso del material compuesto con fibra acetilada, la deformación incrementó en todos sus valores con respecto al material hecho sólo de poliuretano. Esto probablemente se debe a que el tratamiento químico influye y mejora la afinidad entre la matriz de poliuretano y la fibra de raquis de palma africana ya que existe una mayor transferencia de carga de la matriz a la fibra [6]. Deformación Máxima (%) Las variables con las que se trabajó en el proyecto fueron: la concentración de carga en peso de la fibra en el material compuesto y el tratamiento químico a las fibras. Se trabajaron con tres valores de concentración de carga en peso de fibra: 5 %, 10 % y 15 %. Respecto al tratamiento químico a las fibras se tuvo: material compuesto con fibras sin tratamiento químico y material compuesto con fibras químicamente tratadas. En la Tabla 1 se detallan las variables y sus respectivos niveles para el desarrollo del material compuesto de matriz de poliuretano rígido. 91 6% 5% 4% 3% 2% 1% 0% 0 5 10 Concentración en Peso (%) Sin Acetilación 15 Con Acetilación Figura 1. Deformación en el punto de fluencia en el ensayo de flexión Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 92 Proaño A.*; Bonilla O.**; Aldás M.* _______________________________________________________________________________________________________________________________ Esfuerzo Máximo (MPa) El aumento en la resistencia a la flexión se debe a la absorción de esfuerzo por parte de la fibra. Con fibra químicamente tratada los valores de esfuerzo máximo son mayores ya que en los materiales con fibra acetilada existe mayor cantidad de enlaces uretanos. Al existir mayor cantidad de enlaces, los grupos OH de las fibras con los isocianatos forman una red cohesiva para que absorba y distribuya la carga de manera uniforme por todo el material [10]. 0,6 0,5 0,4 Se observa que mayores módulos se obtienen cuando se formula el material compuesto con raquis de palma africana sin acetilar. El módulo de flexión indica que tan rígido es un material, a medida que su módulo aumenta el material se vuelve más rígido. La tendencia muestra que el módulo aumenta cuando se incorpora la fibra, esto quiere decir que la rigidez de la fibra contribuye a la rigidez del material compuesto. Los materiales compuestos con fibra químicamente tratada poseen cadenas más largas lo que les proporciona mayor flexibilidad al material [10]. 3.1.2 Ensayo de tracción 10 8 6 4 2 0 0 5 0,3 0,2 Sin Acetilación 0,1 0,0 15 Con Acetilación Figura 4. Deformación al pico en el ensayo de tracción 5 10 15 Concentración en Peso (%) Sin Acetilación Con Acetilación Figura 2. Esfuerzo máximo en el ensayo de flexión 20 16 12 8 4 0 0 10 Concentración en Peso (%) 0 Módulo (MPa) acetilada, el módulo de flexión aumenta sólo cuando se formula con 10 % en peso de fibra ya que para las demás concentraciones el módulo la variación es mínima. Deformación Máxima (%) En la Fig. 2 se muestran los resultados para el esfuerzo máximo o resistencia a la flexión. La tendencia del esfuerzo es que aumenta cuando se formula el material con 5 % en peso de fibra. Se obtuvieron los mayores resultados cuando se utilizó 10 % en peso de fibra y, cuando se formuló con 15 % en peso de fibra los resultados del esfuerzo máximo son similares al del material puro. La tendencia es similar para el material compuesto formulado con fibra tratada químicamente así como formulado con fibra sin tratar, se observa que los resultados con fibras tratadas químicamente son ligeramente mayores que los presentados por los materiales compuestos formulados con fibras sin tratar. 5 10 15 En la Fig. 4 se observa la deformación al pico en el ensayo de tracción para el material compuesto. En este parámetro se observa que la tendencia es decreciente. Todos los materiales compuestos presentan una disminución en la deformación con respecto al material puro, excepto por el material formulado con 5 % en peso con fibra tratada. A medida que se aumenta el porcentaje de fibra la deformación al pico tiende a disminuir. Esto quiere decir que la fibra vuelve al material más rígido y pierde la elasticidad original de poliuretano. Además, se observa que material compuesto formulado con raquis de palma africana acetilada tiene mayor elasticidad y menor rigidez que el material compuesto formulado con raquis sin tratamiento químico. Esto se debe, a que el tratamiento químico proporciona mayor deformación al material ya que se forman cadenas más largas [11]. Concentración en peso (%) Sin Acetilación Con Acetilación Figura 3. Módulo de flexión En la Fig. 3 se muestran los resultados obtenidos para el parámetro de módulo de flexión del material compuesto. La tendencia para el material compuesto con fibra sin acetilar es el aumento del módulo de flexión cuando se añade 5 % y 10 % en peso de fibra, mientras que cuando se formula con 15 % en peso de fibra el material presenta un módulo similar al del material puro. Para el material compuesto con fibra En la Fig. 5 se muestran los resultados que se obtuvieron de esfuerzo máximo para los materiales compuestos con raquis de palma africana. La resistencia a la tracción presenta mayores resultados cuando se formula el material compuesto con fibra químicamente tratada, esto sucede para todas las distintas concentraciones en peso de fibra. El aumento del esfuerzo sucede porque los grupos OH de la lignocelulosa de la fibra, activados en la acetilación, actúan como poliol para la correspondiente reacción con el isocianato formándose más cadenas para absorber el esfuerzo [10]. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Desarrollo de un Material Compuesto de Matriz de Poliuretano Rígido Reforzado con Fibra de Raquis De Palma Africana __________________________________________________________________________________________________________________________ Existe un aumento en la resistencia la tracción cuando se formula al material con una carga en peso del 5 %, la resistencia a la tracción aumenta porque hubo una buena dispersión de la fibra a través de la matriz lo que ayudó a la transferencia y distribución uniforme del esfuerzo por todo el material. Los valores de esfuerzo máximo decrecen en relación con el material de poliuretano en consecuencia que el exceso de fibra ocasiona una pobre adhesión con la matriz lo que se ve reflejado en la disminución de las propiedades del material compuesto. Esfuerzo Máximo (MPa) 0,5 0,4 0,3 3.2 Selección del material compuesto Para el presente proyecto, con base en los ensayos mecánicos se seleccionó un material compuesto cuya concentración de carga en peso de fibra haya presentado mayores o iguales resultados que el poliuretano en los parámetros de resistencia a la tracción y resistencia a la flexión. De los ensayos mecánicos se observa que los materiales compuestos formulados con fibra químicamente tratada presentan propiedades de tracción y de flexión superiores debido a las reacciones adicionales producidas por los grupos OH de la fibra tratada con el isocianato. A pesar del aumento en los resultados, el proceso de acetilación no es justificable por el incremento de costos de producción que representa. Desde el punto de vista económico, no es conveniente realizarlo ya que el material compuesto formulado con fibra sin tratamiento químico presenta propiedades mecánicas similares. 0,2 0,1 0,0 0 5 10 15 Concentración en Peso (%) Sin Acetilación De los ensayos de flexión se seleccionó el material compuesto cuya concentración de carga en peso de fibra es del 15 % ya que los resultados de esfuerzo máximo se encuentran muy cercanos a los valores que presentó el material de poliuretano. A pesar de que los materiales compuestos con 10 % de concentración de carga de fibra presentaron los mayores resultados en el parámetro antes mencionado, éstos no fueron seleccionados ya que el objetivo del proyecto es incorporar la mayor cantidad de fibra sin que las propiedades del poliuretano sean disminuidas, por lo tanto se seleccionó al material compuesto con 15 % de fibra. Con Acetilación Figura 5. Esfuerzo máximo en el ensayo de tracción Módulo de Young (MPa) 93 15 12 9 6 3 0 0 5 10 15 Concentración en peso (%) Sin Acetilación Con Acetilación Figura 6. Módulo de tracción En la Fig. 6 se observan los resultados obtenidos del módulo de Young para el material compuesto. Se observa que en todas las concentraciones en peso de raquis de palma africana e independientemente del tratamiento químico a las fibras, el módulo de Young aumenta en comparación con el material puro de poliuretano. Esto se debe a la contribución de la rigidez propia de la fibra de raquis de palma africana. Se puede apreciar que el incremento del módulo de Young es mayor cuando se formula el material compuesto con fibra de raquis no acetilada ya que el tratamiento químico aumenta la movilidad de las fibras en la matriz, por lo que movilidad se traduce en mayor flexibilidad para el material compuesto [10]. El material compuesto cuya concentración en peso es de 15 % con fibra no acetilada presenta un aumento de 28.87 % mientras que el material compuesto cuya concentración en peso es de 15 % con raquis de palma africana acetilado presenta un aumento de 22.65 % en comparación con el material hecho sólo de poliuretano. Al igual que en el ensayo de flexión, para el ensayo de tracción se tomó en cuenta los resultados obtenidos para el parámetro de esfuerzo máximo. Los materiales compuestos presentaron la misma tendencia que en los ensayos de flexión, pero en este caso el material formulado con 5 % de fibra presentó los mayores resultados. Se seleccionó al material cuya concentración de carga en peso es del 15 % de fibra debido a que presenta resultados de esfuerzo máximo próximos a los resultados presentados por el material hecho sólo de poliuretano, se considera también que la adición de una mayor cantidad de fibra favorece a una disminución de costos de producción del material. 3.3 Ensayo de densidad La densidad del material rígido de poliuretano tiene un valor de 42.971 kg/m³ mientras que la hoja técnica del producto indica un valor de 40 kg/m³, por lo que se demuestra que el material obtenido en el presente proyecto posee una densidad muy próxima al valor bibliográfico. Los resultados indican que los materiales compuestos desarrollados presentan un aumento de densidad en comparación con el material puro. Esto se debe a que la fibra de raquis de palma africana es más densa que el material de poliuretano. La densidad de la fibra utilizada en el presente proyecto es de 1 100 kg/m³. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 94 Proaño A.*; Bonilla O.**; Aldás M.* _______________________________________________________________________________________________________________________________ En la Fig. 7 se comparan los resultados cuando se formula el material con fibra químicamente tratada con respecto al material formulado con fibra sin tratar, se observa que la diferencia entre estos materiales es mínima. Comparando las tablas se observa que para la producción del material compuesto de raquis de palma africana existe una disminución de costos del 16 %. 70 Densidad (Kg/m3) encuentran en fincas alejadas de las ciudades. El análisis de costos se realizó para la producción de un material compuesto con 15 % en peso de fibra. 60 50 4. CONCLUSIONES 40 30 20 10 0 Material de Poliuretano Material Compuesto Material Compuesto con Raquis con Raquis Acetilado Figura 7. Densidad de los materiales compuestos de poliuretano rígido reforzado con fibras naturales 3.4 Análisis de costos de producción del material compuesto Se realizó una comparación del análisis de costos de producción del material de poliuretano y del material compuesto con 15 % en peso de fibra. Se tomó como base una producción de 1 000 kg de material. En la Tabla 2 se enlistan los costos de la materia prima para una producción de 1 000 kg de poliuretano rígido. En la Tabla 3 se detallan los costos de materia prima para la producción de un lote de 1 000 kg de material compuesto de matriz de poliuretano rígido reforzado con fibra de raquis de palma africana. Tabla 2. Costos de materia prima para la producción de poliuretano rígido Materia Costo Cantidad Materia Costo de Materia Prima Unitario Prima por Lote Prima por Lote Poliol (kg) $ 4.50 500.0 $ 2 250.0 Isocianato (kg) $ 4.50 500.0 $ 2 250.0 Total $ 4 500.0 Tabla 3. Costos de materia prima para la producción del material compuesto de poliuretano rígido con fibra de raquis de palma africana Materia Costo Cantidad Materia Costo de Materia Prima Unitario Prima por Lote Prima por Lote Poliol (kg) $ 4.50 425.0 $ 1 912.50 Isocianato (kg) $ 4.50 425.0 $ 1 912.50 Fibra (kg) $ 0.0 150 $ 0.00 Energía $ 0.10 $ 17.05 $ 1.71 Eléctrica (kW) Transporte 150 $ 60.00 (kg) Total $ 3 886.71 La fibra de raquis de palma africana no tiene costo ya que se trata de un desecho. Además se enumeran los costos de preparación de la fibra de raquis de palma africana. Estos costos se refieren al consumo energético que se gasta para la molienda, tamizado y secado de 150 kg de fibra. La recolección de la fibra de raquis de palma africana implicó costos de traslado, dado que se la debió recolectar de las plantas extractoras de aceite de palma africana que se En los ensayos de flexión el material formulado con 10 % en peso de fibra presentó las mayores propiedades mecánicas. En los ensayos de tracción el material formulado con 5 % en peso de fibra presentó las mayores propiedades mecánicas. En la evaluación de las propiedades mecánicas, tanto de flexión y tracción, los resultados en el parámetro de esfuerzo máximo del material formulado con 15 % en peso son similares a los que presenta el material de poliuretano rígido. El material formulado con fibra acetilada presenta resultados ligeramente mayores a los presentados por el material formulado con fibra sin tratamiento químico en los ensayos de propiedades mecánicas. Se presentó una disminución de costos para la producción del material compuesto de matriz de poliuretano rígido reforzado con fibra de raquis de palma africana en comparación con el material hecho sólo de poliuretano. Existió una disminución de costos del 16 %. REFERENCIAS [1] H. Abdul Khalil y C. Hill. (2000). Effect of fiber treatments on mechanical properties of coir or oil palm fiber reinforced polyester composites. Journal of Applied Polymer Science, 78(9), 1685-1697. [2] M. Amin, K. Anuar, & K. Haji Badri, K. (2007). Palm based bio composites hybridized with kaolinite. Journal of Applied Polymer Science, 105(5), 2488-2496. [3] ASTM D 1622. (2014). Standard test method for apparent density of rigid cellular plastics. Estados Unidos. [4] ASTM D 3039. (2014). Standard test method for tensile properties of polymer matrix composite materials. Estados Unidos. [5] ASTM D 7264. (2007). Standard test method for flexural properties of polymer matrix composite materials. Estados Unidos. [6] G. Barra, M. Fredel, H. Al Qureshi, A. Taylor, y C. Clemenceau. (2006). Properties of chemically treated natural amorphous silica fibers as polyurethane reinforcement. Polymer composites, 27(5), 582590. [7] B. Gnauck, y P. Fründt. (1992). Iniciación a la química de los plásticos. (3ra Edición). Barcelona, España: Hanser, Barcelona. [8] A. Hassan, A. Salema, F. Ani, & A. Bakar. (2010). A review on oil palm empty fruit bunch fiber-reinforced polymer composite materials. Polymer Composites. doi:10.1002/pc.21006 [9] M. Mejía (2012). Elaboración de tableros aglomerados auto-adheridos a partir de fibra de raquis de palma africana. (Proyecto de titulación previo a la obtención de título de Ingeniero Químico publicado). Escuela Politécnica Nacional, Quito, Ecuador. [10] H. Rozman, K. Ahmadhilmi, & A. Abubakar. (2004). Polyurethane (PU) - oil palm empty fruit bunch (EFB) composites: the effect of EFBG reinforcement in mat form and isocyanate treatment on the mechanical properties. Polymer testing, 23(5), 559-565. [11] H. Rozman, K. Ahmad Hilme & A. Abubakar. (2007). Polyurethane composites based on oil palm empty fruit bunches: Effect of isocyanate/hydroxyl ratio and chemical modification of empty fruit Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Desarrollo de un Material Compuesto de Matriz de Poliuretano Rígido Reforzado con Fibra de Raquis De Palma Africana __________________________________________________________________________________________________________________________ bunches with toluene diisocyanate and hexamethylene diisocyanate on mechanical properties. Journal of applied polymer science, 106(4), 2290-2297. [12] H. SinghH. (2011). Calculation of Blowing Agents and the Flammability Characteristics of rigid Polyurethane Foam (RPUF). En 95 conferencia sobre los desafíos y las aplicaciones de las técnicas de modelos matemáticos en Ciencia de la Construcción y Tecnología. (pp. 511520). Roorkee, India: Central Building Research Institute Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 96 Synthesis and Characterization of Six Novel Samarium (III) Complexes with L-aspartic Acid, L-glutamic Acid, Glycine and O-phenanthroline, Bipiridile as Ligands ______________________________________________________________________________________________________________________ Synthesis and Characterization of Six Novel Samarium (III) Complexes with L-aspartic Acid, L-glutamic Acid, Glycine and O-phenanthroline, Bipiridile as Ligands Cardozo E.*; Contreras RR.*; Bellandi F.*; Lopez-Rivera A.**; Avendaño J.*; Araque C.*; Vielma J.* Universidad de Los Andes, Facultad de Ciencias, *Laboratorio de Organometálicos. **Laboratorio de Física Aplicada. Núcleo Pedro Rincón Gutiérrez, La Hechicera, Mérida -5101 - Venezuela e-mail: [email protected]; [email protected] _____________________________________________________________________________________________ Resumen: Se sintetizaron dos series diferentes de complejos de samario (III): Sm(o-Phen) (Ln)3 y Sm (bipy) (Ln)3 (n = 1-3; L1 = ácido l-aspártico, L2 = ácido l-glutámico, L3 = glicina) mediante métodos clásicos. Se utilizó una relación molar 3:1 de los ligandos Ln, de o-fenantrolina o de bipiridilo en cada caso. Los espectros UV-Vis muestran bandas agudas cerca de los 190 nm que corresponden al enlace Sm-O de alta energía así como transiciones f→f. Los espectros FT-IR confirman enlaces Sm-O en las señales situadas entre 490 - 450 cm-1. Los análisis de TGA-DTA, EA y MS revelan una buena correlación con la propuesta estructural. Los cálculos computacionales utilizando métodos semiempíricos facilitan algunas propiedades que sirven de guía para el desarrollo de aplicaciones fotoluminiscentes y antibacteriales. Palabras clave: Complejos de samario, tierras raras, aminoácidos, o-fenantrolina, bipiridilo, cálculo computacional. Abstract: Two different series of samarium (III) complexes, Sm (o-Phen) (Ln) 3 and Sm (bipy) (Ln) 3 (n = 1-3; L1 = L-aspartic acid, L2 = L-glutamic acid, L3 = glicine), were synthesized by classic methods. Ligands Ln and ophenanthroline or bipiridile were used on a molar ratio 3:1 for each compound. UV-Vis reveals sharp signals near 190 nm corresponding to a high energy bond Sm-O and f→f transitions. FT-IR spectra shows itself a band between 490 - 450 cm-1 indicative of Sm-O bond. Analytical analysis of TGA-DTA, EA and MS, resolve structural proposals. Computational calculation using semiempirical methods, show properties as a guide to approach better applications for these compounds like photoluminescence devices and antibacterial drugs. Keywords: Samarium complexes, rare earth, amino acid, o-phenanthroline, bipiridile, computational calculations. 1. INTRODUCTION The number of studies witch implies rare earth complexes have rapidly increased on last ten years because a wide capabilities of these metals as a photon receptors via antenna effects. High coordination numbers favor binding and catalyst functions. Ligands as o-phenantroline and bipiridile provide antenna effect [1-3] and L-aspartic, Lglutamic acids are natural proved chromophores [4-6]. In this paper, alternative synthesis method is used in order to reduce environmental impact caused by traditional ones used for this purposes. Characterization was carried out using UV-Vis, FT-IR, TGA-DTA, EA, MS and computational calculations using sparkle method [7, 8]. 2. MATERIALS AND METHODS 2.1. Materials All samarium complexes were prepared from samarium acetate monohydrate (SIGMA-ALDRICH). All reagents were analytical grade, and used without further purification. 2.2. Measurement of properties of samarium complexes UV/Vis spectra were recorded on a Shimadzu Mini 1240 UV/Vis Spectrophotometer on 1 x 10-4 M solutions. FT-IR spectra were measured with a PE Spectrum RX1 on KBr pellets 5 % w/w. MS data were obtained from a HP GCMS 5988A. TGA/DTA measurements were recorded on TA INSTRUMENTS SDT–Q600. EA were obtained from a Thermo Scientific Fison EA-1108 CHNS-O. Computational calculations were carried out using MOPAC2009 as interface for AM1, PM3 and PM6 semi-empirical methods. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Cardozo E.*; Contreras RR.*; Bellandi F.*; Lopez-Rivera A.**; Avendaño J.*; Araque C.*; Vielma J.* ____________________________________________________________________________________________________________________________ GABEDIT and AVOGADRO were also used for molecular modeling and visualization. earth ion. Besides, νas - νs, on complexes are lower than this difference on the free ligand showing that carboxylate have a bidentate chelating coordination. 2.3. Preparation of samarium complexes Accurately weighted each amino acid (L-aspartic, Lglutamic, glycine) with o-phenantroline on different reaction mixtures on 3:1 molar ratio for the Sm(ophen)(Ln)3 series. Same way for Sm(bipy)(L1)3 but weighting and mixing amino acids with bipiridile. A 70 % ethanolic solution (30 mL) was used as solvent heated to 74 °C (reflux temperature) [9, 10]. Samarium acetate was added to the mixture and stirred for 24 hours. Resulting solution was vacuum evaporated. Solid was washed three times with acetone (5 mL), dried and stored on dark glassware without light exposure. 3. RESULTS AND DISCUSSION 3.1. Uv/vis spectra Characteristic absorption band of –NH3+ appears both, complex and ligands around the 3000 cm-1 suggesting that the coordination do not proceed this way [12]. Table 1. Resume of most important FT-IR signals of ligands: L1 = aspartic acid; L2 = glutamic acid and L3 = glycine o-phen = o-phenantroline; bipy = bipyridyl. Ligands Vibration Type L1 L1 L1 o-phen bipy vNH3+ 3026 m 2966 m 2898 m cCOOH 1690 s 1700 s vasCOO1608 s 1644 s 1612 s vsCOO1420 s 1422 s 1412 s vC=C 1504 s vC=N 1420 s vC=C 1580 s vC=N 1454 s 3330 – 3320 – vO-H 3422 s,b 3428 s,b Table 2. Resume of most important FT-IR signals of samarium (III) complexes: Sm (o-phen)(Ln)3 series and Sm(bipy)(L1)3 series. The shape and intensity of the peaks on the complexes mainly depends of the ligands [11]. Characteristic absorption of o-phenantroline is 201, 227 and 267 nm; bipyridyl is 202, 233, 283 nm. The spectra show some blue shift indicating coordination of two nitrogens with the metal ion and a decrement of the conjugacy of benzene. Samariun (III) complexes Vibration Sm Sm Sm Sm Sm Sm Type (o-phen) (o-phen) (o-phen) (bipy) (bipy) (bipy) (L1)3 (L2)3 (L3)3 (L1)3 (L2)3 (L3)3 vNH3+ 3018 m 3078 m 2915 m 3006 m 3077 m 2975 m 1692 s 1704 s - 1697 s 1700 s - cCOOH vasCOO - vsCOO- 1592 s 1618 s 1596 s 1614 s 1588 s 1618 s 1416m,b 1417 m,b 1434 m 1488 m - 1438 m - vC=C 1502 m 1509 m 1518 m - - vC=N 1416m,b 1417 m,b 1416 m - - - vC=C - - - 1572 w - 1536 m 1452 s vC=N vO-H vSm-O - - - 1412 m 1412 m 3337 – 3338 – 3116 – 3328 – 3340 – 3354 – 3390 s, b 3388 m, b 3378 s, b 3390 s,b 3400 s,b 3413 s,b 480 w 500 w 470 w 480 w 472 w 482 w 3.3. Mass spectrometry Figure 1. UV/Vis spectra of two series of samarium (II) complexes: (1) Sm(o-phen)(Ln)3 series and (2) Sm(bipy)(L1)3 series. L1 = aspartic acid; L2 = glutamic acid and L3 = glycine; o-phen = o-phenantroline; bipy = bipyridyl. 3.2. FT-IR spectra Table 1 and 2 shows the IR spectra for which ternary compound. Two bands between 1600 - 1700 cm-1 and 1450 - 1550 cm-1 can be attributed to symmetric and asymmetric vibrations of COO- indicating coordination with the rare Taking into account preliminary structures of complexes and obtained mass spectra, there is a good compliant with the initial structural proposals due to a molecular ion (P +) observed and fragments due to loss of ligands or part of them. This can lead to propose a fragmentation pattern for each structure. Peaks observed on spectra will vary in a range between ±1 uma and ±6 uma due to action of different isotope 13 distributions on ligands C (1.1122 %), 144 147 ( Sm (11.6540 %); Sm (56.7670 %); 148 Sm (42.4810 %); 149Sm (52.2560 %); 150Sm (27.8200 %); 152 Sm (100 %); 154Sm (84.9620 %)). Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 97 Synthesis and Characterization of Six Novel Samarium (III) Complexes with L-aspartic Acid, L-glutamic Acid, Glycine and O-phenanthroline, Bipiridile as Ligands ______________________________________________________________________________________________________________________ Table 2 and 3 resumes fragment losses and Fig. 2 show a fragmentation pattern. Table 3. Main peaks (uma) observed for complexes Sm (o-phen)(Ln)3. L1 = aspartic acid; L2 = glutamic acid and L3 = glycine; o-phen = ophenantroline. Sm (o-phen)(L1)3 Sm (o-phen)(L2)3 Sm (o-phen)(L3)3 + + P 907.05 P 949.85 P+ 732.90 P+-(Asp) 774.60 P+-2(Glu) 802.30 P+-2(Gly) 656.85 P+-2(Asp) + P -3(Asp) P+-3(OAc) P+-(o-phen) 641.35 508.20 730.20 727.96 P+-3( Glu) + P -3( Glu) P+-3(OAc) P+-(o-phen) 654.95 P+-2(Gly) 582.20 508.15 772.20 769.95 P+-3(Gly) P+-3(OAc) P+-(o-phen) 507.50 555.10 552.55 Table 4. Main peaks (uma) observed for complexes Sm(bipy)(Ln)3. L1 = aspartic acid; L2 = glutamic acid and L3 = glycine; bipy = bipyridyl. Sm (bipy)(L1)3 Sm (bipy)(L2)3 Sm (bipy)(L2)3 + P 882.95 P+ 925.25 P+ 708.55 + + + P -(Asp) 749.20 P -2(Glu) 778.50 P -2(Gly) 633.20 P+-2(Asp) 616.20 P+-3( Glu) 632.85 P+-2(Gly) 556.90 P+-3(Asp) P+-3(OAc) P+-(bipy) 483.65 705.40 725.90 P+-3( Glu) P+-3(OAc) P+-(bipy) 485.15 748.10 769.45 P+-3(Gly) P+-3(OAc) P+-(bipy) 482.85 530.60 551.95 Figure. 2. Fragmentation pattern for Sm (o-phen)(L1)3 complex. (L1 = aspartic acid) 3.4. TGA-DTA analysis Table 5. Loss of mass on the compounds and water of hidratation. L1 = aspartic acid; L2 = glutamic acid and L3 = glycine; o-phen = ophenantroline; bipy = bipyridyl. %W Complex %W T (°C) Fragment remaining 8.573 123.42 4 H2O Sm(o-phen) 38.654 (L1)3 52.773 234.35 Organic 8.735 109.28 5 H2O Sm(o-phen) 31.179 (L2)3 60.086 185.58 Organic 2.096 60.59 1 H2O Sm(o-phen) 40.014 (L3)3 57.890 201.27 Organic 9.143 125.87 5 H2O Sm (bipy) 41.447 (L1)3 49.410 230.01 Organic 11.020 105.20 6 H2O Sm (bipy) 32.261 (L2)3 56.719 188.29 Organic 10.660 85.94 4 H2O Sm (bipy) 36.029 (L3)3 53.311 156.26 Organic 3.5 Elemental analysis Carbon, hydrogen and nitrogen analysis exhibit a good agreement with initial stoichiometry and all other analysis specially with TGA-DTA in which it is possible to establish exactly the same quantity of hydration water on each molecule: Sm (o-phen) (L1)3 .4H2O (Calc. SmC30H38N5O18: %C, 36.801 %H, 4.706; %N, 7.15): Found. %C, 36.931; %H, 4.966; %N = 7.879. Sm (o-phen) (L2)3.5H2O (Calc. SmC33H44N5O18: %C, 41.723; %H = 5.195; %N, 6.744 %): Found. %C, 41.911; %H, 5.425; %N, 6.948. Sm (o-phen) (L3)3.H2O (Calc. SmC24H32N5O12: %C, 35.780; %H, 4.973; %N, 8.705): Found. %C, 36.005; %H, 5.333; %N, 9.139. Sm (bipy) (L1)3.5H2O (Calc. SmC28H38N5O18: %C, 34.530; %H, 4.945; %N, 7.190): Found. %C, 34.775; H = 5.246 %; N = 7.503. Sm (bipy) (L2)3.6H2O (Calc. SmC31H44N5O18: %C, 36.005; %H, 5.424; %N, 6.777): Found. %C, 36.402; %H, 5.686; %N, 6.913. 3.6. Computational calculations. The weightlessness of these compounds exhibited a good agreement with the stoichiometry. o-Phenantroline losses it crystal water under the 100 oC. There are two main steps when complexes being heated. First one correspond to loss of cristal water on the complexes accompanied by a endothermic effect. The second refers to a decomposition of the anhydrous complexes to oxides accompanied by an exothermic effect with one, (or) two peaks. The Table 6 shows the methods used for all calculations, and results are showed on Table 7. Most relevant result shown on this Table 7 is the COSMO volume, this can give a clear idea for select the most affordable complex to have further use as antibacterial agent. This selection represents a molecule with large surface that can be surrounded by many molecules of solvent like water. Sm (o-phen)(L2)3 and Sm(bipy)(L2)3 has higher volumes. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 98 Cardozo E.*; Contreras RR.*; Bellandi F.*; Lopez-Rivera A.**; Avendaño J.*; Araque C.*; Vielma J.* ____________________________________________________________________________________________________________________________ Table 6. Methods used for calculations of samarium (III) complexes. L1 = aspartic acid; L2 = glutamic acid and L3 = glycine; o-phen = ophenantroline; bipy = bipyridyl. Complex/method AM1 PM3 PM6 Sm(o-phen) (L1)3 + + Sm(o-phen) (L2)3 + + Sm(o-phen) (L3)3 + + + Sm (bipy) (L1)3 + + Sm (bipy) (L2)3 + + Sm (bipy) (L3)3 + Table 7. Results of computational calculations for samarium (III) complexes. COSMO* = Solvation model: COnductorlikeScreeningMOdel. L1 = aspartic acid; L2 = glutamic acid and L3 = glycine; o-phen = o-phenantroline; bipy = bipyridyl. Complex Method ∆Hf (kJ) E1 (eV) COSMO* area (A2) Sm(ophen)(L1)3 Sm(ophen)(L2)3 PM3 PM6 AM1 PM6 AM1 PM3 PM6 AM1 PM3 AM1 PM6 -2184.43 -2215.99 -2434.41 -2237.91 -1206.09 -1064.27 -978.29 -2655.46 -2430.72 -2460.02 -2234.28 7.68 9.00 7.59 8.64 8.70 6.81 6.75 9.19 9.30 6.77 7.38 530.08 497.01 581.04 548.52 382.43 403.75 400.32 517.26 536.89 556.72 532.41 PM6 -1017.92 8.55 371.74 Sm(ophen)(L3)3 Sm (bipy) (L1)3 Sm (bipy) (L2)3 Sm (bipy) (L3)3 COSMO* volumen (A3) 700.47 686.08 757.83 749.51 495.99 507.07 499.00 681.94 686.08 727.14 728.77 468.48 Analyzing the HOMO-LUMO surface for each complex (see Fig. 3), it is clearly that only one complex possibly have a good quantum yield because its HOMO and LUMO surface distribution around the molecule. Both surfaces are over the chromosphore ligand on Sm (bipy) (L3)3. Table 8. Results for catalytic trials. Conditions: substrate catalyst ratio 100/1, 150 °C, 1000 psi and 24 h, a biphasic medium aqueous/ciclohexane. Percent (%) Complex cistrans1-Hexene Hexane 2-hexene 2-hexene Sm (o-phen)(L1)3 70.73 15.23 8.66 5.38 Sm (o-phen)(L2)3 92.62 3.77 2.49 1.05 Sm (o-phen)(L3)3 83.40 8.69 5.02 2.89 Sm (bipy)(L1)3 96.78 2.50 0.53 0.20 Sm (bipy)(L2)3 69.05 15.62 9.17 6.15 Sm (bipy)(L3)3 95.75 2.68 1.06 0.52 4. CONCLUSION All six complex were successfully synthetized with a classic method that represent a low cost and low environmental impact synthesis. UV, IR, MS, EA and TGA-DTA technics reveals good accuracy with initial stoichiometry planned showing an effective bonding between O and N donors and RE(III) metal ion. Two complex possibly have better behavior as antibacterial agents: Sm (Glu)3Phen, Sm (Glu)3Bipy. Sm(Gly)3Bipy could have some photoluminiscent activity because a close distance between HOMO-LUMO surface on the chromosphore ligand. AKNOWLEDGEMENT To CDCHTA and Universidad de Los Andes, for the knowledge. REFERENCES Figura 3. (a) HOMO surface and (b) LUMO surface for Sm (bipy) (L3)3 complexes. 3.7. Catalytic trials Proved catalytic activity of samarium (III) salts and complexes [13 – 15] leads to make some trials as follow. All six complexes were tested with 1-hexene as substrate in presence of hydrogen for hydrogenation reactions. The conditions for all trials were: substrate catalyst ratio 100/1, 150 °C, 1000 psi and 24 h, a biphasic medium aqueous/ciclohexane was used. All complexes shows hydrogenation and isomerization capabilities. Results are shown on the Table 8. [1]. Liu, L. Xu, X. Lou, Z. et al. Luminiscent properties of a novel terbium complex Tb(o-BBA)3(Phen). Journal of Rare Earths. 2006, 21(1): 253 – 256. [2]. R. Kumar, U. Singh. Molecular structure, photophysical and thermal properties of samarium (III) complexes. Journal of Molecular Structure. 2008. 872: 427 – 434. [3]. L. Armelao, S. Quici, F. Barigelletti, et al. Design of luminiscent lanthanide complexes: From molecules to highly efficient photoemitting materials. Coordination Chemistry Reviews. 2010. 254: 487 – 505. [4]. P. Gawryszewska, J. Lisowski. Lanthanide (III) complexes of N4O4 Schiff base macrocycle: Luminescense and formation of heterodinuclear d-f complexes. Inorganica Chimica Acta. 2012. 383: 220 – 229. [5]. Z. Ahmed, K. Iftikhar. Synthesis, luminescence and NMR studies of lanthanide (III) complexes with hexafluoroacetylacetone and phenanthroline. Part II. Inorganica Chimica Acta. 2012. 392: 165 – 176. [6]. Z. Piskula, J. Czajka, K. Staninski, et al. Luminescense properties of calcium tungstate activated by lanthanide (III) ions. Journal of Rare Earths. 2014. 32(3): 221 – 225. [7]. K. Monahan, B. Kumari, G. Rijulal. “Microwave assited synthesis, spectroscopic, thermal and antifungal studies of some lanthanide (III) complexes with a heterocyclic bishydrazone”. Journal of Rare Earths. 2008. 26:16 – 21. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 99 Synthesis and Characterization of Six Novel Samarium (III) Complexes with L-aspartic Acid, L-glutamic Acid, Glycine and O-phenanthroline, Bipiridile as Ligands ______________________________________________________________________________________________________________________ [8]. Kremer, C. Torres, J. Dominguez, S. et al. Structure and thermodynamic stability of lanthanide complexes with aminoacids. Coordination Chemistry Reviews. 2005, 249(1): 567 – 590. [9]. Freire, R. da Costa, N. Rocha, G. et al. Sparkle/AM1 Parameters for the modeling of samarium (III) and promethium (III) complexes. Journal of Chemical Theory and Computation. 2006, 2(1): 64 – 74. [10]. Freire, R. da Costa, N. Rocha, G. et al. Sparkle/PM3 Parameters for the modeling of samarium (III) and promethium (III) complexes. Journal of Chemical Theory and Computation. 2007, 3(1): 1588 – 1596. [11]. Hui, Y. Qizhuang, H. Jing, Y. et al. Syntheses, characterization and antibacterial properties of rare earths (Ce3+, Pr3+, Nd3+, Sm3+, Er3+) complexes with L-aspartic acid and ophenanthroline. Journal of Rare Earths. 2006, 24(1): 4. [12]. Y. Sun, M. Machala, F. Castellano. “Controlled microwave synthesis of RuII synthons and chromosphores relevant to solar energy conversion”. Inorganica Chimica Acta. 2010. 363; 283 – 287. [13]. Qizhuang, H. Jing, Y. Hui, M. et al. Studies on the spectra and antibacterial properties of rare earth dinuclear complexes with L-phenylalanine and o-phenanthroline. Materials Letters. 2006, 60(1): 317 – 320. [14]. Woznicka, E. Kopacz, M. Umbreit, M. et al. New complexes of La(III), Ce(III), Pr(III), Nd(III), Sm(III), Eu(III) and Gd(III) ions with morin. Journal of Inorganic Biochemistry. 2007, 101(1): 774 – 782. [15]. Yan, Z. Tang, Y. Liu, W. et al. Syntheses, characterization and luminiscent properties of lanthanide complex with an unsymmetrical tripodal ligand. Journal of Luminescence. 2008, 128(1): 1394 – 1398. [16]. Silverstein, R. Bassler, G. Morrill, T. Spectrometric Identification of Organic Compounds. 5th Edition. New York: John Wiley & Sons Inc., 1991. [17]. D. Evans, A. Hoveyda. Samarium-Catalyzed Intramolecular Tischenko Reduction of β-Hydroxy Ketones. A stereoselective Approach to the Synthesis of Differentiated Anti 1,3-Diol Monoesters. Journal of American Chemical Society. 1990. 121: 6447 – 6449. [18]. D. Evans, S. Nelson, M. Gagné et al. A chiral Samarium-Based Catalyst for the Asymmetric Meerwein-Ponndorf-Verley Reduction. Journal of American Chemical Society. 1993. 115: 9800 – 9801. [19]. D. Chen, J. Zou, W. Xiang. Synthesis, crystal structure and catalytic property of a samarium complexes with [Hpytza = 5(3-pyridyl) tetrazole-2-acetic acid]. Inorganic Chemistry Communications. 2014. 40: 35 – 38. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 100 Implementación de Medidas de Prevención y Control de Ruido para los Trabajadores del Centro de Generación de Energía de la Empresa Dipor S.A. _________________________________________________________________________________________________________________________ Implementación de Medidas de Prevención y Control de Ruido para los Trabajadores del Centro de Generación de Energía de la Empresa Dipor S.A. Villacis W. *; Andrade C.* * Escuela Politécnica Nacional, Facultad de Ingeniería Química y Agroindustria Quito, Ecuador e-mail: [email protected]; [email protected] Resumen: El presente trabajo tuvo por objeto el diseño e implementación de medidas preventivas y de control de ruido para el Centro de Generación de Energía de la empresa Dipor S.A., con el fin de salvaguardar la salud auditiva de los trabajadores. Se identificaron fuentes de ruido y actividades capaces de causar daño a la audición o molestias para realizar actividades intelectuales. Con este antecedente se evaluaron los niveles de ruido mediante un sonómetro calibrado y bajo la norma UNE-EN ISO 9612:2009. Los resultados de la medición demostraron que los niveles de ruido del generador y oficinas, sobrepasan el límite permisible de 85 dB(A) y 70 dB(A) respectivamente; límites establecidos por el Decreto Ejecutivo 2393. Para minimizar el riesgo de pérdida auditiva, se dictaminó el uso obligatorio de protectores auditivos mediante señales de seguridad; así como un plan de rotación del personal para disminuir el tiempo de exposición a ruido. En las oficinas se modeló un acondicionamiento acústico para bajar el nivel de ruido y evitar que las actividades de carácter intelectual se vean interferidas por el ruido excesivo en el ambiente. Se ejecutó un plan de capacitación para dar a conocer los efectos del ruido en la salud y las medidas de control implementadas por la empresa. Para verificar el estado de salud auditiva se realizaron audiometrías a los trabajadores antes y después de la implementación del proyecto; esto con el fin de constatar si a lo largo del tiempo sufrieron alteraciones. Adicional, se implementó un plan de mantenimiento preventivo de los elementos mecánicos susceptibles de causar ruido dentro del generador. Palabras clave: Evaluación de ruido, medidas preventivas, medidas correctivas, riesgos físicos, nivel de potencia sonora. Abstract: The objective of this work was the design and implementation of preventive measures and noise control for the Energy Generation Center of the company Dipor S.A., to safeguard the hearing health of workers. It began with the identification of noise sources and activities that can cause hearing damage or inconvenience for intellectual activities, to further evaluate the noise levels using a calibrated sound level meter and under the UNE-EN ISO 9612: 2009. The measurement results showed that the noise of the generator and offices exceed the permissible limit of 85 dB(A) and 70 dB(A), respectively; limits established by Executive Order 2393. To minimize the risk of hearing loss, the use of hearing protectors was established by safety signals; and a plan to reduce the noise exposure time of workers. An acoustic conditioning was modeled inside the office to reduce the noise level and prevent the intellectual activities being interfered by excessive noise in the environment. A training plan was implemented to show the effects of noise on health and control measures implemented by the company. Audiograms were performed to workers before and after implementation of the project to check the status of hearing health and determine whether over time suffered hearing impairment. Additional, a plan of preventive maintenance of mechanical elements capable of causing noise inside the generator was implemented. Keywords: Noise assessment, preventive measures, corrective measures, physical hazard, sound power level. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 101 102 Villacis W.*; Andrade C.* _______________________________________________________________________________________________________________________________ 1. INTRODUCCIÓN Entre los factores de riesgo a los que un trabajador está expuesto, en áreas industriales, el ruido es uno de los más comunes. Sus efectos en el trabajador van “desde un deterioro temporal de la audición con recuperación parcial o total al cesar la exposición, hasta la pérdida permanente e irreversible de la audición” [1]. Existen también otros efectos fisiológicos como afectaciones en los aparatos circulatorio, muscular, digestivo, respiratorio; y efectos a nivel psicológico, como disminución de la concentración, la efectividad, la productividad, interferencias en las conversaciones y aumento en la frecuencia de accidentes [1]. En el Ecuador se han expedido normativas como el Reglamento de Seguridad y Salud de los Trabajadores y Mejoramiento del Medio Ambiente de Trabajo 2393, que regula los límites máximos de exposición a los que un trabajador puede estar sometido durante su jornada laboral. En este contexto legal, es obligación de las empresas, controlar regularmente los niveles de ruido a los que están expuestos los trabajadores e implementar medidas de prevención. DIPOR S.A. preocupada por la salud y seguridad de sus trabajadores busca, preservar la salud auditiva y capacidad de concentración de quienes laboran en el Centro de Generación de Energía, a través de la implementación de medidas de prevención y control de ruido en sus puestos de trabajo, que cumplan con esta normativa legal de salud y seguridad. 2. PARTE EXPERIMENTAL Para facilitar la implementación de medidas correctivas y preventivas con el fin de controlar los niveles de ruido existentes dentro del centro de Generación de Energía de la Empresa Dipor S.A. y minimizar al máximo el riesgo de pérdida de la audición de los trabajadores, se comenzó con la identificación de fuentes de ruido y puestos de trabajo. El objetivo de esta etapa previa fue conseguir la mayor información posible sobre todas las condiciones de trabajo. El análisis de esta información ayudó a decidir si es necesario o no la medición de los niveles de ruido en todos los puestos de trabajo [2]. trabajo. Este método se basa en hacer mediciones en los puestos de trabajo críticos, los mismos que, según la identificación de fuentes de ruido y puestos de trabajo previa arrojó que era necesaria una medición y evaluación de ruido [2]. Figura 1. Esquema conceptual sobre la identificación de fuentes de ruido y puestos de trabajo El esquema adoptado para el inicio de este trabajo se lo describe en la Fig. 1, donde se indican las fases de la identificación de fuentes de ruido y puestos de trabajo [3]. Una vez que se obtuvieron los datos de las mediciones realizadas tanto en el horario matutino como vespertino, fue necesario garantizar que la diferencia de las medias poblacionales sea insignificante para cada una de las mediciones; esto se prueba a través de la diferencia de medias para lo que se aplicó la “Prueba t de Student” [4]. El resultado debió estar dentro del rango de aceptación de hipótesis nula de varianzas iguales. Los resultados obtenidos fueron comparados con los valores mínimos indicados en el Capítulo V, artículo 55 referente a Ruidos y Vibraciones del Reglamento de Seguridad y Salud de los Trabajadores y Mejoramiento del Medio Ambiente de Trabajo 2393 [1, 5]. En la Fig. 2 se presenta un esquema general sobre la medición y evaluación de ruido. Se resalta la toma de datos sobre las fuentes generadoras de ruido, características inherentes del puesto de trabajo, características de los trabajadores y la ubicación física de los diferentes puestos. Una vez que se obtuvo toda la información acerca de la identificación de fuentes de ruido y puestos de trabajo, se definió una estrategia de medición para que con un número mínimo de mediciones garantice la representatividad de las condiciones de trabajo del puesto a evaluar [2]. Para realizar la medición de ruido laboral, se utilizó el procedimiento específico DP.PEE.MAS.5.4.04, cumpliendo la norma UNE-EN ISO 9612:2009 título Acústica, que refiere sobre la determinación de la exposición al ruido en el Figura 2. Esquema conceptual sobre la medición de ruido Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Implementación de Medidas de Prevención y Control de Ruido para los Trabajadores del Centro de Generación de Energía de la Empresa Dipor S.A. __________________________________________________________________________________________________________________________ Después de la evaluación de los niveles de ruido en los distintos puestos de trabajo, se diseñaron e implementaron medidas de prevención y control de ruido con el fin de garantizar que los trabajadores no tengan daños en su salud a causa de este riesgo físico. Adicional, el diseño permite que los trabajadores ejecuten tareas que requieran cierto grado de concentración sin que se vean afectadas por causa del nivel de ruido elevado que existe en el ambiente [6]. Dentro de las medidas determinadas e implementadas se destacan controles de ingeniería, administrativo y equipos de protección personal: 1. Diseño de un acondicionamiento acústico en la oficina ubicada en el segundo piso. 2. Plan de mantenimiento preventivo del generador. 3. Plan de rotación de turnos del personal que trabaja en los puestos de electricista, mecánico y supervisión del tablero de control y que por sus actividades tienen que permanecer en el área del generador por un lapso de tiempo. 4. Plan de capacitación sobre los efectos del ruido en la salud para los trabajadores del Centro de Generación de Energía y trabajadores que visitan periódicamente el lugar. 5. Protectores auditivos acordes al tipo de ruido existente dentro del área donde se encuentra el generador. 6. Señalización de obligatoriedad de utilizar equipos de protección personal ubicados en lugares estratégicos a lo largo del Centro de Generación de Energía. 7. Realización de audiometrías a los trabajadores con prolongados tiempo de exposición. 3. RESULTADOS Y DISCUSIÓN Una vez obtenidos los valores de nivel de presión sonora en los puestos de trabajo críticos, éstos se analizaron y compararon con valores de referencia que sirvieron para poder implementar medidas de prevención y control de ruido y así prevenir una posible pérdida de la audición de los trabajadores expuestos así como el normal desenvolvimiento de las tareas en ambientes donde se requiere un grado de concentración. 103 Tabla 1. Análisis de los puestos de trabajo Puesto de Trabajo Núm Personal Expuesto Tiempo Exposición Conclusión 7.5 h Supervisión de tablero de control 2 Electricista 2 Medir y Evaluar 0.5 h 7.5 h Medir y Evaluar 0.5 h 7.5 h Mecánico 2 Medir y Evaluar 0.5 h Cuarto transformadores 1 0.12 h por persona No evaluar Bodega de repuestos 1 1 h al mes No Evaluar Área generador 6 0.5 h por persona Medir y Evaluar 3.2 Medición de ruido en los puestos de trabajo de Centro de Generación de Energía de la empresa DIPOR S.A. Las tomas se basaron en el procedimiento DP.PEE.MAS.5.4.04, cumpliendo la norma UNE-EN ISO 9612:2009; la mismas que se basa en hacer mediciones en los puestos de trabajo críticos; previo a la medición se debió hacer una verificación en terreno del instrumento ya que condiciones ambientales como temperatura, velocidad del aire y humedad relativa, pueden afectar parcialmente la respuesta del instrumento. Además, fue indispensable la calibración mediante el pistófono en áreas donde no exista ruido ya que este interfiere en la calibración [7]. Una vez que se comprobó y calibró el sonómetro se realizaron las mediciones; las mismas que se efectuaron en 2 horarios, matutino y vespertino, con el fin de garantizar que se representen los valores de nivel de presión sonora tanto para el turno de la mañana como para el turno de la tarde y noche. El sonómetro, específicamente el micrófono, fue ubicado en una posición orientada hacia la fuente, a la altura del oído del trabajador cuando se encuentra sentado (1.35 m), además se verificó que no se entorpezcan las tareas realizadas por el trabajador. En el caso del generador de energía, se colocó el sonómetro a una distancia de 1 m de la fuente y a una altura de 1.55 m. Cabe recalcar que el tiempo de medición fue de 10 minutos por puesto de trabajo. A continuación la Tabla 2 muestra los resultados de las mediciones de ruido realizadas en los diferentes puestos de trabajo dentro del Centro de Generación de Energía. 3.1. Identificación de fuentes de ruido y puestos de trabajo En esta etapa, se identificaron las distintas fuentes de ruido existentes en los lugares de trabajo, se caracterizó el tipo de ruido proveniente de las máquinas y se analizaron los puestos de trabajo. El análisis de esta información ayudó a decidir si era necesaria o no la medición de los niveles de ruido de cada área. Estos puntos relevantes se pueden apreciar en la Tabla 1. Tabla 2. Valor promedio nivel de presión sonora equivalente en dB(A) Ubicación Nivel de Presión Sonora Prueba Límite dB(A) “T” permisible dB(A) Leq Lmax Lmin Sup. tablero 73.6 75.4 52.9 0.05 70.0 de control Electricista 74.4 76.1 55.3 0.05 70.0 Mecánico 75.0 76.2 54.1 0.02 70.0 Cuarto 106.8 107.3 99.4 0.01 85.0 Generador Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 104 Villacis W.*; Andrade C.* _______________________________________________________________________________________________________________________________ Para poder representar en un plano los niveles medidos, la Fig. 3 detalla mediante un mapa de ruido los valores Leq de cada puesto de trabajo. materiales propuestos en el diseño del acondicionamiento acústico. SUPERVISOR TABLERO CONTROL 73,8 BODEGA REPUESTOS MECÁNICO 75,0 ELECTRICISTA 74,4 PARED HORMIGÓN CUARTO TRANSFORMADOR 106,9 GENERADOR PARED Figura 3. Mapa de ruido distributivo del Centro de Generación de Energía 3.3 Determinación de medidas preventivas y correctivas para el control de ruido Figura 5. Cortina 0.475 kg/m2 fruncida al 50 % dispuesta para las ventanas Se tiene como base que la prioridad de actuación para el control de ruido es en la fuente, luego en el medio de transmisión y por último en el receptor; bajo este concepto se diseñaron e implementaron una serie de medidas de prevención y control de ruido. 3.3.1 Acondicionamiento acústico En la oficina ubicada en el segundo piso fue indispensable modelar matemáticamente un diseño y acondicionamiento acústico. Esto se lo hizo con el fin de aumentar el coeficiente de absorción de la sala, de tal manera que se disminuya el tiempo de reverberación de la sala (T60) y así aumentar el valor de la reducción del nivel sonoro (R). Figura 6. Sonex de 35 mm dispuesto como material absorbente entre el techo y el cielorraso La Fig. 4, Fig. 5, Fig. 6 y Fig. 7 muestran las fotografías de los diferentes materiales elegidos para el diseño y acondicionamiento acústico referentes a las posibles vías de transmisión del ruido proveniente del generador. Figura 7. Panel cielorraso de gypsum dispuesto para el techo Figura 4. Alfombra de lana de 1.2 kg/m2 dispuesta para el piso Una vez que se seleccionaron los materiales que se van a utilizar en el diseño acústico, fue importante calcular el aporte en la reducción de ruido en la oficina. En la Fig. 8, Fig. 9 y Fig. 10 se comparan de manera general los coeficientes de absorción por banda de frecuencias de cada uno de los materiales existentes en la oficina versus los Figura 8. Comparación de coeficientes de absorción de los materiales existentes y propuestos en el diseño acústico para el piso de la oficina. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Implementación de Medidas de Prevención y Control de Ruido para los Trabajadores del Centro de Generación de Energía de la Empresa Dipor S.A. __________________________________________________________________________________________________________________________ 105 Tabla 3. Reducción del nivel sonoro de los puestos de trabajo ubicados en la oficina del segundo piso después del diseño acústico REDUCCIÓN DEL NIVEL SONORO Nivel de Leq sin Leq con reducción A.C. A.C. sonoro por el dB(A) dB(A) A.C. dB(A) Límite máx permisible dB(A) Cumple Norma Supervisor tablero control 73.8 15.4 66.5 70.0 Si Mecánico 75.0 15.4 68.0 70.0 Si Electricista 74.4 15.4 67.4 70.0 Si Puesto trabajo Figura 9. Comparación de coeficientes de absorción de los materiales existentes y propuestos en el diseño acústico para el techo de la oficina. de 3.3.2 Plan de mantenimiento preventivo del generador El plan de mantenimiento preventivo del generador debe asegurar el correcto funcionamiento de la máquina con el fin de evitar problemas eléctricos y/o mecánicos que aumenten el nivel de ruido emitido. La empresa Dipor S.A. por medio del Departamento de Seguridad y Salud Ocupacional implementó un formato para el registro de mantenimiento del generador, en el mismo consta el control y verificación del mantenimiento de los elementos susceptibles de provocar ruido; así como el responsable del seguimiento. Las imágenes del mantenimiento se muestran en la Fig. 12. Figura 10. Comparación de coeficientes de absorción de los materiales existentes y propuestos en el diseño acústico para las ventanas de la oficina. De igual manera, se representaron gráficamente los niveles de ruido mediante un Mapa de ruido Distributivo, el mismo que denota los niveles de ruido después del acondicionamiento acústico propuesto. En la Fig. 11 se puede apreciar el mapa con los diferentes valores. SUPERVISOR TABLERO CONTROL 73,8 --> 66,5 BODEGA REPUESTOS MECÁNICO 75 --> 68 ELECTRICISTA 74,4 --> 67,4 PARED HORMIGÓN CUARTO TRANSFORMADOR Figura 12. Personal en mantenimiento del generador La manera de controlar si el mantenimiento es efectivo fue mediante el formato de control, el mismo que fue diseñado para verificar que los elementos sensibles de provocar ruido dentro del generador han recibido un correcto mantenimiento. La Fig. 13 muestra el porcentaje de cumplimiento de los trabajos efectuados a los elementos sensibles de provocar ruido antes y después de la implementación del plan. 106,9 GENERADOR PARED Figura 11. Reducción de los niveles de ruido en los puestos de trabajo acondicionados acústicamente. La Tabla 3 detalla los resultados obtenidos donde se puede apreciar que la atenuación del nivel de ruido del diseño del acondicionamiento acústico llegó hasta valores inferiores a 70 dB(A) con lo que se cumple con el límite máximo permisible dispuesto en el Reglamento de Seguridad y Salud de los Trabajadores y Mejoramiento del Medio Ambiente de Trabajo 2393. Figura 13. Porcentaje de cumplimiento del mantenimiento a los elementos sensibles de provocar ruido del generador antes y después de la implementación del plan. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 106 Villacis W.*; Andrade C.* _______________________________________________________________________________________________________________________________ 3.3.3 Plan de rotación del personal El objetivo del plan de rotación de personal en el Centro de Generación de Energía fue no sobrepasar los valores dictaminados en la norma que dispone 30 min como el tiempo máximo permitido de exposición para un nivel de 106.9 dB(A). Los trabajadores del Centro de Generación de Energía deben ejecutar actividades cotidianas mandatorias en el área del generador por un corto lapso de tiempo, razón por la cual se diseñó un plan de rotación mensual con el fin de disminuir el tiempo de exposición a ruido de cada trabajador a máximo 30 min. La Fig. 14 muestra la comparación entre las horas trabajadas antes y después de la implementación del plan de rotación por cada uno de los colaboradores. Figura 14. Comparación de las horas trabajadas dentro del área del generador para cada uno de los empleados del Centro de Generación de Energía. 3.3.4 Plan de capacitación del personal La capacitación es fundamental para obtener los resultados esperados en el proyecto ya que ayuda a sensibilizar al personal sobre el riesgo físico al que están expuestos y los roles que desempeñan, tanto la empresa como cada uno de los colaboradores para minimizar el riesgo a niveles tolerables. Tabla 4. Cronograma de capacitación MESES TEMAS Nov Dic Ene Feb Efectos físicos y psicológicos del ruido en la salud Hipoacusia y trauma acústico Medidas de control de ruido implementadas por la empresa Mar X X X Protectores auditivos Exposición a ruido fuera de la jornada laboral X X La Fig. 15 muestra el promedio general de los participantes en cada curso y hace énfasis en el puntaje mínimo para aprobar cada curso. Figura 15. Comparación del promedio de calificaciones de los cursos sobre ruido del personal del Centro de Generación de Energía con la nota mínima para aprobar los cursos. 3.3.5 Implementación de protectores auditivos Debido a que el nivel de ruido en el área del generador es alto, resulta necesario que todas las personas que ingresan a esta área deben utilizar equipos de protección personal de manera obligatoria. Una vez que se decidió el tipo, se analizó la atenuación por banda de octava que ofrecen ciertos protectores auditivos que existen en el mercado, con el fin de verificar si son idóneos o no para el tipo de ruido existente en el ambiente. La implementación del plan de capacitación continua de seguridad industrial y salud ocupacional para el personal del Centro de Generación de Energía, tuvo como base evitar enfermedades profesionales a causa del ruido y las diferentes técnicas de control implementadas por parte de la empresa para brindar seguridad y enfatizar el compromiso de la empresa con los trabajadores. Para poder garantizar que el nivel de ruido emitido por el generador y que le llega al trabajador se ha visto atenuado por el protector auditivo elegido, fue indispensable realizar un análisis espectral del ruido versus el nivel de atenuación del protector. La Tabla 4 muestra el cronograma de capacitación diseñado desde noviembre hasta marzo. La Tabla 5 muestra los resultados donde se puedo evaluar la atenuación del protector auditivo por banda de octava en el área evaluada. El requisito para aprobar el plan fue alcanzar un promedio igual o mayor a 7 puntos sobre 10, caso contrario, tenían que repetir los cursos no aprobados. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 107 Implementación de Medidas de Prevención y Control de Ruido para los Trabajadores del Centro de Generación de Energía de la Empresa Dipor S.A. __________________________________________________________________________________________________________________________ Tabla 5. Análisis espectral de los protectores auditivos Peltor Optime 101 MÉTODO DE ANÁLISIS ESPECTRAL PARA SELECCIÓN DE PROTECTORES AUDITIVOS Frec (Hz) 63.5 125 250 500 1k 2k 3.1 k 4k 6.3 k 8k NPS dB(A) 61.1 75.8 94.8 101.5 103 95.8 93.0 90.2 87.0 82.1 Att orejeras (Peltor) 18.5 20.8 27.6 31.5 35.6 34.7 34.3 33.4 30.2 2 Desviació n Standard 6.0 4.0 4.8 5.6 5.2 5.6 6.4 5.4 7.8 60.3 76.0 76.3 75.2 62.8 61.1 59.1 56.3 55.8 Leq bajo el protector dB(A) 61.1 Leq Ruido emitido por el generador dB(A) Figura 17. Comparación del porcentaje de entendimiento del tipo de señal con el mínimo requerido para aceptar los resultados de la encuesta 3.4 Audiometrías 106.9 Nivel de ruido estimado bajo el 80.9 protector dB(A) 3.3.6 Señales de seguridad Una medida complementaria para el control de ruido fue la implementación de señales de seguridad, las mismas que fueron realizadas y colocadas según lo dispuesto en la norma INEN 439 que hace referencia a las señales y símbolos de seguridad. Las señales se instalaron a una altura promedio de 1.9 m y en una posición apropiada en relación al ángulo visual de los trabajadores que ingresan a las diferentes áreas. La Fig. 16 muestra el diseño implementado. Para verificar el estado de la salud auditiva de los trabajadores que realizan actividades en el Centro de Generación de Energía, se realizaron audiometrías en el mes de noviembre previo a la implementación de medidas de prevención y control de ruido. Los resultados obtenidos como los índices ELI (Early Loss Index) y SAL (Speech Average Loss), permitieron cuantificar si los trabajadores tenían o no pérdida auditiva. La Tabla 6 muestra los resultados de las audiometrías de cada uno de los trabajadores. Tabla 6. Resultados de las audiometrías de los trabajadores Trabajadores Índices 1 2 3 4 5 6 Eearly Loss Index (ELI) C Normal C Normal C Normal C Normal Speech Average Loss (SAL) B Casi A Normal Normal A Normal B Casi B Casi B Casi Normal Normal Normal Pérdida Binaural 1.60 % 0.00 % 0.00 % 0.00 % C Normal 0.00 % C Normal 0.00 % 4. CONCLUSIONES Figura 16. Pictograma de la señal de obligatoriedad utilizado Para poder evaluar y verificar el entendimiento correcto de dichas señales, se pidió, aleatoriamente, a gente de distintas áreas de la empresa que se acerquen a observar las señales y describan el significado de la señal observada y mencionen si ese tipo de señal era de obligatoriedad o advertencia. Para poder garantizar que el resultado de la encuesta es válido, al menos el 70 % de los entrevistados debían definir correctamente a la señal; es así que la Fig. 17 muestra los resultados obtenidos. Se identificó al generador de energía como la principal fuente de ruido a lo largo del Centro de Generación de Energía, el nivel de presión sonora que emite la máquina es de 106.8 dB(A). Las actividades que realizan los trabajadores dentro del área administrativa del Centro de Generación de Energía demanda cierto grado de concentración, la misma que se ve afectada por el excesivo nivel de ruido en el ambiente proveniente del generador de energía. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 108 Villacis W.*; Andrade C.* _______________________________________________________________________________________________________________________________ La medición y evaluación del nivel de ruido efectuada en cada uno de los puestos de trabajo evidenció que tanto en el área del generador como en el área administrativa del Centro de Generación de Energía no se cumple con el límite máximo permisible de 85 dB(A) y 70 dB(A) respectivamente; establecidos por el Reglamento de Seguridad y Salud de los Trabajadores y Mejoramiento del Medio Ambiente de Trabajo 2393. Una de las medidas de control implantadas para la prevención y control de ruido, que permitió minimizar el riesgo de pérdida auditiva dentro del área del generador fue la selección con criterio técnico e implementación de protectores auditivos, los mismo que permitieron reducir el nivel del ruido que le llega al trabajador hasta un valor menor a 85 dB(A), tal como lo indica el Reglamento de Seguridad y Salud de los Trabajadores y Mejoramiento del Medio Ambiente de Trabajo 2393. REFERENCIAS [1] Henao, F., Riesgos Físicos I: Ruido, Vibraciones y Presiones Anormales. Bogotá, Colombia: ECOE ediciones, 2007, p. 24, 48. [2] Cortés, J., Seguridad e Higiene del Trabajo, Madrid, España: Tébar, 2007, p. 441. [3] Harris, C., Manual de Medidas Acústicas y Control de Ruido, New York, Estados Unidos: McGraw-Hill, 1991, p. 22.2. [4] Lind, D., Marchal, W. y Mason, R., Estadística para Administración y Economía, New York, Estados Unidos: McGraw-Hill, 2004, p. 384. [5] Gobierno del Ecuador, Reglamento de Seguridad y Salud de los Trabajadores Mejoramiento del Medio Ambiente de Trabajo. Decreto Ejecutivo 2393, Quito, Ecuador: Talleres de la Corporación de Estudios, 1986, p.34. [6] Gómez, G., Manual para la Formación en Prevención de Riesgos Laborales, 2006, p. 270. [7] Quest, J., Manual de Usuario Sonómetro tipo II. New York, Estados Unidos, 2004, p. 7. Una medida de prevención complementaria fue la implementación de un plan de mantenimiento preventivo de los elementos mecánicos del generador propensos a causar ruido, con esta medida se pudo garantizar que dichos elementos van a recibir un mantenimiento mensual por parte de la empresa proveedora. Para disminuir el tiempo de exposición a ruido por parte de los trabajadores, se implementó un plan de rotación, el mismo que permitió bajar el número de horas de exposición dentro del área del generador, especialmente para el turno de la mañana. La ejecución del plan de capacitación para los trabajadores expuestos, permitió formar a los trabajadores en temas referentes al ruido y sus efectos en la salud así como exponer las medidas de prevención que implementó la empresa para salvaguardar la integridad de los trabajadores. La evaluación de la salud auditiva mediante audiometrías, tanto al inicio como luego de haber implementado las medidas de prevención y control de ruido, determinaron que no hubo cambios significativos en la audición de cada uno de los trabajadores Bajo el criterio del médico ocupacional de la empresa, se verificó que todos los empleados se encontraban aptos para trabajar en ambientes ruidosos siempre y cuando se les proteja de una manera adecuada. Debido a que el nivel de ruido en el área administrativa se encontraba fuera del límite permisible, se modeló y diseñó un acondicionamiento acústico específico para el área, el mismo que fue evaluado matemáticamente y los resultados determinaron que si se implementa el diseño, el nivel de ruido bajaría a un valor menor a 70 dB(A) en cada uno de los puestos de trabajo Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Obtención de Bioetanol Anhidro a Partir de Paja (Stipa ichu) 109 ___________________________________________________________________________________________________ Obtención de Bioetanol Anhidro a Partir de Paja (Stipa ichu) Albarracín K.*; Jaramillo L.*; Albuja M.* Escuela Politécnica Nacional, Facultad de Ingeniería Química, Laboratorio de Operaciones Unitarias Quito, Ecuador *e-mail: {katty.albarracin; lorena.jaramillo; marcelo.albuja} @epn.edu.ec Resumen: En vista de la existencia de una dependencia energética mundial hacia los combustibles fósiles se han desarrollado fuentes alternativas de energía; una de estas alternativas es el bioetanol. El bioetanol se produce a partir de varios tipos de residuos y varias materias primas renovables como plantas. La secuencia de operaciones unitarias empleadas para la obtención de bioetanol a partir de biomasa consiste en cinco etapas principales: pretratamiento, hidrólisis, fermentación, filtración o decantación y recuperación o purificación del etanol. En el presente proyecto tiene como finalidad obtener bioetanol de concentración igual al 99.6 % v/v a partir de paja Stipa ichu. Se caracterizó la materia prima obteniéndose como resultado la siguiente composición en base seca: 45.9 % celulosa, 18.2 % lignina, 5.5 % pentosanos y 5.6 % cenizas. La humedad en base seca es igual a 57.7 % y el contenido de ceras, resinas y grasas igual a 6.7 %. Para obtener un sustrato rico en azúcares, la materia prima se sometió a hidrólisis ácida, como resultado de estos ensayos se determinaron las mejores condiciones de la hidrólisis: concentración de ácido sulfúrico de 8 % w/w y tiempo de reacción de 6 horas. Estos parámetros permitieron alcanzar un rendimiento de 0.41 gramos de azúcares reductores por cada gramo de paja hidrolizada, empleando una relación sólido/líquido de 50 g de paja por cada litro de solución ácida. El producto de la hidrólisis se sometió a fermentación alcohólica, se obtuvo en rendimiento de 0.46 g etanol/g azúcares reductores. El fermentado se destiló y rectificó hasta alcanzar una concentración cercana al 85 % de etanol w/w; luego fue sometido a destilación extractiva empleando glicerina como agente de separación. Se logró obtener bioetanol anhidro en los ensayos de destilación extractiva empleando una relación molar glicerina:etanol igual a 3:1. Palabras clave: Bioetanol; biomasa; hidrólisis; fermentación; destilación extractiva. Abstract: Because of the existence of a global energy dependence on fossil fuels, alternative energy sources have been developed; one of these alternatives is bioethanol. Bioethanol is produced from various types of wastes and renewable raw materials. The sequence of unit operations used to obtain bioethanol from biomass consists of five steps: pretreatment, hydrolysis, fermentation, filtration and recovery of ethanol or purification. The present project aims to obtain bioethanol (99.6 % v/v) from straw Stipa ichu. The composition of the material was analyzed, the cellulose content was determinated to be 45.9 %; lignin 18.2 %, pentosans 5.5% and the ash to be 5.6 % of the dry weight material. Dry basis moisture is equal to 57.7 % and the content of waxes, resins and fats equal to 6.7 %. Hydrolysis of the straw was studied in order to find suitable conditions for hydrolysis of the cellulose parts. The highest yield of hydrolysis (0.41 grams of reducing sugar per gram of straw) were found at a sulfuric acid concentration of 8 % w/w and reaction time of 6 hours using a solid / liquid ratio of 50 g straw/L of acid solution. The hydrolysis product was subjected to alcoholic fermentation, the fermentation yield obtained is of 0.46 g ethanol / g reducing sugars. The fermentation product was rectified to an ethanol concentration around 85 % w/w; then was subjected to extractive distillation using glycerine as separating agent. It was possible to obtain anhydrous ethanol by distillation assays employing a molar ratio glycerol: ethanol of 3:1. Keywords: Bioethanol; biomass; hydrolysis; fermentation; extractive distillation Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 110 Albarracín K.*; Jaramillo L.*; Albuja M.* ___________________________________________________________________________________________________ 1. INTRODUCCIÓN El bioetanol puede emplearse como combustible o como aditivo en la gasolina. La producción mundial de bioetanol alcanza los 100 billones de litros anuales, convirtiéndose en el biocombustible más utilizado. Estados Unidos es el país que posee mayor producción de etanol, generando alrededor de 50 billones de litros anuales de etanol a partir de maíz; seguido por Brasil que produce 35 billones de litros de bioetanol empleando caña de azúcar como materia prima [1, 2, 11, 19]. El uso de combustibles fósiles y el aumento de las emisiones de los gases de efecto invernadero ha dado lugar a un creciente interés en la producción y uso de combustibles alternativos, lo que a nivel nacional se refleja en el Reglamento Ambiental para las Operaciones Hidrocarburíferas en Ecuador, artículo 67, que cita: “Se preferirá y fomentará la producción y uso de aditivos oxigenados, tal como el etanol carburante, a partir de materia prima renovable”. Además en el Decreto Ejecutivo No. 2332, artículo 1, se cita: “Se declara de interés nacional la producción, comercialización y uso de los biocombustibles” [7, 17]. La producción actual de bioetanol en Ecuador corresponde a 100 mil L/día, para el año 2020 se pretende alcanzar una producción de 800 millones L/año, empleando principalmente caña de azúcar como materia prima [7]. El proceso de obtención de bioetanol comprende principalmente un pretratamiento seguido por la hidrólisis de la estructura lignocelulósica de la biomasa para la obtención de azúcares fermentables, la fermentación y finalmente la destilación del producto fermentado [20]. Una mezcla típica obtenida en un proceso de fermentación alcohólica tiene alrededor de 10 % en peso de etanol y esta mezcla se puede concentrar hasta alcohol de alta pureza. El etanol forma una mezcla azeotrópica con el agua por lo que no es posible la obtención de etanol anhidro mediante procesos de destilación simple, por esta razón, se requiere la implementación de procesos de separación no convencionales como la destilación extractiva, en la que se añade un tercer componente a la mezcla con el fin de desplazar el azeótropo formado por el etanol y el agua [22]. Por otra parte, la importancia de la etapa de hidrólisis del material lignocelulósico hacia azúcares fermentables radica en que este proceso determina la eficiencia global del proceso. Para la obtención de azúcares a partir de biomasa se emplean también varios métodos enzimáticos [20]. La biomasa amilácea, como el maíz, trigo, arroz y otros cereales, contiene almidón en sus granos, tallos y raíces a partir de los que se pueden obtener azúcares. El bioetanol producido a partir de este tipo de biomasa es conocido como bioetanol de primera generación; mientras que el bioetanol de segunda generación se obtiene a partir de la estructura vegetal completa de los materiales lignocelulósicos [4]. El uso de biomasa azucarada como materia prima en la producción de bioetanol ha generado controversias respecto a su competencia con el mercado alimentario. El bioetanol producido a partir de materiales lignocelulósicos como la paja Stipa ichu se muestra como una alternativa que no afecta la disponibilidad de productos comestibles [18]. Ecuador posee 12 500 km2 de páramo. Tres cuartas partes de todos los páramos sufren algún grado de intervención humana y dentro la intervención se incluye principalmente la quema de los pajonales. Si la paja Stipa ichu se emplea como materia prima para la obtención de bioetanol, se convierte en un recurso aprovechable. El uso de este recurso es de relevancia para el cumplimiento de las metas establecidas por organismos estatales, de forma que contribuya a los objetivos presentados en el ámbito del desarrollo energético, sostenibilidad y demanda de bioetanol en el país [7]. La importancia de este trabajo radica en que se determinaron las condiciones bajo las que se puede obtener bioetanol a partir de la paja Stipa ichu, otorgándole un uso a un material lignocelulósico abundante en el país que no tiene utilidad productiva. 2. PROCEDIMIENTO 2.1 Caracterización de la Paja de Páramo (Stipa ichu) Se determinaron las siguientes características de la paja de páramo de acuerdo con los métodos estandarizados en las normas TAPPI (Technical association of the pulp and paper industry): Contenido de celulosa, lignina, pentosanos, humedad, ceras y grasas y cenizas [24]. 2.2 Evaluación del Efecto de la Concentración de Ácido y del Tiempo de Reacción en la Hidrólisis Ácida de la Paja de Páramo (Stipa ichu) 2.2.1 Hidrólisis El material vegetal seco se trituró en un molino de martillos perteneciente al Laboratorio de Operaciones Unitarias de la Escuela Politécnica Nacional. Luego se tamizó la paja triturada para obtener un producto con diámetro de partícula promedio menor a 2 mm correspondiente a la malla estándar ASTM E-1195 número 10, con el que se realizaron los ensayos de hidrólisis. El material vegetal seco se sometió a hidrólisis ácida mediante ebullición a reflujo a presión atmosférica. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 111 Obtención de Bioetanol Anhidro a Partir de Paja (Stipa ichu) ___________________________________________________________________________________________________________________________ segunda de hidróxido de sodio y tartrato doble de sodio y potasio (solución de Fehling B). La mezcla de soluciones de Fehling A y B se mantiene a ebullición, se añade como indicador azul de metileno y se titula con la muestra de hidrolizado obtenida experimentalmente que ha sido clarificada con crema de alúmina. 2.3 Estudio de la Cinética de Producción de Etanol en la Fermentación Alcohólica del Hidrolizado de Paja (Stipa ichu) 2.3.1 Fermentación Figura 1. Hidrólisis Ácida de Paja de Mediante Ebullición a Reflujo El equipo se muestra en la Fig. 1. Para evaluar la influencia de la concentración de ácido sulfúrico y del tiempo de reacción en los ensayos de hidrólisis, se experimentó con ácido sulfúrico a diferentes concentraciones: 2, 4, 6, 8 y 10 % en peso y con distintos tiempos de reacción: 2, 4, 6 y 8 horas de reacción; generando un ensayo 5x4 del que se realizaron tres repeticiones. Se definió como variable de respuesta la concentración de azúcares reductores obtenida en cada experimento. Los resultados obtenidos se examinaron mediante el análisis de varianza (ANOVA) en el programa STATGRAPHICS Centurion XVI. Para determinar diferencias estadísticamente significativas entre pares de medias se realizó una prueba de rangos múltiples LSD. La hidrólisis se realizó en un balón de vidrio de tres bocas y de 1 L de capacidad, equipado con una camisa de calentamiento, un agitador y un refrigerante para condensación de vapores como se observa en la Fig. 1. El reactor se cargó con 800 mL de solución ácida a distintas concentraciones (2, 4, 6, 8 y 10 % en peso) y se utilizó una relación de 1 g de paja/20 mL de solución. Las soluciones ácidas se prepararon con agua destilada y ácido sulfúrico grado técnico de densidad igual a 1.8063 g/mL y de pureza igual al 88 % en peso. La temperatura de ebullición en cada experimento corresponde a 92 °C y se tomaron muestras líquidas de hidrolizado a las 2, 4, 6 y 8 horas de hidrólisis. 2.2.2 Cuantificación de Azúcares Reductores Se determinó el contenido de azúcares reductores en las muestras tomadas durante el proceso de hidrólisis. Las muestras fueron clarificadas empleando crema de alúmina acorde a lo indicado en el Método Oficial 925.46 de la AOAC (Association of Official Analytical Chemists) para la “Preparación y uso de agentes clarificadores”. La determinación de azúcares reductores se realizó según el procedimiento descrito en el Método Oficial 923.09 de la AOAC [3]. Este método consiste en la valoración volumétrica de una mezcla de soluciones, la primera de sulfato cúprico (solución de Fehling A) y la Como sustrato para la fermentación se usó el hidrolizado con mayor concentración de azúcares obtenido en la hidrólisis de la paja. El sustrato se esterilizó a 121 °C durante 15 minutos en un autoclave. En la etapa de preparación del inóculo, se añadieron 5 g de levadura a 100 mL de sustrato, el inóculo permaneció bajo agitación durante 4 horas y luego se añadió a 900 mL de sustrato; la fermentación se realizó en matraces Erlenmeyer de 1 L que se sellaron con silicona para asegurar condiciones anaerobias, el CO2 producido se recogió en agua destilada. La fermentación se desarrolló a una temperatura de 30 ± 0.5 °C, sin aireación y sin agitación durante 72 horas; el sustrato se ajustó a pH 4.5 con ácido sulfúrico al 72 % en peso y amoniaco concentrado; se utilizó como microorganismo levadura comercial Saccharomyces cerevisiae [14]. 2.3.2 Cuantificación de Metanol, Etanol y Acetaldehído por Cromatografía de Gases Se tomaron asépticamente 10 mL de sustrato a las 12, 24, 36, 48, 60 y 72 horas de fermentación. Las muestras se recolectaron en frascos estériles donde se añadieron 2 mL de sorbato de potasio 0.01 M para inhibir la fermentación [16]. Las muestras se refrigeraron a 4 °C y luego se centrifugaron a 3 000 rpm durante 20 minutos en una centrífuga marca THERMO ELECTRON. Los sobrenadantes obtenidos en la centrifugación se filtraron a través de filtros CROMAFIL XTRA de tamaño de poro de 0.45 μm, para su posterior análisis por cromatografía de gases. Se empleó un cromatógrafo de gases marca AGILENT 7890 A. Se realizaron curvas de calibración con estándares de concentraciones de 1, 2, 4, 6 y 8 g/L de metanol, etanol y acetaldehído. Los reactivos utilizados en la preparación de los estándares se señalan a continuación: -Metanol, grado CROMASOLV HPLC, ALDRICH -Etanol absoluto, grado HPLC, PANREAC -Acetaldehído, 99.5 %, BHD Chemicals Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 SIGMA 112 Albarracín K.*; Jaramillo L.*; Albuja M.* ___________________________________________________________________________________________________ En la Tabla 1 se observan las condiciones de operación del cromatógrafo de gases AGILENT 7890 A. La inyección de las muestras se efectuó en una torre de inyección automática. El control del instrumento, adquisición de datos y reporte de resultados se llevaron a cabo utilizando el software ChemStation. Tabla 1. Condiciones de Operación del Cromatógrafo de Gases AGILENT 7890 A VF-WAXms 30 m, 0.25 mm, Columna 0.25 um 0.5 μL Volumen de inyección 35 ºC (4 minutos) Temperatura del horno 250 ºC Temperatura de inyección FID Detector 300 ºC Temperatura del detector Nitrógeno Gas portador 160 mL/min 1:80 Split 2.4.1 Evaluación del Efecto de la Glicerina Como Agente de Separación en la Obtención de Etanol Anhidro por Destilación Extractiva Se realizaron ensayos de destilación extractiva con relaciones molares de glicerina:etanol 85 % en peso de 3:1, 4:1 y 5:1. El etanol rectificado se sometió a destilación extractiva con glicerina como agente de separación. Se empleó el sistema indicado en la Fig. 2. El balón se cargó con 25 mL de etanol rectificado y la cantidad requerida de glicerina acorde cada relación molar. Se utilizó glicerina, grado PA, marca Panreac. En los ensayos de destilación extractiva se siguió el procedimiento empleado para la rectificación indicado anteriormente. La composición de los productos de la destilación extractiva se determinó por cromatografía de gases donde se cuantificó etanol, metanol y acetaldehído. 3. RESULTADOS 2.4 Destilación Simple y Rectificación El producto obtenido en la fermentación se sometió a destilación simple. Se realizaron destilaciones sucesivas en lotes de 1 L en un rotavapor perteneciente al Laboratorio de Investigaciones Aplicadas de la Escuela Politécnica Nacional, se recolectó el destilado mientras la temperatura de destilación se mantuvo constante. El producto de la destilación simple se rectificó en un equipo conformado por un balón con chaqueta de calentamiento, una columna de fraccionamiento tipo Vigreux marca NORMSCHIFF GLASGERATE de 24 cm de longitud y 2 cm de diámetro con una llave de tres vías acoplada en la cabeza y un refrigerante de agua; el esquema del equipo se muestra en la Fig. 2. Figura 2. Sistema de Rectificación El procedimiento empleado en la rectificación es el siguiente: Se inició el calentamiento y se ajustó la llave de tres vías de la cabeza de la columna para que todo el destilado se revierta al balón hasta que el sistema estuvo en equilibrio, dicho equilibrio se alcanzó cuando no se registraron variaciones de temperatura en la cabeza de la columna de destilación. Se recolectó el destilado mientras la temperatura de destilación se mantuvo constante. Se cuantificó metanol, etanol y acetaldehído en el producto de destilación y rectificación por cromatografía de gases. 3.1 Caracterización de la Paja (Stipa ichu) La composición de la paja Stipa ichu en base seca que se determinó experimentalmente se indica en la Tabla 2. La paja Stipa ichu posee un contenido de celulosa de 45.9 % y su contenido de lignina es de 18.2 %. Estos resultados coinciden con lo enunciado por Sims et al. [21] quienes señalan que la celulosa representa entre 40 y 50 % del peso seco del material vegetal y la lignina representa entre el 15 y 20 %. El contenido de lignina presenta una variación entre los resultados obtenidos en este trabajo (18.2 % de lignina) y los resultados de Coûteaux et al. La composición de lignina presentada por Coûteaux et al. corresponde al 6.64 %, siendo este valor menor al obtenido en este proyecto. En cuanto al contenido de cenizas, existe una diferencia entre los resultados de Coûteaux et al y los indicados en este trabajo; el contenido de cenizas señalado por Coûteaux et al. es igual a 16.1 %; este valor es mayor al contenido de cenizas encontrado en este trabajo que corresponde a 5.6 % [15]. La composición de la paja Stipa ichu no puede ser determinada con total exactitud debido a que depende de la parte de la planta que se analiza, de su localización geográfica y condiciones de crecimiento [10, 15]. La paja caracterizada por otros autores pertenece al Antiplano Bolviano, mientras que la materia prima utilizada en este proyecto corresponde a los páramos ecuatorianos; posiblemente esta es una de las razones por las que existe una variación entre los resultados encontrados en diferentes trabajos [10, 15]. Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 113 Obtención de Bioetanol Anhidro a Partir de Paja (Stipa ichu) ___________________________________________________________________________________________________________________________ 3.2 Evaluación del Efecto de la Concentración de Ácido y del Tiempo de Reacción en la Hidrólisis Ácida de la Paja de Páramo (Stipa ichu) Una vez conocido el contenido de azúcares reductores (AR) (g/L) obtenido en cada ensayo de hidrólisis, se determinó la conversión de celulosa a azúcares. En la Fig. 3 se puede observar la variación de la conversión de celulosa a azúcares reductores respecto al tiempo para cada una de las concentraciones de ácido usadas experimentalmente. Tabla 2. Composición de la Paja Stipa ichu como Porcentaje del Peso Seco Parámetro Composición promedio (%) Humedad 57.7 Celulosa 45.9 Lignina 18.2 Pentosanos 5.5 Cenizas 5.6 Resinas, ceras y grasas 6.7 Para el tratamiento con ácido al 8 % en peso con 6 horas de reacción, se obtienen alrededor de 20 g/L de azúcares reductores lo que equivale a un rendimiento de 0.41 g de Figura 3. Conversión de Celulosa a Azúcares Reductores en Función del Tiempo para Distintas Concentraciones de Ácido AR/ g de paja, la fermentación se realizó con el sustrato obtenido bajo estas condiciones ya que esta es la mejor combinación de tiempo y concentración de ácido sulfúrico. Se realizó un análisis de varianza ANOVA, para determinar la existencia de diferencias estadísticamente significativas entre los grupos de datos obtenidos experimentalmente. La hipótesis nula se planteó de la siguiente forma: el tiempo de reacción y la concentración de ácido no tienen efecto en la producción de azúcares reductores. Como resultado del análisis se obtuvieron valores P menores que 0.05 por lo que se rechaza la hipótesis nula y se acepta la hipótesis alternativa, por tanto, se dice que los valores obtenidos como resultados no son producto del azar, sino que dichos resultados se deben a que el tiempo y la concentración de ácido tienen un efecto estadísticamente significativo sobre la conversión de paja a azúcares reductores. El efecto del tiempo de reacción y la concentración de ácido se observa en el diagrama de Pareto que se muestra en la Fig. 4, donde la longitud de las barras es proporcional al efecto de cada factor sobre la variable de respuesta. La barra de mayor longitud corresponde a la concentración de ácido, por tanto este es el factor que tiene mayor influencia en la conversión, obteniéndose mayores conversiones con el aumento de dicha concentración del ácido. El tiempo de reacción también presenta un efecto significativo sobre la variable de respuesta, de la misma manera, la conversión aumenta con el tiempo de reacción. Las mejores condiciones de hidrólisis ácida obtenidas experimentalmente se resumen en la Tabla 3 indicada a continuación. Existen estudios de hidrólisis de materias primas similares a la paja Stipa ichu, por ejemplo, para la paja de arroz se ha conseguido el mejor rendimiento en la obtención de azúcares con hidrólisis a una temperatura de 100 °C, 10 % H2SO4 y 4 horas de reacción; estos resultados se asemejan a las mejores condiciones de hidrólisis encontradas en este trabajo [13]. 3.3 Estudio de la Cinética de Producción de Etanol en la Fermentación Alcohólica del Hidrolizado de Paja (Stipa ichu) En los análisis por cromatografía de gases no se identificaron metanol ni acetaldehído como subproductos aunque en ensayos de fermentación donde se emplean sustratos azucarados provenientes de papel se ha Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 114 Albarracín K.*; Jaramillo L.*; Albuja M.* ___________________________________________________________________________________________________ obtenido alrededor de 0.20 g/L de metanol y 1.23 g/L de acetaldehído [5]. En la Figura 5 se observa la producción Diagrama de Pareto Estandarizada CONVERSIÓN durante la promedio de etanol respecto al paratiempo fermentación alcohólica del hidrolizado de paja Stipa ichu + - B:CONCENTRACIÓN DE ÁCIDO Tabla 3. Mejores Condiciones de Hidrólisis Ácida de Paja Stipa ichu Parámetro Valor Unidad 92 (°C) Temperatura de reacción 6 (Horas) Tiempo de reacción Relación sólido / Líquido Concentración de ácido sulfúrico A:TIEMPO DE REACCIÓN ( 8 (% en peso) 0.41 Rendimiento g paja 50 ) L solución ácida ( g AR g paja ) AB 0 2 4 6 8 10 12 EFECTO ESTANDARIZADO Figura 4. Diagrama de Pareto Figura 5. Producción Promedio de Etanol Durante la Fermentación Alcohólica del Hidrolizado de Paja Stipa ichu (20 g/L de Azúcares Reductores) En el gráfico se puede identificar una disminución en la concentración de etanol entre las 48 y 72 horas de fermentación. Esta disminución pudo ser causada por una ligera volatilización de etanol ocurrida a la temperatura de 30 °C bajo la que se desarrolló la fermentación. En la Figura 5 también se puede observar que la producción de etanol se detiene a las 48 horas de fermentación. La finalización de la producción de etanol se debe a que las levaduras consumieron los azúcares presentes en el sustrato y no por un efecto inhibitorio del etanol sobre la fermentación; ya que la inhibición de la fermentación alcohólica por etanol se produce con concentraciones de alcohol etílico de alrededor de 55 g/L [8]. Los resultados del rendimiento de azúcares reductores a etanol se indican en la Tabla 4, en esta tabla se observan también los valores de rendimiento de paja a etanol. El máximo rendimiento obtenido experimentalmente es igual 0.46 g etanol / g azúcares reductores, lo que corresponde al 89.78 % del máximo rendimiento teórico, por lo que se ratifica que los resultados obtenidos son coherentes con lo estipulado en bibliografía y se encuentran en el rango esperado. El máximo rendimiento alcanzado es igual a 0,18 g etanol /g paja hidrolizada, como se observa en la Tabla 4. Tabla 4. Rendimientos del Proceso de Fermentación Tiempo de fermentación (horas) Rendimiento promedio (g etanol/g azúcares reductores) Eficiencia (%) Rendimiento promedio (g etanol/g paja) 12 24 36 48 60 72 0.13 0.20 0.35 0.46 0.46 0.45 26.24 40.00 68.48 89.78 89.78 87.51 0.05 0.08 0.14 0.18 0.18 0.18 Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 115 Obtención de Bioetanol Anhidro a Partir de Paja (Stipa ichu) ___________________________________________________________________________________________________________________________ 3.3.1 Productos de la Destilación Los resultados de la cuantificación de metanol, etanol y acetaldehído por cromatografía de gases en los productos de la destilación de fermentado alcohólico y de la rectificación se indican en la Tabla 5 aquí también se presenta la cantidad de producto obtenido en cada ensayo de destilación. Tabla 5. Resultados de la Cuantificación de Etanol por Cromatografía de Gases para el Destilado Ensayo Concentración de etanol (g/L) % Etanol (w/w) % Etanol (v/v) Destilación Rectificación 473.22 707.50 51.78 84.59 59.63 89.15 3.4 Evaluación del Efecto de la Glicerina Como Agente de Separación en la Obtención de Etanol Anhidro por Destilación Extractiva En la Tabla 6 se señalan los resultados del análisis de cromatografía de los productos de la destilación extractiva. Tabla 6. Contenido de Etanol en los Productos de Destilación Extractiva para Diferentes Relaciones Molares Etanol:Glicerina Relación molar Contenido de etanol promedio (g/L) % Etanol w/w (promedio) % Etanol v/v (promedio) 3:1 4:1 5:1 790.55 ± 1.55 721.16 ± 4.13 535.90 ± 1.69 99.40 86.81 59.84 99.63 90.87 67.54 Como se observa en la Tabla 6, en este trabajo se logra obtener bioetanol de una concentración igual a 99.63 % en volumen como producto de la destilación extractiva que se realizó con una relación molar de glicerina:etanol igual a 3:1 [9, 12]. El etanol y la gasolina son miscibles entre sí, pero la presencia de pequeñas cantidades de agua en el etanol promueven la separación de fases lo que puede provocar un rendimiento deficiente del vehículo y dañar el motor, por esta razón el etanol combustible debe tener una pureza mínima de 99.6 % en volumen de acuerdo a lo requerido en la Norma Técnica Ecuatoriana INEN 2478 [9, 19]. En la Fig. 6 se observa la concentración de etanol alcanzada en los experimentos de destilación extractiva con relaciones molares de glicerina:etanol de 3:1, 4:1 y 5:1; la composición de etanol en el producto de la destilación extractiva se representa con las barras del gráfico, mientras que la composición del azeótropo es indicada por una línea horizontal. Se consigue una concentración de etanol superior a la del azeótropo solamente con una relación molar de glicerina:etanol igual a 3:1. Mediante este resultado se puede considerar que la glicerina es un agente útil para la ruptura del azeótropo formado por el etanol y el agua, además de ser una alternativa económica frente a otros agentes de mayor costo comercial como el etilén glicol y las sales iónicas [23]. Navarrete et al. [23] deshidrataron etanol por destilación extractiva utilizando glicerina como agente de separación; en este estudio se emplearon relaciones molares de glicerina:etanol de 0.4:1 y 0.9:1 y se alcanzaron composiciones de destilado mayores a 98 % en peso de etanol para ambas proporciones, las cantidades de glicerina empleadas por Navarrete et al. son evidentemente menores a las empleadas en este trabajo; cabe aclarar que durante esta investigación se emplearon relaciones de glicerina:etanol mayores a 3:1 debido a que según Treybal la relación entre el agente de separación y la alimentación debe ser mayor a 3:1, de esta forma es posible obtener separaciones efectivas que no se vean afectadas por las pérdidas del agente de separación durante su recirculación al proceso [22]. En la Tabla 7 se indica el volumen de producto obtenido en cada ensayo de destilación extractiva siendo evidente que a medida que aumenta la relación molar glicerina:etanol disminuye la cantidad de producto obtenido. Respecto al tiempo de calentamiento, conforme se aumenta la cantidad de glicerina alimentada a la destilación existe un ligero incremento en el tiempo de destilación lo que era deesperarse, ya que se requiere aumentar la temperatura de una mayor cantidad de mezcla de glicerina y etanol. Los procesos de obtención de biocombustibles generan grandes cantidades de glicerina como subproducto que pueden constituirse como materia prima valiosa para la industria. Actualmente se encuentran en estudio varios procesos para la obtención de biocombustibles que emplean glicerina como materia prima, uno de estas técnicas consiste en la producción de bioetanol por fermentación del glicerol [6]. Pese a esto, en este trabajo se propone emplear glicerina como agente de separación en la destilación extractiva por ser una sustancia accesible respecto a su costo, no tóxica ni corrosiva y estable químicamente; esto permite sureutilización: una vez empleada en el proceso de destilación extractiva es retirada como un producto de fondo y se envía a una columna de regeneración para luego ser recirculada al proceso [22]. Entre las diferentes alternativas para la obtención de biocombustibles, se considera que los procesos de menor coste e impacto ambiental serán los que tengan preferencia en el mercado. 4. CONCLUSIONES La paja Stipa ichu tiene un contenido en base seca de 45.9 % celulosa, 18.2 % lignina, 5.5 % pentosanos y 5.6 % cenizas. Su base en base seca es igual a 57.7 % Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 116 Albarracín K.*; Jaramillo L.**; Albuja M.*** _______________________________________________________________________________________________________________________________ Figura 6. Concentración de Etanol (% w/w) en el Producto de la Destilación Extractiva para cada Relación Molar Glicerina:Etanol Tabla 7. Volúmenes de Productos, Tiempos y Temperaturas de Destilación Extractiva Relación molar glicerina:etanol Volumen producto (mL) Temperatura de destilación (°C) Tiempo de destilación (min) 3:1 4:1 5:1 21 19 18 70 70 70 20 23 25 Es posible obtener bioetanol anhidro mediante destilación extractiva empleando una relación molar glicerina:etanol igual a 3:1. En los ensayos experimentales de destilación extractiva donde se empleó relaciones molares de glicerina:etanol de 4:1 y 5:1 no se consiguió desplazar el azeótropo formado por el etanol y el agua. AGRADECIMIENTOS y su contenido de ceras, resinas y grasas igual a 6.7 %. Las mejores condiciones de hidrólisis ácida de la paja Stipa ichu corresponden a una concentración de ácido sulfúrico de 8 % en peso y tiempo de reacción de 6 horas. Bajo estos parámetros se obtiene un rendimiento de 0.41 gramos de azúcares reductores por cada gramo de paja hidrolizada cuando el proceso de hidrólisis se realiza bajo una relación sólido/líquido de 50 g de paja por litro de solución ácida. El rendimiento del proceso de hidrólisis ácida si se ve influenciado por la concentración del ácido, el tiempo de reacción y la interacción entre ambas variables. No existe diferencia representativa entre los resultados obtenidos al hidrolizar paja Stipa ichu con ácido sulfúrico de concentración igual a 4 y 6 % en peso. Tampoco existe una diferencia entre los resultados obtenidos en la hidrólisis con ácido sulfúrico de concentración igual a 8 y 10 % en peso. El máximo rendimiento obtenido experimentalmente en los ensayos de fermentación alcohólica es igual a 0.46 g etanol/g azúcares reductores, lo que corresponde al 89.78 % del máximo rendimiento teórico. Este resultado se obtiene al fermentar el hidrolizado de paja con un pH de 4.5 y bajo una temperatura de 30 °C. El rendimiento de paja Stipa ichu a etanol es igual a 0.18 g etanol/g paja hidrolizada. Los autores dejan constancia de su agradecimiento a la Escuela Politécnica Nacional y a la Facultad de Ingeniería Química y Agroindustria por el apoyo brindado a este trabajo de investigación y al personal del Departamento de Ingeniería Química por su colaboración profesional. REFERENCIAS [1] A. Demirbas. (2005). Bioethanol from Cellulosic Materials: A Renewable Motor Fuel from Biomass. Energy Sources, 27(1), pp. 327, Available: doi 10.1080/00908310390266643 [2] A. Faik, “Plant Cell Wall Structure-Pretreatment the Critical Relationship in Biomass Conversion to Fermentable Sugars”, en Green Biomass Pretreatment for Biofuels Production, G. Tingyue, Ed. Ohio:Athens, 2013, pp. 2. [3] AOAC INTERNATIONAL, Official Methods of Analysis, 18va. ed., Maryland:AOAC International, 2005, pp. 9-4. [4] D. Rutz y R. Janssen, BioFuel Technology Handbook, 1ra. edición, München:WIP Renewable Energies, 2013, pp. 40. [5] E. Palacios, “Obtención de etanol a partir de papel bond de desecho”, Tesis previa a la obtención del título de Ingeniero Químico, Escuela Politécnica Nacional, Quito, Ecuador, p. 65. [6] F. Bauer y C. Hulteberg. (2013, Nov.). Is there a future in glycerol as a feedstock in the production of biofuels and biochemicals?. Biofuels, Bioprod,. Bioref., 7(2013), pp. 43-51. Available: doi: 10.1002/bbb.l370 [7] F. Figueroa. (2008). "Tablero de comando para la promoción de los biocombustibles en Ecuador”, Available: http://www.eclac.org/publicacion es /xml/9/33219/lcw189e.pdf [8] G. Moulin, H. Boze y P. Galzy. (1984). Inhibition of alcoholic fermentation. Biotechnology and Genetic Engineering Reviews, 2(Octubre), pp. 365. Available: http://www.nottingham.ac.uk/ncmh/BGER/pdf/Volume%20 2/B GER2pdf (Agosto, 2014). Revista Politécnica - Septiembre 2015, Vol. 36, No. 2 Obtención de Bioetanol a Partir de Paja (Stipa ichu) _________________________________________________________________________________________________________________________ [9] Instituto Ecuatoriano de Normalización. (2009). “Etanol anhidro. Requisitos”, Available: https://law.resource.org/pub/ec/ibr/ec.ne.2478.2009.pdf [10] J. Brigham, W. Adney y M. Himme, “Hemicellulases: diversity and applications”, en Wyman, C.E., Handbook on Bioethanol: Production and utilization, Washington DC:Taylor & Francis, 1989, pp. 119. [11] J. Sánchez y C. Cardona. (2008). Trends in biotechnological of fuel ethanol from different feedstocks. Bioresource Technology. 99(13). pp. 5270. Available: doi: 10.1016/j.bi ort och. 2007.11.013 [12] . Saucedo, A. Castro, M. Martínez y J. Campos. (2010). Diseño de un bioproceso para la obtención de etanol carburante a partir de bagazo del agave tequilana weber. Ciencia Nicolaita, Especial, 1, Available: http://www.cic.umich.mx/documento/ciencia_nicolaita/2010/especial/A 25.pdf [13] K. Karimi, S. Kheradmandinia y M. Taherzadehb. (2006). Conversion of rice straw to sugars by dilute-acid hydrolysis. BIOMASS AND BIOENERGY, 30(3), pp. 247, Available: doi: 0.1016/j.biombioe.2005.11.015 [14] L. Serna, D. Mera, J. López y A. Gómez. (2011). “Cinética de fermentación alcohólica utilizando como fuente de nitrógeno harina de semilla de guayaba Psidium guajava”, Ponencia presentada en V Congreso Internacional de Biofáfricas, Palmira, Colombia. Available: http://www.unalmed.edu.co/biofab/memorias/Ponencias/Bioetanol_lign ocelulosicos/Cinetica_fermentacion_alcoholica.pdf [15] M. Coûteaux, D. Hervé y S. Beck. (2006). Descomposición de hojarasca y raíces en un sistema de descanso largo (Altiplano de Bolivia). Ecología en Bolivia, 41(3), pp. 85, Available: http://www.scielo.org.bo/ pdf/reb/v41n3/v41n3a07.pdf [16] M. Dharmadhikari. (1992). Sorbic Acid. Missouri State Fruit Experiment Station, 7(6), p. 1, Available: https://www.extension.iastate.edu/NR/rdonlyres/173729E4-C734-486AAD16-78678B3E1CF/73968/Sorbic Acid1.pdf [17] M. Mora, M. Albuja, O. Proaño. (2014, Ene.). Evaluación de la pirólisis térmica de aceite vegetal de desecho en un reactor batch. Revista Politécnica.[Online].33(2),pp. 1-15. Available: http://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd= 1&cad=rja&uact=8&ved=0CB4QFjAA&url=http%3A%2F%2Fwww.re vistapolitecnica.epn.edu.ec%2Fojs2%2Findex.php%2Frevista_politecni ca2%2Farticle%2Fdownload%2F137%2Fpdf&ei=6l2PVfmVJYzbggSw xIGgAg&usg=AFQjCNF80eMAIj_vazcG86tdpckzjswwWg&bvm=bv.9 6783405,d.eXY [18] M. Tomás. (2010). “Bioetanol de paja de trigo: estrategias de integración de las etapas del Proceso”, Memoria para optar al grado de doctor, Recuperado de la base de datos E-Prints Complutense. (Código ID 10802). [19] M. Walker. (2010). “Bioethanol: Science and Technology of fuel alcohol”. Available: http://bookboon.com/es/bioethanol-science-andtechnology-of-fuel-alcohol-ebook [20] P. Binod, K. Janu, R. Sindhu, y A. Pandey, “Hydrolysis of Lignocellulosic Biomass for Bioethanol Production”, en Biofuels: alternative feedstocks and conversion processes, A. Pandey, C. Larroche, S. Ricke, C.O Dussap y E. Gnansounou, Ed. Burlington: Academic Press, 2011, pp. 229. [21] R. Sims y M. Taylor, International Energy Agency, J. Saddler y E. Mabee. From 1st. To 2nd. Generation Biofuel Technology. 1ra. edición, París:Editorial OECD/IEA, París, München, 2008, pp. 35. [22] R. Treybal, Operaciones de transferencia de masa. México, México: Mc. Graw-Hil, 1988, pp. 509-510. [23] S. Navarrete, M. Sánchez, F. Barroso, S. Hernández y A. Castro. (2014). Use of glicerol as entrainer in the dehydration of bioetanol using extractive batch distillation: Simulation and experimental studies. Chemical Engineering and Processing: Process Intensification, 77(Marzo), pp. 38. Available: doi:10.1016/j.cep.2014.01.003 [24] Technical association of the pulp and paper industry. (2013). “Fibrous materials and pulp testing”, Available: http://www.tappi.org/content/pdf/stan dards/ numeric_index_tms.pdf Revista Politécnica-Septiembre 2015, Vol. 36, No. 2 117 Preparación de Artículos para la Revista Politécnica _________________________________________________________________________________________________________________________ Preparación de Artículos para la Revista Politécnica Utilizar Mayúsculas en Cada Palabra en el Caso del Título Apellido A.*; Apellido B.**; Apellido C.**;Apellido D.***; Apellido E.*** *Escuela Politécnica Nacional, Facultad de Ingeniería Mecatrónica, Quito, Ecuador e-mail: [email protected] ** Escuela Politécnica del Litoral, Facultad de Ingeniería Industrial, Guayaquil, Ecuador e-mail: {autorB; authorC}@espol.edu.ec *** Universidad de Cuenca, Facultad de Ciencias Exactas, Cuenca, Ecuador e-mail: [email protected]; [email protected] Resumen: Las siguientes instrucciones establecen las pautas para la preparación de artículos para la Revista Politécnica. Los artículos pueden ser escritos en español o en inglés, pero tendrán un resumen en ambos idiomas. Los autores pueden hacer uso de este documento como una plantilla para componer su artículo si están utilizando Microsoft Word 6.0 o superior. Caso contrario, este documento puede ser utilizado como una guía de instrucciones. El número máximo de páginas será 10. La versión completa del formato así como para el envío de los artículos, los autores deben ingresar al sitio web de la Revista Politécnica (www.revistapolitecnica.epn.edu.ec). Palabras clave: Incluir una lista de 5-10 palabras clave. Abstract: These instructions give you guidelines for preparing papers for EPN Journal. Papers can be written in Spanish or English; however, an abstract in both languages is required. Use this document as a template to compose your paper if you are using Microsoft Word 6.0 or later. Otherwise, use this document as an instruction set. The maximum number of pages will be 10. For submission guidelines, follow instructions on paper submission system from the EPN Journal website(www.revistapolitecnica.epn.edu.ec). Keywords: Include a list of 5-10 keywords. Artículo recibido el XX, 2013; revisado XX julio de 2013. (Escriba la fecha en que presentó su documento para su revisión). Esta sección puede ser utilizada para colocar información adicional de los autores. Esta 99.9 98 Weibull Breakdown Probability (%) 11. INTRODUCCION Este documento es una plantilla para versiones Microsoft Word 6.0 o posteriores. Si está leyendo una versión impresa de este documento, por favor descargue el archivo electrónico, revistapolitécnicaplantilla.doc. Por favor, no coloque ningún número consecutivo, encabezado / pie de páginaen el documento presentado. Puede escribir sobre las secciones de la revistapolitécnicaplantilla.doc o cortar y pegar de otro documento y, luego usar estilos de marcado. El menú desplegable de estilo está a la izquierda de la barra de herramientas de formato, en la parte superior de la ventana de Word (por ejemplo, el estilo en este punto del documento es "Texto"). Resalte una sección que usted quiera designar con un cierto estilo, y luego seleccione el nombre apropiado en el menú de estilo. El estilo ajustará los tipos de letra y espaciado de línea. No cambie los tamaños de fuente o espaciado de renglones para ajustar el texto a un número limitado de páginas. Utilice cursiva o negrita para dar énfasis a un texto, no subrayado. 90 70 50 30 20 10 5 2 1 0.5 0.2 0.1 100 101 102 Breakdown Voltage (kV) Figura 1. Distribución Weibull de 60 Hz voltajes de ruptura11 cables α = 45.9 kV picoβ = 5.08.Intervalo de Confidencia 95% Las figuras y tablas deben estar en la parte superior e inferior de las columnas. Evite colocarlas en medio de las columnas. Las figuras y tablas grandes pueden extenderse a lo largo de ambas columnas. Las leyendas de las figuras deben estar centradas debajo de las figuras, los títulos de las tablas deben estar centrados arriba. Evite colocar figuras y tablas antes de su primera mención en el texto. Use la abreviación "Fig. 1", incluso al principio de una frase. Revista Politécnica – Septiembre 2015, Vol. 36, No. 2 Preparación de Artículos para la Revista Politécnica _________________________________________________________________________________________________________________________ 1.2 Acerca de los Autores Formato básico para libros: Como sugerencia, es importante tomar en cuenta que, el primer autor es el investigador que hizo la mayor parte del trabajo, y probablemente va a colocar esto en su tesis y destacarlo en su CV, mientras que el último autor suele ser el profesor quien es el líder intelectual y, a menudo edita y presenta el borrador final del documento. [1] W.K.Chen,LinearNetworksandSystems.Belmont, 1993, pp. 123–135. [2] G.O.Young,―Syntheticstructureofindustrial plastics,‖in Plastics, 2nd ed., vol. 3, J. Peters, Ed. New York: McGraw-Hill,1964,pp.15–64. Formato básico para periódicos: [3] J. U. Duncombe, ―Infrared navigation—Part I: An assessment of feasibility,‖ IEEE Trans. Electron Devices, vol. ED-11, no. 1, pp. 34–39, Jan. 1959. [4] E. H. Miller, ―A note on reflector arrays,‖ IEEE Trans. Antennas Propagat., to be published. E. P. Wigner, ―Theory of traveling-wave optical laser,‖Phys. Rev., vol. 134, pp. A635–A646, Dec. 1965. 2. PROCEDIMIENTO PARA LA PRESENTACIÓN DEL ARTÍCULO 2.1Etapa de Revisión Por favor, utilice este documento como una "plantilla" para preparar el manuscrito. Para las pautas de presentación, siga las instrucciones emitidas por el sistema del sitio web de la revista de la EPN. La presentación inicial debe tomar en cuenta todas las indicaciones que se presentan en esta plantilla, para de esta manera tener una buena estimación de la longitud del artículo a publicarse. Además, de esta manera el esfuerzo necesario para la presentación final del manuscrito será mínimo. CA:Wadsworth, [5] Formato básico para manuales: [6] Motorola Semiconductor Data Manual, Motorola Semiconductor Products Inc., Phoenix, AZ, 1989. [7] Transmission Systems for Communications, 3rd ed., Western Electric Co., Winston-Salem, NC, 1985, pp. 44–60. Formato básico para libros (cuando están habilitados en línea): 2.2 Etapa Final [8] Los autores deben trabajar activamente con los márgenes solicitados. Los documentos de la revista serán marcados con los datos del registro de la revista y paginados para su inclusión en la edición final. Si la sangría de los márgenes en su manuscrito no es correcta, se le pedirá que lo vuelva a presentar y esto, podría retrasar la preparación final durante el proceso de edición. J. Jones.(1991, May Available:http://www.atm.com 10). Networks.(2ndEd.)[Online]. Formato básico para revistas (cuando estén habilitadas en línea): [9] R. J. Vidmar. (1992, Aug.). On the use of atmospheric plasmasaselectromagneticreflectors. IEEETrans. PlasmaSci.[Online].21(3),pp. 876–880. Available: http://www.halcyon.com/pub/journals/21ps03-vidmar Formato básico para artículos presentados en conferencias (cuando estén habilitadas en línea): 3. CONCLUSIONES Una sección de conclusiones es requerida. Aunque una conclusión puede repasar los puntos principales del documento, no repita lo escrito en el resumen como conclusión. Una conclusión podría extender la importancia del trabajo o podría hacer pensar en aplicaciones y extensiones futuras. REFERENCIAS La lista de referencias deben estar ordenadas alfabéticamente de acuerdo con el primer autor de la lista de referencia, las siguiente líneas deben tener sangría. No debe existir números de referencia de la publicación por los mismos autores, deben estar listadas en orden del año de la publicación si hay más de un artículo del mismo autor(es) y con la misma fecha se organiza a,b, etc., por ejemplo Morris et al. (1990a, b). Por favor nótese que todas las referencias listadas aquí deben estar directamente citadas en el cuerpo del texto usando []. [10] PROCESS Corp., MA. Intranets: Internet deployedbehindthefirewall forcorporateproductivity. INET96AnnualMeeting.[Online]. http://home.process.com/Intranets/wp2.htp technologies Presentedat Available: Formato básico para las actas de congresos (publicadas): [11] D. B. Payne and J. R. Stern, ―Wavelength-switched passivelycoupledsingle-mode opticalnetwork,‖in Proc. IOOCECOC,1985, pp.585–590. Basic format for theses (M.S.) and dissertations (Ph.D.): [12] G. Brandli and M. Dick, ―Alternatingcurrent fed power supply,‖ U.S.Patent 4 084 217,Nov.4,1978. Formato básico de las tesis (MS) y disertaciones (Ph.D.): [13] N. Kawasaki, ―Parametric study of thermal and chemical nonequilibrium nozzle flow,‖ M.S. thesis, Dept. Electron. Eng., Osaka Univ., Osaka, Japan, 1993. [14] J. O. Williams, ―Narrow-band analyzer,‖ Ph.D. dissertation, Dept. Elect. Eng., Harvard Univ., Cambridge, MA, 1993. Formato básico para los tipos más comunes de referencias no publicadas: Revista Politécnica – Septiembre 2015, Vol. 36, No. 1 Preparación de Artículos para la Revista Politécnica _________________________________________________________________________________________________________________________ [15] A. Brahms, ―Representation error for real numbers in binary computer arithmetic,‖ IEEE Computer Group Repository, Paper R-67-85. [18] IEEE Criteria for Class IE Electric Systems, IEEE Standard 308, 1969. [19] Letter Symbols for Quantities, ANSI Standard Y10.5-1968. [16] A. Harrison, private communication, May 1995. [17] B. Smith, ―An approach to graphs of linear forms,‖ unpublished. Formato básico de las normas: Apéndice A. PRIMER APÉNDICE Apéndice B. SEGUNDO APÉNDICE INFORMACIÓN ADICIONAL Fecha Límite de Recepción de Artículos: Diciembre 2015 Fecha Límite de Aceptación de Originales: Enero 2016 Sistema de Arbitraje: Todos los artículos cumple con una revisión por pares, la cual consiste en: Selección de dos o tres árbitros, actualmente la Revista Politécnica cuenta con revisores internos, externos e internacionales, quienes envían al editor su evaluación del artículo y sus sugerencias acerca de cómo mejorarlo. El editor reúne los comentarios y los envía al autor Con base en los comentarios de los árbitros, el editor decide si se publica el manuscrito. Cuando un artículo recibe al mismo tiempo evaluaciones tanto muy positivas como muy negativas, para romper un empate, el editor puede solicitar evaluaciones adicionales, obviamente a otros árbitros. Otra manera de desempate consiste en que los editores soliciten a los autores que respondan a las críticas de los árbitros, a fin de refutar una mala evaluación. En esos casos el editor generalmente solicita al árbitro que comente la respuesta del autor. Toda la evaluación se realiza en un proceso ciego, es decir los autores no conocen quienes son sus revisores, ni los revisores conocen los autores del artículo. Instructivo para publicar un Artículo 1. 2. 3. 4. Solicitar usuario y contraseña para acceder al portal web de la Revista Politécnica al correo [email protected] Ingresar al portal web e iniciar el proceso de envío Comenzar el envío Colocar requisitos de envío Lista de comprobación de preparación de envíos Como parte del proceso de envío, se les requiere a los autores que indiquen que su envío cumpla con todos los siguientes elementos, y que acepten que envíos que no cumplan con estas indicaciones pueden ser devueltos al autor. - La petición no ha sido publicada previamente, ni se ha presentado a otra revista (o se ha proporcionado una explicación en Comentarios al Editor). El fichero enviado está en formato OpenOffice, Microsoft Word, RTF, o WordPerfect. Se han añadido direcciones web para las referencias donde ha sido posible. El texto tiene interlineado simple; el tamaño de fuente es 10 puntos; se usa cursiva en vez de subrayado (exceptuando las direcciones URL); y todas las ilustraciones, figuras y tablas están dentro del texto en el sitio que les corresponde y no al final del todo. El texto cumple con los requisitos bibliográficos y de estilo indicados en las Normas para autoras/es, que se pueden encontrar en "Acerca de la Revista". Nota de copyright Los autores que publican en esta revista están de acuerdo con los siguientes términos: Revista Politécnica – Septiembre 2015, Vol. 36, No. 1 Preparación de Artículos para la Revista Politécnica _________________________________________________________________________________________________________________________ - Los autores conservan los derechos de autor y garantizan a la revista el derecho de ser la primera publicación del trabajo al igual que licenciado bajo una Creative Commons Attribution License que permite a otros compartir el trabajo con un reconocimiento de la autoría del trabajo y la publicación inicial en esta revista. - Los autores pueden establecer por separado acuerdos adicionales para la distribución no exclusiva de la versión de la obra publicada en la revista (por ejemplo, situarlo en un repositorio institucional o publicarlo en un libro), con un reconocimiento de su publicación inicial en esta revista. - Se permite y se anima a los autores a difundir sus trabajos electrónicamente (por ejemplo, en repositorios institucionales o en su propio sitio web) antes y durante el proceso de envío, ya que puede dar lugar a intercambios productivos, así como a una citación más temprana y mayor de los trabajos publicados (Véase The Effect of Open Access) (en inglés). Declaración de privacidad - 5. 6. 7. 8. Los nombres y direcciones de correo-e introducidos en esta revista se usarán exclusivamente para los fines declarados por esta revista y no estarán disponibles para ningún otro propósito u otra persona. Subir el envío Introducir metadatos Subir ficheros adicionales Confirmar el envío Revista Politécnica – Septiembre 2015, Vol. 36, No. 1
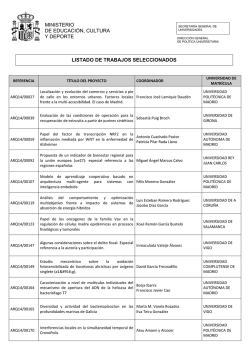

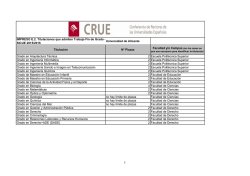

© Copyright 2026